

Significance
Apolipoprotein E−/− (ApoE−/−) mice deficient in nucleotide binding oligomerization domain-containing protein 2 (NOD2) and subjected to an oral gavage of Porphyromonas gingivalis developed elevated serum inflammatory cytokines, cholesterol, alveolar bone loss, and atherosclerosis. Stimulation of NOD2 by Muramyl DiPeptide (MDP) in ApoE−/− mice reduced P. gingivalis-induced inflammatory cytokines, cholesterol, alveolar bone loss, and atherosclerosis by reducing the expression of inhibitor of NF-κB kinase-β, NF-κB, JNK mRNA, and TNF-α protein levels. A reduction in body weight gain was observed in ApoE−/− mice fed a high-fat diet (HFD) and injected with MDP compared to ApoE−/− mice fed a HFD but saline injected. MDP activation of NOD2 should be considered in the treatment of inflammatory processes affecting atherosclerosis, bone loss, and possibly, weight gain.
Keywords: TLRs, animal models
Abstract
The purpose of this study was to elucidate the role of nucleotide binding oligomerization domain-containing protein 2 (NOD2) signaling in atherosclerosis and periodontal bone loss using an Apolipoprotein E−/− (ApoE−/−) mouse model based on the proposed role of NOD2 in inflammation. NOD2−/−ApoE−/− and ApoE−/− mice fed a standard chow diet were given an oral gavage of Porphyromonas gingivalis for 15 wk. NOD2−/−ApoE−/− mice exhibited significant increases in inflammatory cytokines, alveolar bone loss, cholesterol, and atherosclerotic lesions in the aorta and the heart compared with ApoE−/− mice. In contrast, ApoE−/− mice injected i.p. with Muramyl DiPeptide (MDP) to stimulate NOD2 and given an oral gavage of P. gingivalis displayed a reduction of serum inflammatory cytokines, alveolar bone loss, cholesterol, and atherosclerotic lesions in the aorta and aortic sinus compared with ApoE−/− mice orally challenged but injected with saline. A reduction in body weight gain was observed in ApoE−/− mice fed a high-fat diet (HFD) and injected with MDP compared with ApoE−/− mice fed a high-fat diet but injected with saline. MDP treatment of bone marrow-derived macrophages incubated with P. gingivalis increased mRNA expressions of NOD2, Toll-like receptor 2, myeloid differentiation primary response gene 88, and receptor-interacting protein-2 but reduced the expressions of inhibitor of NF-κB kinase-β, NF-κB, c-Jun N-terminal kinase 3, and TNF-α protein levels compared with saline control, highlighting pathways involved in MDP antiinflammatory effects. MDP activation of NOD2 should be considered in the treatment of inflammatory processes affecting atherosclerosis, periodontal bone loss ,and possibly, diet-induced weight gain.
The nucleotide binding and oligomerization domain 2 protein (NOD2) is an intracellular protein containing leucine-rich repeats similar to the repeats found in Toll-like receptors (TLRs) that are capable of sensing bacteria-derived muramyl dipeptide (MDP), and it was initially described as a susceptibility gene for Crohn disease and intestinal inflammatory diseases (1–3). NOD2 is expressed in various cell subsets, including myeloid cells (particularly macrophages, neutrophils, and dendritic cells), as well as Paneth cells in the small intestine (4), and it was found to process inflammatory signals (5). Immune cells express receptors that recognize a broad range of molecular patterns foreign to the mammalian host but commonly found on pathogens. These molecules trigger immune responses through interactions with members of the toll-like receptor family (TLRs) at the cell membrane and NACHT, neuronal apoptosis inhibitor protein (NAIP), CIITA, HET-E and TP-1 domain–Leucine-rich repeat (LRR) proteins (NLRs) in the cytosol (6, 7). Cells expressing NOD2 can activate NF-κB after intracellular recognition of MDP (8, 9). The recognition of MDP is mediated through the LRR domain of NOD2, leading to downstream signaling through interaction between the caspase recruitment domain (CARD) of Receptor-interacting serine/threonine-protein kinase 2 (RIP2) and the CARDs of NOD2. In vitro, NOD2 has been found to be involved in bacterial clearance (10). NOD2-deficient mice display increased susceptibility to Staphylococcus aureus because of, in part, defective neutrophil phagocytosis, elevated serum levels of Th1 cytokines, and a higher bacterial tissue burden (11). However, stimulation of NOD2 with MDP was found to enhance host antibacterial function in vitro (12).
Atherosclerosis is a chronic inflammatory condition that can lead to an acute clinical event by plaque rupture and thrombosis. It is a multifactorial disease characterized by the accumulation of cells from both the innate and acquired immune system within the intima of the arterial wall (13). Triglyceride-rich lipoproteins and free fatty acids are important factors involved in fatty streak formation and advanced atherosclerosis (14). Microorganisms have also been implicated as aggravating factors in atherosclerosis (15). In atherosclerosis, normal homeostatic functions of the endothelium are altered, promoting an inflammatory response that results in an increased expression of adhesion molecules. This increased expression leads to the recruitment of leukocytes, including monocytes, that penetrate the intima, predisposing the vessel wall to lipid deposits (13). Reportedly, mast cells also contribute to coronary plaque progression and diet-induced obesity and diabetes through the secretion of vasoactive mediators, cytokines, and proteinases (16).
Evidence is accumulating that distant bacterial infection is involved in the pathophysiology of local chronic inflammatory processes underlying atherosclerosis (17). The transfer of bacteria into the blood or lymph system from barrier organ surfaces has been suggested as a possible mechanism of atherosclerosis. Advanced gum infection (periodontitis) is known to induce local inflammation, often leading to gingival ulcerations and local vascular changes, which have the potential to increase the incidence and severity of transient bacteremia. Porphyromonas gingivalis, an important microorganism associated with periodontitis, has been identified in atherosclerotic plaques of patients suffering from atherosclerosis, suggesting that this pathogen might be critical for atheroma formation (18). Indeed, we showed that endothelial dysfunction associated with repetitive exposure by P. gingivalis can exacerbate the development of atherosclerosis (19).
NOD2 expression and unique functions have also been described in other cell types, including adipocytes, gingival, pulp and periodontal fibroblasts, oral epithelial cells, and vascular endothelial cells (20–25). However, the precise role of NOD2 in chronic inflammatory diseases remains unclear.
We showed previously that, in Apolipoprotein E+/− (ApoE+/−) mice, TLR2 deficiency reduces pathogen-associated atherosclerosis (26). In this report, we tested the role of NOD2 in two chronic inflammatory diseases, atherosclerosis and alveolar bone loss, by capitalizing on our model of P. gingivalis-associated atherosclerosis in ApoE−/− mice.
Results
P. gingivalis Counts.
P. gingivalis was only detected in mouse groups that received P. gingivalis oral gavage (refer to Fig. 1 for detailed animal grouping and experimental time scheduling). Colony forming unit (CFU) counts at all time points were about 50% greater in NOD2−/−ApoE−/− mice than ApoE−/− mice (P < 0.05) (Fig. 2A), showing that NOD2-deficiency promoted greater P. gingivalis counts in the murine oral cavity.
Fig. 1.

Animal grouping and time scheduling. ApoE−/− and NOD2−/−ApoE−/− mice involved in the NOD2 loss-of-function study were fed an SCD and randomly assigned to six groups. ApoE−/− mice involved in the NOD2 gain-of-function study with MDP treatment were fed either an SCD or an HFD and assigned randomly to six groups (10 mice/group). The mice were challenged with oral gavage of either P. gingivalis (109 CFU/100 µL) or 2% CMC sham control (100 µL) or i.p. injected with either 200 µg MDP in 100 µL PBS or 100 µL saline (sham) control three times per week accordingly. The experiments all were started with mice at 8 wk of age after a 2-wk antibiotic pretreatment and a 3-d rest and terminated at the 15th week. The week 0 control groups (10 mice/group) were euthanized right before the experiment at an age of 8 wk.
Fig. 2.
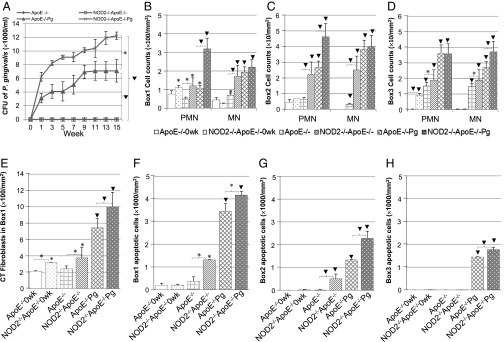
P. gingivalis counts and cell infiltrate in periodontal tissues. (A) CFUs of P. gingivalis (CFU × 1,000/mL). The plaques from the experimental mice were collected and cultured biweekly for 15 wk. NOD2−/−ApoE−/− mice showed statistical significance of CFU throughout the 15-wk time window compared with the ApoE−/− group. (B–D) Assessment of PMN and MN infiltrated in periodontal soft tissue assigned as (B) box 1 (top area), (C) box 2 (middle area), and (D) box 3 (bottom area; in cell counts per millimeter2). (E) Assessment of CT fibroblasts in periodontal box 1 region (in cell counts per millimeter2). (F–H) Assessment of TUNEL-positive apoptotic cells in periodontal soft tissue assigned as regions of (F) box 1, (G) box 2, and (H) box 3 (in cell counts per millimeter2). The longitudinal sections of the right side of palatal tissues containing three molars in the same occlusal plane were stained with either (B–E) H&E or (F–H) TUNEL. Images were captured and counted five regions per box area at 400× amplification. Data are shown as mean ± SE. *P < 0.05; ▾P < 0.01.
Counts of Inflammatory Cells, Connecting Tissue Fibroblasts, and Apoptotic Cells.
A very similar distribution pattern of polymorphonuclear leukocyte (PMN) and mononuclear cell (MN) infiltration was noted throughout the three interdental areas of interest, boxes 1, 2, and 3 (Fig. S1 shows anatomical landmarks and describes the boxes) (27), over time in all groups (Fig. 2 B–D). An increased number of inflammatory cells was observed in boxes 1, 2, and 3 when mice were subjected to an oral gavage with P. gingivalis irrespective of genotype. At week 15, in boxes 1, 2, and 3, NOD2 deficiency resulted in a 1.5- to 3.0-fold increase in PMN counts in uninfected groups. P. gingivalis exposure elicited a threefold increase of PMN cell counts (Fig. 2 B and C) in the NOD2−/−ApoE−/−Pg compared with the ApoE−/−Pg group (P < 0.01) in boxes 1 and 2. At week 15 in boxes 1 and 2, in uninfected groups, NOD2 deficiency resulted in a two- to fivefold increase in MN cell counts. In infected groups, this difference was significant only in box 3 (Fig. 2 B–D, ▾). Thus, NOD2 deficiency increased the inflammatory cell infiltration in proximity to the alveolar bone.
For connective tissue (CT) fibroblasts, statistical significance was observed only in box 1 (Fig. 2E). NOD2 deficiency resulted in an increase of CT fibroblast counts at weeks 0 and 15 (P < 0.05). P. gingivalis exposure elicited a 1.5-fold increase in CT fibroblast counts in NOD2−/−ApoE−/−Pg compared with ApoE−/−Pg (Fig. 2E, *).
There were no significant changes in apoptotic cell counts in all boxes between the two strains at week 0 (Fig. 2 F–H). At week 15, in boxes 1 and 2 regions and uninfected groups, NOD2 deficiency resulted in a 3.5- to 25.0-fold increase of apoptotic cell counts. P. gingivalis exposure induced a significant increase in apoptotic cells in all boxes, regardless of the animal genotype; however, greater apoptotic cell counts were obtained in NOD2−/−ApoE−/−Pg compared with ApoE−/−Pg animals. (Fig. 2 F–H, ▾).
Quantification of Alveolar Bone Loss, Osteoclast Number, and Epithelial Tissue Down-Growth.
Alveolar bone loss measured (morphometrically or histomorphometrically) by the Cemento-Enamel Junction-Alveolar Bone Crest distance (CEJ-ABC) was increased at week 15 compared with week 0, regardless of the animal genotype and the bacterial infection. P. gingivalis oral gavage induced a greater increase in CEJ-ABC distance in the NOD2−/−ApoE−/−Pg group compared with the ApoE−/−Pg group by morphometric or histomorphometric assessment (P < 0.05) (Fig. 3 A–N). No significant differences were observed at baseline between the two groups. However, at week 15, NOD2 deficiency resulted in an increase of osteoclast numbers, regardless of the bacterial infection. P. gingivalis exposure induced increased osteoclast numbers compared with uninfected groups, but a significant increase was observed in NOD2−/−ApoE−/−Pg animals compared with ApoE−/− animals (P < 0.05) (Fig. 3O).
Fig. 3.

Alveolar bone loss, EP down-growth, and osteoclast count in NOD2-deficient mice compared with control. (A, B, E, F, I, and J) Representatives of the left side of palatal bones stained with methylene blue. Images were captured at 3.45× amplification. (C, D, G, H, K, and L) Representatives of cross-sections of the right side of palatal tissues stained with H&E. Images were captured at 100× amplification. Arrows in the images indicate the defined CEJ and ABC. (M) Assessment of macromorphometric alveolar bone loss by buccally measuring the linear distances from CEJ to ABC on methylene blue-stained palatal bone and teeth (in micrometers). (N) Assessment of micromorphometric alveolar bone loss by measuring the distance of CEJ-ABC on the H&E-stained sections (in micrometers). (O) Assessment of osteoclasts by counting histochemical tartrate-resistant acid phosphatase (TRAP)-stained molar tissue sections. TRAP-positive multinucleated cells located in or near Howship lacunae were counted as osteoclast (cells per millimeter). Osteoclasts within bone marrow spaces were not included. (P) Assessment of EP down-growth by measuring the distance of apical epithelial tissue migration to to CEJ on H&E-stained sections (in micrometers). Images were captured on three molar teeth in the same occlusal plane for micromorphometric analysis. A and C are from the ApoE−/−0wk group, B and D are from the NOD2−/−ApoE−/− 0 wk group, E and G are from the ApoE−/− group with 2% CMC sham oral gavage for 15 wk, F and H are from the NOD2−/−ApoE−/− group with 2% CMC sham oral gavage for 15 wk, I and K are from the ApoE−/−Pg group challenged with P. gingivalis oral gavage for 15 wk, and J and L are from the NOD2−/−ApoE−/−Pg group challenged with P. gingivalis oral gavage for 15 wk. Data are represented as mean ± SE. *P < 0.05; ▾P < 0.01.
Epithelial (EP) down-growth assessed by the distance of apical migration of EP relative to the CEJ was increased at week 15 compared with week 0, regardless of the animal genotype and the bacterial infection (Fig. 3 A–L and P). At week 0, no significant differences were observed between the two groups, whereas at week 15, NOD2 deficiency resulted in a twofold increase in EP down-growth irrespective of the bacterial infection (P < 0.01). P. gingivalis exposure induced a greater EP down-growth in NOD2−/−ApoE−/−Pg mice compared with ApoE−/− mice (Fig. 3 C, D, G, H, K, L, and P, ▾).
Quantification of Serum Cholesterol, Aorta, and Aortic Sinus Lesions Stained with Oil Red O and Monocyte–Macrophage Marker-2.
Total serum cholesterol levels increased over time in both groups. At week 0, NOD2−/−ApoE−/−0wk animals had a twofold increase in total cholesterol compared with the ApoE−/−0wk group. NOD2 deficiency resulted in an increase in total cholesterol levels from week 0 to 15, regardless of the infection bacteria (Fig. 4A), but exposure of P. gingivalis resulted in a 1.5-fold increase in total cholesterol in the NOD2−/−ApoE−/−Pg compared with the ApoE−/−Pg group (P < 0.05) (Fig. 4A). NOD2 deficiency resulted in a three- to fourfold increase of Oil Red O (ORO) -stained lesions (quantified by en face preparation of aortic surface area) from week 0 to 15 irrespective of the infection status. The exposure to P. gingivalis stimulated a threefold increase of ORO-stained lesions in the NOD2−/−ApoE−/−Pg compared with the ApoE−/−Pg group (P < 0.05) (Fig. 4 B and C, ▾). ORO-stained histological sections of proximal aortic sinus had a similar trend as the en face lesions. Notably, at week 0, 17% of the aortic sinus was already covered with ORO-stained lesions in NOD2−/−ApoE−/−0wk animals, whereas no lesions were apparent in ApoE−/−0wk animals. NOD2 deficiency promoted a threefold increase in development of ORO-stained lesions from week 0 to 15, regardless the bacterial infection (Fig. 4 D and E, ▾).
Fig. 4.
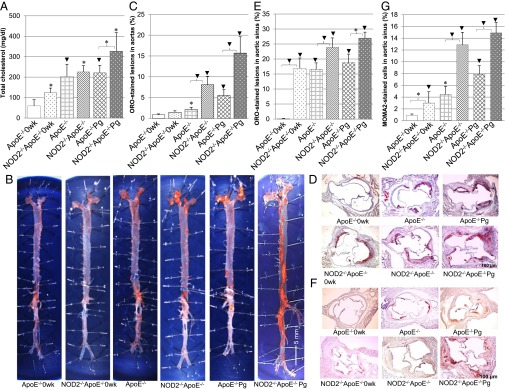
Serum total cholesterol and ORO and MOMA-2 staining in NOD2-deficient lesions compared with the controls. (A) Assessment of total blood cholesterol (milligrams per deciliter). (B) Representatives of morphometric en face ORO-stained lesions (red) in the aorta. (Magnification: 0.3×.) (C) Assessment of percentage of ORO-stained atheroma lesions in the aorta surface. (D) Representatives of ORO-stained proximal aortic sinus cross-sections. (Magnification: 40×.) (E) Assessment of the percentage of ORO-stained lesions in aortic sinus. (F) Representatives of MOMA-2–stained monocytes and macrophages (brown) in atherosclerotic lesions in aortic sinus. (Magnification: 40×.) (G) Assessment of percentage of MOMA-2–stained monocytes and macrophages in atherosclerotic lesions in aortic sinus. Data are represented as mean ± SE. *P < 0.05; ▾P < 0.01.
Aortic sinus histological sections immunostained for monocyte–macrophage marker-2 (MOMA-2 revealed macrophage staining in atheroma lesions (Fig. 4F, brown). NOD2 deficiency resulted in a significant increase of MOMA-2–stained macrophages in the lesions of aortic sinus at week 0 and a two- to threefold increase at week 15, regardless the bacterial infection (P < 0.05) (Fig. 4G). At week 15, P. gingivalis infection induced a greater macrophage staining in atheroma lesions present in the aortic sinus, regardless of the animal genotype (Fig. 4 F and G).
Quantification of Serum Cytokines.
At week 0, NOD2 deficiency induced cytokine expressions of proinflammatory cytokines, such as TNF-α, IL-12p70, IL-1β, IL-6, IL-2, IL-5, IL-7, IL-17, and GM-CSF, and antiinflammatory cytokine IL-10, except IFNγ-induced protein-10 (IP-10). At week 15, NOD2 deficiency resulted in a significant increase in expressions of proinflammatory cytokines of the IL-12p70, IL-2, IL-6, TNF-α, IL-5, IL-7, IP-10, and GM-CSF groups (P < 0.05) (Fig. 5). TNF-α, IL-6, IL-7, IP-10, and GM-CSF levels in the uninfected NOD2−/−ApoE−/− mice were similar to or higher than in the infected ApoE−/−Pg group at week 15. P. gingivalis oral gavage resulted in a 2- to 10-fold increase of the assayed cytokines in the NOD2−/−ApoE−/−Pg group compared with the ApoE−/−Pg group (P < 0.05). NOD2 deficiency resulted in a two- to threefold induction of the antiinflammatory cytokine IL-10, regardless the bacterial infection at week 15 (P < 0.05) (Fig. 5).
Fig. 5.

Serum inflammatory cytokines in NOD2-deficient mice compared with control. The assessment of 11 cytokine expressions was carried out by Milliplex MAP Mouse-Cytokine/Chemokine Premixed 32plex Immunoassay Kits (in picograms per milliliter). Data are represented as mean ± SE. *P < 0.05; ▾P < 0.01.
MDP Effects on Cholesterol, ORO Lesions, and Alveolar Bone Loss.
NOD2 gain of function was achieved by i.p. injection of MDP to ApoE−/− mice maintained on standard chow diet (SCD) and exposed to P. gingivalis oral gavage for 15 wk. On completion of the experiment (week 15), MDP injection did not significantly affect the P. gingivalis CFU counts in ApoE−/−Pg+MDP compared with untreated ApoE−/−Pg animals. Furthermore, MDP did not have any effects on unchallenged animals for the bone loss (CEJ-ABC), total cholesterol, or ORO-stained lesions (ApoE−/−MDP animals) (Fig. 6 A–E). However, MDP treatment reduced alveolar bone loss by 30%, en face ORO-stained lesions by 50%, and total cholesterol by 20% in infected ApoE−/−Pg+MDP compared with infected but untreated ApoE−/−Pg animals (Fig. 6 B–E, ▾). When ApoE−/−mice were fed a high-fat diet (HFD), 15 wk of MDP treatment (group HFD-MDP) resulted in a 25% reduction in body weight gain compared with the control group (HFD-Sham) (Fig. 6F, *). In contrast, MDP did not affect body weight when mice were maintained on SCD.
Fig. 6.
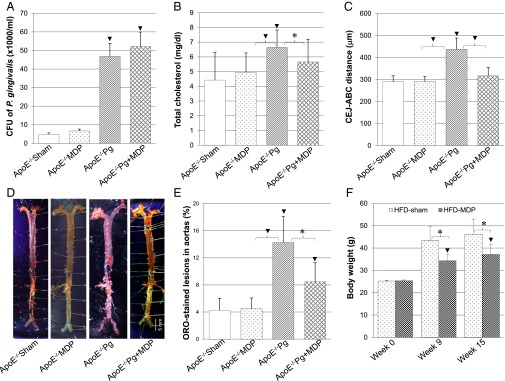
Effects of MDP treatment on bacterial counts, alveolar bone loss, total cholesterol, ORO staining, and body weight. ApoE−/− mice were i.p. injected with either MDP or saline (i.p. control) with oral gavage of either P. gingivalis or 2% CMC (sham control). Data are represented as mean ± SE. *P < 0.05; ▾P < 0.01. (A) CFU of P. gingivalis in ApoE−/− mice groups on SCD (CFU × 1,000/mL). (B) Assessment of blood total cholesterol level (milligrams per deciliter). (C) Assessment of alveolar bone loss by macromorphometrically measuring the distance of CEJ-ABC in ApoE−/− mice fed on SCD (in micrometers). (D) Representatives of morphometric en face ORO staining lesions (red) in aorta of ApoE−/− mice fed on SCD. (Magnification: 0.3×.) (E) Assessment of percentage of ORO-stained atheroma lesions in aorta surface of ApoE−/− mice fed on SCD. (F) Assessment of body weight of ApoE−/− mice fed on HFD. ApoE−/− mice were i.p. injected with either MDP or saline (i.p. control). Data are represented as mean ± SE. *P < 0.05; ▾P < 0.01.
Quantitative RT-PCR of Cell Signaling Mediators and TNF-α ELISA.
Three hours after P. gingivalis exposure in vitro, RNA expressions of TLR2, TNF receptor-associated factor 6 (TRAF6), and c-Jun N-terminal kinase-3 (JNK-3) and TNF-α protein levels were found statistically elevated in bone marrow-derived macrophages (BMMs) from NOD2−/−ApoE−/−Pg compared with control ApoE−/−Pg BMMs (Fig. 7 A and C, *). RNA expression of NOD2 and RIP2 in NOD2-deficient BMMs was significantly reduced compared with ApoE−/− BMMs, regardless of the bacterial infection, whereas levels of inhibitor of NF-κB kinase-β (IKK-β) and NF-κB were slightly reduced in NOD2-deficient compared with ApoE−/− BMMs (Fig. 7B, *). TNF-α protein levels were slightly elevated in NOD2-deficient compared with ApoE−/− BMMs (Fig. 7C, *). MDP treatment did not have any significant effect in NOD2-deficient BMMs for all genes tested in the absence of infection. However, MDP treatment with P. gingivalis exposure reduced significantly RNA levels for JNK3, IKK-β, and NF-κB and TNF-α protein level, whereas it increased NOD2 and RIP2 RNA levels in ApoE−/−Pg+MDP compared with ApoE−/−Pg BMMs (P < 0.01). No significant differences were observed for MDP at 50 and 200μg/mL in vitro compared with 100 μg/mL.
Fig. 7.

RT-PCR analysis of NOD2 signal transduction genes. BMMs from male mice of ApoE−/− and NOD2−/−ApoE−/− strains were incubated with P. gingivalis and MDP or saline (sham) for 3 h after maturely differentiated. Data are represented as mean ± SE. *P < 0.05; ▾P < 0.01. (A) Quantitative RT-PCR analysis of inflammatory mediators involved in the TLR2 pathway. Enhanced TLR2 signaling pathway is observed in NOD2-deficient BMMs especially TLR2, TRAF6 and JNK3 in response to P. gingivalis infection. MDP treatment down-regulated JNK3 RNA expression in P. gingivalis-infected ApoE−/−BMMs. (B) Quantitative RT-PCR analysis of the mediators involved in the inflammatory NOD2/RIP2 signaling pathway. MDP treatment up-regulated RNA expressions of NOD2 and RIP2 but down-regulated the RNA expressions of IKKβ and NF-κB in P. gingivalis-infected ApoE−/−mouse BMMs. (C) ELISA analysis of TNF-α expression in culture media of ApoE−/−BMM and NOD2−/−ApoE−/−mouse BMMs. TNF-α was significantly hindered by MDP treatment in the infected ApoE−/−mouse BMMs. (D) Proposed pivotal role of MDP/NOD2 signaling pathways in P. gingivalis-induced periodontitis/atherosclerosis.
Discussion
In the present study, loss-of-function experiments by the generation of NOD2-deficient animals in an ApoE-KO mouse background led to an aggravation of inflammatory processes associated with two chronic diseases: atherosclerosis and periodontitis. NOD2 gain-of-function experiments achieved by MDP treatment in ApoE−/− mice showed significant decrease in ORO-stained plaque accumulations, alveolar bone loss, serum cytokines, and serum cholesterol.
Exposing NOD2−/−ApoE−/− mice to P. gingivalis leads to greater bacterial counts in the murine oral cavity, resulting in heightened periodontitis, which was evidenced by high inflammatory cell infiltration, severe alveolar bone resorption, and loss of epithelial attachment. These pathological consequences are typically associated with a host response to persistent bacterial accumulation at the tooth surface. NOD2-deficient mice present elevated numbers of resorbing osteoclasts, further supporting the role of NOD2 in preventing bacterial-induced inflammatory bone loss. Therefore, loss of NOD2 function plays an important role in aggravating periodontitis. We, thus, conclude that NOD2 is required in vivo to surveil P. gingivalis infection, which ultimately affects the host response (28).
Interestingly, our study also shows that periodontal inflammation, alveolar bone resorption, and atherosclerosis are observed even in mice lacking NOD2 function but unchallenged. Possibly, NOD2 deficiency impairs the host bacterial surveillance involved in maintaining a proper balance between host and pathogens, resulting in a shift of the opportunistic bacterial flora to a more pathogenic form, which in turn, is capable of triggering the immune responses associated with both periodontitis and atherosclerosis (29).
Current evidence suggests that oral manipulation procedures are the most common cause of bacterial dissemination in the bloodstream from oral niches (30). Clearly, a higher microbial load would facilitate such dissemination, because it is known that individuals with poor oral hygiene are at a higher risk of developing bacteremia during periodontal surgeries (30).
Current literature supports the assertion that periodontal pathogens can contribute to atherosclerosis (31). Therefore, we asked whether NOD2 function is an important link in the complex relationship between P. gingivalis-induced periodontitis and infection-associated atherosclerosis. NOD2 deficiency leads to elevated blood cholesterol level together with the accumulation of monocytes/macrophages into the heart and aortic intima space in P. gingivalis-challenged animals. This accumulation promotes atherosclerosis in our ApoE−/− animal model. Particularly, lesions in the aortic sinus start as early as 8 wk of age in the NOD2−/−ApoE−/−0wk group, with 17% coverage at the inception of the experiment. In contrast, it took 15 wk for the ApoE−/− control group to develop a similar extent of ORO lesions (17%) in the aortic sinus, assigning to NOD2 an important role in the signaling pathway associated with the prevention of deleterious inflammation and atherosclerosis, even in the absence of bacterial challenge (32). Hence, in the presence of bacterial challenge or HFD, TLR activation, known to play a role in macrophage polarization to an M1 phenotype (33), recruited macrophages that exert diverse functions in atherosclerosis, including altered lipid metabolism, production of inflammatory cytokines, endothelial cell interactions, and matrix degradation (34). It is, therefore, conceivable given the functional relationship of TLR and NOD2 (35) that, in addition to the immune dysregulation, mice deficient in both NOD2 and ApoE may have an imbalance of lipoprotein components that are vital to the development of atherosclerosis.
Mutations involving loss of function of NOD2 were linked to Crohn disease, another chronic inflammatory disease, possibly as a result of heightened TLR2 responses (36). Interestingly, inflammatory bowel disease patients are reported to be at greater risk of developing atherosclerosis (37, 38) and possibly, periodontal disease (39, 40). Our study concurs with these observations: in response to the invading microbes, NOD2 loss of function in mice stimulates TLR2 signaling by up-regulation of TRAF6 and JNK3, leading to the production of NF-κB and resulting in the overexpression of serum cytokines, which play an important role in alveolar bone resorption in periodontitis and atherosclerosis. The elevated levels of cytokines further substantiate the role of NOD2 in modulating the P. gingivalis effects in this murine periodontitis model and atherosclerosis, especially knowing that NOD2 is expressed in gingival, pulp and periodontal fibroblasts, oral epithelial cells, and vascular endothelial cells (20, 22–25). Surprisingly, IL-10, an antiinflammatory and antiatherogenic cytokine produced by macrophages, Th1, and B cells, was also significantly elevated in NOD2-deficent mice. This IL-10 elevation may be a response to the important proinflammatory process observed in NOD2-deficient animals (41).
Gain of function studies revealed that MDP/NOD2 recruits RIP2 and counteracts the TLR2 inflammatory signaling pathway by inhibiting TRAF6 and suppressing IKKs, NF-κB (42) and JNK3, which leads to a significant reduction of TNF-α protein levels and possibly, other inflammatory mediators (5). This reduction may help explain the reduced alveolar bone loss, serum cholesterol level, and atherosclerosis observed after MDP treatment. Indeed, MDP did not have any effect in modulating P. gingivalis CFUs; its effect on inflammation may be solely directed on the NF-κB pathway.
A surprising observation in our present study was that MDP treatment resulted in reduced body weight of mice fed HFD, with no deleterious effects on SCD-fed animals, suggesting that the MDP/NOD2 axis may reduce the HFD stimulation of TLRs through the NF-κB pathway (5) and/or production of endogenous ligands (43).
The present findings show that the NOD2 ligand (MDP) administration to ApoE−/− mice reduces experimental atherosclerotic lesions and periodontal bone loss. It should be noted that MDP or certain derivatives of MDP have been shown to have adjuvant properties, possibly because activation of NOD2 can transiently enhance certain immune responses before it exerts a more dominant inhibitory response. The use of MDP remains possible given that all MDP preparations do not have these effects, and these immune responses may be dependent on dose or route of administration. Furthermore, at all of the concentrations tested, MDP did not show any overt deleterious effect, possibly implying that NOD2 activation with MDP may be a safe way to modulate exaggerated inflammatory responses to chronic bacterial stimulation. Additionally, MDP may be modulating diet-associated inflammatory process and should be considered in therapeutic approaches aimed at reducing HFD-induced obesity, a major contributor to diabetes and cardiovascular disease.
Material and Methods
Mice and Diets.
The Institutional Animal Care and Use Committee of Boston University approved all animal protocols. Female NOD2−/− and male ApoE−/− mice were purchased from Jackson Laboratory and bred to generate NOD2 and ApoE double deficient homozygotes (NOD2−/−ApoE−/−). The genotypes of the strains were confirmed by PCR. The animals weaned at 4 wk of age were fed SCD containing 0.02% cholesterol and 4.5% (wt/vol) fat (rodent diet 5001). A 2-wk antibiotic pretreatment was carried out with Sulfamethoxazole/Trimethoprim (Hi-Tech Pharmacal Co., Inc.) at 850/170 mg per 1 L drinking water followed by a 3-d rest before the experiment. All in vivo experiments were started with the animals at 8 wk of age. Twenty NOD2−/−ApoE−/− mice were maintained on SCD, and eighty ApoE−/− mice were randomly assigned to either a cholate-free HFD (D12492 Research Diet) or SCD. Because estrogen has been suggested to be cardioprotective in females (44), only male mice were used in this study.
Bacterial Strain, Dose, Route of Inoculation, and MDP Treatment.
P. gingivalis strain 381 (FDC-381; ATCC), a human isolate, was grown on anaerobic agar plates as described previously (45). P. gingivalis was administered at a dosage and delivery route comparable with the bacteremia encountered in humans after dental infection, periodontal surgery, scaling, tooth extraction, or flossing (46–48). Live P. gingivalis was given by oral gavage at a dose of 109 CFU suspended in 100 μL 2% (wt/vol) carboxymethylcellulose (CMC) in PBS three times per week for 15 wk to both ApoE−/− and NOD2−/−ApoE−/− mice fed an SCD, whereas sham-infected control ApoE−/− and NOD2−/−ApoE−/− mice received uninfected broth (2% CMC in PBS). In each gavage administration, one-half (50 μL) of the volume was placed down the throat of the mouse, and one-half (50 μL) was left in its oral cavity. Biweekly, subgingival plaque samples from the left and right maxillary second molars were collected in each group using sterile paper points (Dentsply Maillefer). P. gingivalis colonies were identified both on the basis of known colony morphology and chemically through the bioMerieux Rapid ID 32A Identification System. The total CFUs of P. gingivalis on anaerobic blood agar were counted manually and reported as 1,000 CFUs/mL.
For NOD2 gain-of-function study, the animals were injected three times per week for 15 wk (1 h before P. gingivalis gavage inoculation) with either an optimized dose of MDP (200 μg/mouse; InvivoGen) in 100 μL endotoxin-free water or 100 μL saline (sham).
Animal Grouping and Time Schedule.
For NOD2 loss-of-function study, 60 mice on SCD were randomly assigned to six groups (10 mice/group) by their genotypes: groups 1 and 2, ApoE−/−0wk and NOD2−/−ApoE−/−0wk controls euthanized at the inception of the experiment; groups 3 and 4, ApoE−/− and NOD2−/−ApoE−/− sham controls received 2% CMC/PBS by oral gavage three times per week for 15 wk; and groups 5 and 6, ApoE−/−Pg and NOD2−/− ApoE−/−Pg received P. gingivalis oral gavage three times per week for 15 wk. For NOD2 gain of function by MDP study, 40 ApoE−/− mice fed SCD were randomly grouped (10 mice/group) for a 15-wk experimental course after antibiotics pretreatment: group1, ApoE−/−sham controls received oral gavage of 2% CMC/PBS and i.p. injection of saline; group 2, ApoE−/−MDP received i.p. injection of MDP and oral gavage of 2% CMC/PBS (three times per week); group 3, ApoE−/−Pg received P. gingivalis oral gavage and i.p. injection of saline (three times per week); and group 4, ApoE−/−Pg+MDP received P. gingivalis oral gavage and i.p. injection of MDP (three times per week). For MDP treatment in animals without P. gingivalis challenge but maintained on HFD, 20 ApoE−/− mice were equally divided into two groups: group 1 (HFD-sham), i.p. injection of saline three times per week for 15 wk; group 2 (HFD-MDP), i.p. injection of MDP three times per week for 15 wk. Body weights were measured and recorded weekly. Animal grouping and experimental time scheduling are illustrated in Fig. 1.
Statistical Analysis.
Comparisons of atherosclerotic lesions, alveolar bone loss, cell counts, and cytokine concentrations among the groups were analyzed, and statistical significance of multiplicities was evaluated by ANOVA (two-way) followed by a posthoc Scheffé test. P ≤ 0.05 was considered significant. Data (n = 10/group) are expressed as mean ± SE. Data capture and data analyses were performed in a blinded fashion by two independent investigators. Interexaminer variation was less than 5%.
Tissue Harvesting and Preparation.
Details are provided in SI Materials and Methods.
Culture of BMMs and Treatment.
Details are provided in SI Materials and Methods.
Characterization of Periodontal Inflammation and Tissue Remodeling.
Details are provided in SI Materials and Methods.
Characterization of Periodontal Bone Loss, Osteoclasts, and EP Down-Growth.
Details are provided in SI Materials and Methods.
En Face Preparation and Histomorphometric Quantification of ORO- and MOMA-2–Stained Lesions.
Details are provided in SI Materials and Methods.
Quantification of Serum Total Cholesterol, Chemokines, and Cytokines.
Details are provided in SI Materials and Methods.
Quantitative RT-PCR.
Relative RNA expression levels of inflammatory mediators were carried out by qRT-PCR with primers listed in Table S1. Details are provided in SI Materials and Methods.
Supplementary Material
Acknowledgments
The authors thank Prof. Theoharis C. Theoharides for constructive criticisms of the manuscript. This study was supported by National Institutes of Health/National Heart, Lung, and Blood Institute Grant R01HL076801.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1320862110/-/DCSupplemental.
References
- 1.Ogura Y, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature. 2001;411(6837):603–606. doi: 10.1038/35079114. [DOI] [PubMed] [Google Scholar]
- 2.Hugot JP, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature. 2001;411(6837):599–603. doi: 10.1038/35079107. [DOI] [PubMed] [Google Scholar]
- 3.Abraham C, Cho JH. Inflammatory bowel disease. N Engl J Med. 2009;361(21):2066–2078. doi: 10.1056/NEJMra0804647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lala S, et al. Crohn’s disease and the NOD2 gene: A role for paneth cells. Gastroenterology. 2003;125(1):47–57. doi: 10.1016/s0016-5085(03)00661-9. [DOI] [PubMed] [Google Scholar]
- 5.Zheng S, Abraham C. NFκB1 Inhibits NOD2-Induced Cytokine Secretion through ATF3-Dependent Mechanisms. Mol Cell Biol. 2013 doi: 10.1128/MCB.00797-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4(7):499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 7.Inohara C, Chamaillard, McDonald C, Nuñez G. NOD-LRR proteins: Role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74(15952891):355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- 8.Travassos LH, et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010;11(1):55–62. doi: 10.1038/ni.1823. [DOI] [PubMed] [Google Scholar]
- 9.Mo J, et al. Pathogen sensing by nucleotide-binding oligomerization domain-containing protein 2 (NOD2) is mediated by direct binding to muramyl dipeptide and ATP. J Biol Chem. 2012;287(27):23057–23067. doi: 10.1074/jbc.M112.344283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hisamatsu T, et al. CARD15/NOD2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology. 2003;124(4):993–1000. doi: 10.1053/gast.2003.50153. [DOI] [PubMed] [Google Scholar]
- 11.Deshmukh HS, et al. Critical role of NOD2 in regulating the immune response to Staphylococcus aureus. Infect Immun. 2009;77(4):1376–1382. doi: 10.1128/IAI.00940-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yamamoto-Furusho JK, Barnich N, Hisamatsu T, Podolsky DK. MDP-NOD2 stimulation induces HNP-1 secretion, which contributes to NOD2 antibacterial function. Inflamm Bowel Dis. 2010;16(5):736–742. doi: 10.1002/ibd.21144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anogeianaki A, et al. Atherosclerosis: A classic inflammatory disease. Int J Immunopathol Pharmacol. 2011;24(4):817–825. doi: 10.1177/039463201102400401. [DOI] [PubMed] [Google Scholar]
- 14.Welty FK. How do elevated triglycerides and low HDL-cholesterol affect inflammation and atherothrombosis? Curr Cardiol Rep. 2013;15(9):400. doi: 10.1007/s11886-013-0400-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Libby P, Ridker PM, Hansson GK. Leducq Transatlantic Network on Atherothrombosis Inflammation in atherosclerosis: From pathophysiology to practice. J Am Coll Cardiol. 2009;54(23):2129–2138. doi: 10.1016/j.jacc.2009.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Theoharides TC, et al. Mast cells squeeze the heart and stretch the gird: Their role in atherosclerosis and obesity. Trends Pharmacol Sci. 2011;32(9):534–542. doi: 10.1016/j.tips.2011.05.005. [DOI] [PubMed] [Google Scholar]
- 17.Zelkha SA, Freilich RW, Amar S. Periodontal innate immune mechanisms relevant to atherosclerosis and obesity. Periodontol 2000. 2010;54(1):207–221. doi: 10.1111/j.1600-0757.2010.00358.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cavrini F, et al. Molecular detection of Treponema denticola and Porphyromonas gingivalis in carotid and aortic atheromatous plaques by FISH: Report of two cases. J Med Microbiol. 2005;54(Pt 1):93–96. doi: 10.1099/jmm.0.45845-0. [DOI] [PubMed] [Google Scholar]
- 19.Amar S, Wu SC, Madan M. Is Porphyromonas gingivalis cell invasion required for atherogenesis? Pharmacotherapeutic implications. J Immunol. 2009;182(3):1584–1592. doi: 10.4049/jimmunol.182.3.1584. [DOI] [PubMed] [Google Scholar]
- 20.Hirao K, et al. Roles of TLR2, TLR4, NOD2, and NOD1 in pulp fibroblasts. J Dent Res. 2009;88(8):762–767. doi: 10.1177/0022034509341779. [DOI] [PubMed] [Google Scholar]
- 21.Stroh T, et al. Nucleotide oligomerization domains 1 and 2: Regulation of expression and function in preadipocytes. J Immunol. 2008;181(5):3620–3627. doi: 10.4049/jimmunol.181.5.3620. [DOI] [PubMed] [Google Scholar]
- 22.Hosokawa I, et al. Proinflammatory effects of muramyldipeptide on human gingival fibroblasts. J Periodontal Res. 2010;45(2):193–199. doi: 10.1111/j.1600-0765.2009.01217.x. [DOI] [PubMed] [Google Scholar]
- 23.Oh HM, et al. Induction and localization of NOD2 protein in human endothelial cells. Cell Immunol. 2005;237(1):37–44. doi: 10.1016/j.cellimm.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 24.Tang L, et al. Expression of TRAF6 and pro-inflammatory cytokines through activation of TLR2, TLR4, NOD1, and NOD2 in human periodontal ligament fibroblasts. Arch Oral Biol. 2011;56(10):1064–1072. doi: 10.1016/j.archoralbio.2011.02.020. [DOI] [PubMed] [Google Scholar]
- 25.Sugawara Y, et al. Toll-like receptors, NOD1, and NOD2 in oral epithelial cells. J Dent Res. 2006;85(6):524–529. doi: 10.1177/154405910608500609. [DOI] [PubMed] [Google Scholar]
- 26.Madan M, Amar S. Toll-like receptor-2 mediates diet and/or pathogen associated atherosclerosis: Proteomic findings. PLoS One. 2008;3(9):e3204. doi: 10.1371/journal.pone.0003204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan H, Gupte R, Zelkha S, Amar S. Receptor activator of nuclear factor kappa B ligand antagonists inhibit tissue inflammation and bone loss in experimental periodontitis. J Clin Periodontol. 2011;38(11):1029–1036. doi: 10.1111/j.1600-051X.2011.01780.x. [DOI] [PubMed] [Google Scholar]
- 28.Opitz B, et al. Nucleotide-binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem. 2004;279(35):36426–36432. doi: 10.1074/jbc.M403861200. [DOI] [PubMed] [Google Scholar]
- 29.Petnicki-Ocwieja T, et al. Nod2 is required for the regulation of commensal microbiota in the intestine. Proc Natl Acad Sci USA. 2009;106(37):15813–15818. doi: 10.1073/pnas.0907722106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parahitiyawa NB, Jin LJ, Leung WK, Yam WC, Samaranayake LP. Microbiology of odontogenic bacteremia: Beyond endocarditis. Clin Microbiol Rev. 2009;22(1):46–64. doi: 10.1128/CMR.00028-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reyes L, Herrera D, Kozarov E, Roldán S, Progulske-Fox A. Periodontal bacterial invasion and infection: Contribution to atherosclerotic pathology. J Clin Periodontol. 2013;40(Suppl 14):S30–S50. doi: 10.1111/jcpe.12079. [DOI] [PubMed] [Google Scholar]
- 32.Kwon MY, et al. Nucleotide-binding oligomerization domain protein 2 deficiency enhances neointimal formation in response to vascular injury. Arterioscler Thromb Vasc Biol. 2011;31(11):2441–2447. doi: 10.1161/ATVBAHA.111.235135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kadl A, et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ Res. 2010;107(6):737–746. doi: 10.1161/CIRCRESAHA.109.215715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gleissner CA, Shaked I, Little KM, Ley K. CXC chemokine ligand 4 induces a unique transcriptome in monocyte-derived macrophages. J Immunol. 2010;184(9):4810–4818. doi: 10.4049/jimmunol.0901368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Borm MEA, van Bodegraven AA, Mulder CJJ, Kraal G, Bouma G. The effect of NOD2 activation on TLR2-mediated cytokine responses is dependent on activation dose and NOD2 genotype. Genes Immun. 2008;9(3):274–278. doi: 10.1038/gene.2008.9. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe T, et al. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen-specific colitis. Immunity. 2006;25(3):473–485. doi: 10.1016/j.immuni.2006.06.018. [DOI] [PubMed] [Google Scholar]
- 37.Dagli N, et al. Is inflammatory bowel disease a risk factor for early atherosclerosis? Angiology. 2010;61(2):198–204. doi: 10.1177/0003319709333869. [DOI] [PubMed] [Google Scholar]
- 38.Kayahan H, et al. Evaluation of early atherosclerosis in patients with inflammatory bowel disease. Dig Dis Sci. 2012;57(8):2137–2143. doi: 10.1007/s10620-012-2148-x. [DOI] [PubMed] [Google Scholar]
- 39.Brito F, et al. Subgingival microflora in inflammatory bowel disease patients with untreated periodontitis. Eur J Gastroenterol Hepatol. 2013;25(2):239–245. doi: 10.1097/MEG.0b013e32835a2b70. [DOI] [PubMed] [Google Scholar]
- 40.Habashneh RA, Khader YS, Alhumouz MK, Jadallah K, Ajlouni Y. The association between inflammatory bowel disease and periodontitis among Jordanians: A case-control study. J Periodontal Res. 2012;47(3):293–298. doi: 10.1111/j.1600-0765.2011.01431.x. [DOI] [PubMed] [Google Scholar]
- 41.Srinivasan S, Leeman SE, Amar S. Beneficial dysregulation of the time course of inflammatory mediators in lipopolysaccharide-induced tumor necrosis factor alpha factor-deficient mice. Clin Vaccine Immunol. 2010;17(5):699–704. doi: 10.1128/CVI.00510-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Perez LH, et al. Direct bacterial killing in vitro by recombinant Nod2 is compromised by Crohn’s disease-associated mutations. PLoS One. 2010;5(6):e10915. doi: 10.1371/journal.pone.0010915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin CFR, Flavell RA. Innate sensors of pathogen and stress: Linking inflammation to obesity. J Allergy Clin Immunol. 2013;132(2):287–294. doi: 10.1016/j.jaci.2013.06.022. [DOI] [PubMed] [Google Scholar]
- 44.Bourassa PA, Milos PM, Gaynor BJ, Breslow JL, Aiello RJ. Estrogen reduces atherosclerotic lesion development in apolipoprotein E-deficient mice. Proc Natl Acad Sci USA. 1996;93(19):10022–10027. doi: 10.1073/pnas.93.19.10022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibson FC, 3rd, et al. Innate immune recognition of invasive bacteria accelerates atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109(22):2801–2806. doi: 10.1161/01.CIR.0000129769.17895.F0. [DOI] [PubMed] [Google Scholar]
- 46.Bhanji S, Williams B, Sheller B, Elwood T, Mancl L. Transient bacteremia induced by toothbrushing a comparison of the Sonicare toothbrush with a conventional toothbrush. Pediatr Dent. 2002;24(4):295–299. [PubMed] [Google Scholar]
- 47.Gangloff SC, Guenounou M. Toll-like receptors and immune response in allergic disease. Clin Rev Allergy Immunol. 2004;26(2):115–125. doi: 10.1007/s12016-004-0006-0. [DOI] [PubMed] [Google Scholar]
- 48.Wank HA, Levison ME, Rose LF, Cohen DW. A quantitative measurement of bacteremia and its relationship to plaque control. J Periodontol. 1976;47(12):683–686. doi: 10.1902/jop.1976.47.12.683. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.