

Abstract
Runx proteins are essential for a number of developmental processes and are aberrantly expressed in many human cancers. Runx factors bind DNA and co-factors to activate or repress genes crucial for bone formation, hematopoiesis, and neuronal development. Co-activator activator (CoAA) is a nuclear protein that regulates gene expression, RNA splicing and is overexpressed in many human tumors. In this study, we identified CoAA as a Runx2 binding protein. CoAA repressed Runx factor-dependent activation of reporter genes in a histone deacetylase-independent manner. CoAA also blocked Runx2-mediated repression of the Axin2 promoter, a novel Runx target gene. The carboxy-terminus of CoAA is essential for binding the Runt domains of Runx1 and Runx2. In electophoretic mobility shift assays, CoAA inhibited Runx2 interactions with DNA. These data indicate that CoAA is an inhibitor of Runx factors and can negate Runx factor regulation of gene expression. CoAA is expressed at high levels in human fetal osteoblasts and osteosarcoma cell lines. Suppression of CoAA expression by RNA interference reduced osteosarcoma cell viability in vitro, suggesting that it contributes to the proliferation and/or survival of osteoblast lineage cells.
Keywords: Cbfa1, AML3, Runx1, Runx3, RBM14, CoAM, Axin2, Osteosarcoma
INTRODUCTION
Runt domain transcription factors (Runx1, Runx2 and Runx3) are DNA binding proteins that control the expression of genes involved in numerous development processes. Mice deficient in Runx1 (Cbfa2 or AML1), Runx2 (Cbfa1 or AML3) and/or Runx3 (Cbfa2 or AML2) exhibit severe defects in the differentiation or function of hematopoietic cells, osteoblasts, chondrocytes, gastric epithelial cells, and dorsal root ganglion neurons [Komori et al., 1997; Levanon et al., 2002; Li et al., 2002; Niki et al., 1997; Otto et al., 1997; Taniuchi et al., 2002; Woolf et al., 2003]. Runx factor genes are frequently altered in human cancers by chromosomal translocations, point mutations or epigenetic silencing [Blyth et al., 2005; Ito, 2004]. Runx factors bind to a consensus nucleotide sequence, TGT/c GGTT [Kamachi et al., 1990; Meyers et al., 1993], via a conserved Runt domain [Daga et al., 1992] to control gene expression in many tissues. The Runt domain is more than 90% identical in mammalian Runx factors. In addition to mediating DNA contact, the Runt domain is a protein-protein interaction motif that binds core binding factor (Cbf)-beta and several other proteins [Schroeder et al., 2005]. Runt domain factors were originally described as necessary but insufficient activators of viral and lymphocyte enhancers [Kamachi et al., 1990; Redondo et al., 1992; Redondo et al., 1991] and as repressors of Drosophila pair-rule genes [Manoukian and Krause, 1993]. In the last two decades, it has become clear that Runx factors are crucial organizers of enhancer and promoter complexes that can activate or repress mammalian gene expression depending on cellular and promoter/enhancer context [Lian et al., 2006; Schroeder et al., 2005]. Runx factors interact with other transcription factors and recruit numerous chromatin-modifying proteins to regulate gene expression [Schroeder et al., 2005]. Among the co-factors that interact with Runx proteins are co-activators: p300 and CREB binding protein (CBP); and co-repressors: mSin3A, transducin-like enhancer of split proteins (TLEs), and several histone deacetylases (Hdacs) [Durst and Hiebert, 2004; Schroeder et al., 2005], including Hdac3 [Lamour et al., 2007; Makita et al., 2008; Schroeder et al., 2004].
Co-Activator Activator (CoAA) is a broadly expressed nuclear protein that participates in transcription-coupled RNA splicing and is elevated in some human tumors [Auboeuf et al., 2002; Iwasaki et al., 2001; Sui et al., 2007]. CoAA was originally described as a binding partner of the LXXLL-containing general co-activator, thyroid hormone receptor binding protein (TRBP) [Iwasaki et al., 2001]. CoAA also augments the activity of the co-activators CBP and synovial sarcoma translocation protein (SYT) [Iwasaki et al., 2001; Perani et al., 2005]. CoAA is a potent co-activator for nuclear receptors, including the glucocorticoid, thyroid hormone, progesterone and estrogen receptors [Auboeuf et al., 2004; Iwasaki et al., 2001]. It also enhances mitogen-activated protein kinase kinase (MEKK)-induced activation of NFkB and AP reporters [Iwasaki et al., 2001]. The activator function appears to be context dependent however as CoAA was recently shown to recruit Hdac3 and repress the c-myc proto-oncogene in kidney cells [Kang et al., 2008]. In addition to its role as a transcription co-factor, CoAA regulates RNA splicing of steroid-responsive genes via two RNA recognition motifs (RRM) in it amino terminus [Auboeuf et al., 2002] and is also known as RNA binding motif protein 14 (RBM14). The RRM motifs of CoAA are required for transcriptional activation of some promoters, but the carboxy-terminal region lacking the RRMs are necessary to regulate other promoters and for interacting with TRBP [Auboeuf et al., 2004; Iwasaki et al., 2001].
We previously described an affinity purification/mass spectrometry experiment that identified several Runx2-interacting proteins [Jensen et al., 2008]. Among the Runx2-binding proteins that resolved in the acrylamide gel at approximately 65–75 kd, the DEAD box proteins Ddx5 and Ddx17 (p68 and p72) were identified with the highest confidence. RBM14 (i.e. CoAA) was also identified in this proteomic screen as a potential Runx2 interacting factor. In this manuscript, we verify that CoAA binds Runx2 in vivo and define its functional effects on Runx factor transcriptional activity.
EXPERIMENTAL PROCEDURES
Cell Culture
C2C12 cells were grown in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS), 200 mM L-glutamine, 50 units/ mL penicillin and 50 µg/ mL streptomycin. U2-OS were grown in Dulbecco’s modified Eagle’s/F12 medium containing 10% FBS, 200 mM L-glutamine, 50 units/ mL penicillin and 50 µg/ mL streptomycin. Runx2−/− cells [Pratap et al., 2003] were kindly provided by Dr. André VanWijnen and cultured in minimal essential medium supplemented with 10% FBS, 50 units/ mL penicillin, 50 µg/mL streptomycin and 1% nonessential amino acids. Primary calvarial osteoblasts were isolated from C57Bl/6 mice as previously reported [Schroeder and Westendorf, 2005] and cultured in the same medium as the Runx2-deficient cells.
Plasmids
The Runx1, Runx2 and Hdac3 plasmids were previously described [Schroeder et al., 2004]. The p6OSE-luciferase [Ducy and Karsenty, 1995] and murine osteocalcin gene (mOG)2-luciferase plasmids [Montecino et al., 1996] were obtained from Drs. Gerard Karsenty and Jane Lian, respectively. Dr. Lan Ko generously provided the FLAG-tagged CoAA, CoAM and AxxQ expression plasmids. Dr. Frank Costantini kindly provided the luciferase reporter construct containing the Axin2 promoter, exon1 and intron 1 in pGL3 [Jho et al., 2002]. The Axin2 promoter sequence (Accession number: AF343582) was searched for potential Runx binding sites using the Transcription Element Search System (TESS) [Schug, 2008].
Immunoprecipitations and Immunoblotting
For immunoprecipitations, U2-OS cells were lysed in phosphate buffered saline (PBS) containing 0.5% NP-40 and protease inhibitors (Roche). Equal fractions of the lysates were then incubated with Runx2 (Santa Cruz, C-19, sc-8566), CoAA (Abcam Ab12325) or FHOD1 antibodies [Westendorf et al., 1999] as an IgG control, or with no antibody as a control for non-specific binding to the beads. The immune complexes were collected with Protein G Dynabeads (Invitrogen, 100.03D). Proteins were resolved by SDS-8% PAGE, transferred to PVDF membrane (Immobilon-P, Millipore) and immunoblotted with anti-CoAA antibodies (1:3000, Abcam, Ab12325).
To detect Axin2 proteins levels, wildtype and Runx2−/− calvarial cells were lysed in modified RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 0.25% sodium deoxycholate, protease inhibitors). Proteins (100 µg) were resolved by SDS-8% PAGE, transferred to Immobilon P membranes and immunoblotted with Axin2 (1:1000, Abcam, Ab32197) and mSin3A (1:1000, Santa Cruz Biotechnology, K-20) antibodies.
Immunofluorescence
U2-OS cells were grown on glass coverslips and transiently transfected with pCMV-HA-Runx2 using Lipofectamine (Invitrogen). Two days later, the cells were fixed in 4% paraformaldehyde for 20 minutes, permeabilized with 0.3% TritonX-100 in PBS for 5 minutes, blocked for 30 minutes in immunofluorescence buffer (3% BSA, 20 mM MgCl2, 0.3% Tween20 in PBS), and incubated with anti-CoAA antibody (Abcam, Ab12325) and HA monoclonal antibody (12CA5) in immunofluorescence buffer. Cells were washed three times with 0.1% TritonX-100 in PBS, incubated 30 minutes with Alexa-conjugated secondary antibodies at 1:800 (Invitrogen), washed three times, and mounted in 90% glycerol/ 0.4% N-propyl-gallate. Images were obtained using an Olympus Fluoview 500 confocal microscope and processed using Adobe Photoshop.
GST pulldowns
GST-Runx fusion proteins and pulldowns were previously described [Schroeder et al., 2004]. Briefly, CoAA, CoAM and CoAA-AxxQ were in vitro transcribed and translated with a T7 primer and the TNT kit (Promega, L5010) in the presence of 35S-methionine. GST or the indicated GST-Runx fusion proteins were isolated from E. coli lysates with glutathione beads (GE Healthcare, 17-0756-1) and incubated with in vitro transcribed and translated 35S-labeled CoAA, CoAM or CoAA-AxxQ proteins in 10 nM MES, pH 6.5, 150 nM NaCl, 2mM MgCl2, 0.5 mM EDTA, 0.5% TX-100, 5 mM DTT. Following extensive washing in reaction buffer, the proteins were eluted from the beads by incubating at 100 degrees C for 5 minutes, resolved by SDS-10%-PAGE, and visualized by autoradiography. Gels were incubated in Autofluor (National Diagnostics, LS-315) for 15 minutes prior to drying and exposing to film.
EMSAs
COS cells were transiently transfected with pCMV5-Runx2 expression plasmids. Cells were lysed has previously described [Kahler et al., 2006]. These lysates expressing Runx2 (2 µg) were incubated with increasing amount of in vitro transcribed and translated CoAA (0, 2, 4, 8 µl) on ice for 30 minutes. A double-stranded DNA probe (5’-AATTCGAGTATTGTGGTTAATACG-3’) containing a Runx binding element (underlined) was labeled with [α- 32P] dATP using Klenow polymerase and mixed with the Runx2 expressing COS cell lysates and TnT reactions for 30 minutes on ice. Protein-DNA complexes were resolved on nondenaturing 4% polyacrylamide gel prior to drying and exposing to film. Relative quantities of Runx2-probe complexes were determined by densitometry with Image J software.
Reporter assays
C2C12 cells were transfected using Lipofectamine (Invitrogen) in 12-well plates with 200 ng of p6OSE2-luc, 50 ng of pRL-null, and unless otherwise noted, 300 ng of CMV-Runx1, 2 or 3, CoAA and Hdac3 expression plasmids as indicated. pcDNA3.1 was added to maintain a uniform amount of total DNA per transfection. The Axin2-promoter (200 ng) and murine osteocalcin gene 2 (mOG2) promoter (200 ng) were transfected with pRL-null luciferase in selected experiments. Luciferase activity was measured 24 hours after transfection using the Dual-Luciferase Assay System (Promega). Each transfection was performed in triplicate and normalized to Renilla luciferase activity. The pan-Hdac inhibitor, Trichostatin A (TSA, (20 nM, Sigma T8552), was added 24h post transfection in the indicated experiments.
RT-PCR
Primary calvarial osteoblasts from wildtype and Runx2−/− mice were lysed with Trizol (Invitrogen) to collect RNA. Quantitative RT-PCR was performed using the QuantiTech SYBR Green RT-PCR kit (Qiagen) in an iCycler (BioRad). Briefly, RNA (10 ng) was added to a 20 µl reaction with QuantiTech SYBR Green RT mastermix, QuantiTech RT mix, and 0.5 pmol/µl of each of the primers for murine Axin2 (Forward: 5’-CGCCACCAAGACCTACATACG-3’, Reverse: 5’-ACATGACCGAGCCGATCTGT-3’) or actin (Forward: 5’-AAGGAAGGCTGGAAAAGAGC-3’, Reverse: 5’-GCTACAGCTTCACCACCACA-3’). Data were normalized to mouse actin levels and relative amounts of Axin2 were calculated using the previously described 2−ΔΔCt method [Pfaffl, 2001].
RNA Interference and Proliferation Assays
Pre-designed ON-TARGET plus SMARTpool RBM14 (CoAA) or non-targeting siRNA #1 (Thermo Scientific; L-020144-00 and D-001810-01-05, respectively) were introduced into U2-OS cells using DharmaFECT™ 1 transfection reagent (Thermo Scientific, T-2001-01, 2 µl/ml) as directed by the manufacturer. Protein lysates were harvested after 48 hours from cells cultured in 12 well plates for immunoblot analysis using anti-CoAA (Abcam, ab12325) and anti-Lamin B (sc-6216, Santa Cruz Biotechnology) antibodies. Non-radioactive cell proliferation assays (Promega, G5430) were performed at 24, 48 and 72 hours post-transfection on cells cultured in 96 well plates.
RESULTS
CoAA Interacts with Runx2 In Vivo
We identified CoAA as a potential binding partner of Runx2 in a tandem affinity purification experiment [Jensen et al., 2008]. To verify the proteomic identification of CoAA as a Runx2 interacting protein, native Runx2 complexes were collected by immunoprecipitation from U2-OS human osteosarcoma cells and immunoblotted with CoAA antibodies. As expected, CoAA was present in Runx2 immunoprecipitates (Figure 1A). CoAA was also immunoprecipitated with a CoAA antibody, but not by a control immunoglobulin or beads alone. The in vivo interactions between Runx2 and CoAA were also assessed by immunofluorescence. Both proteins were nuclear and localized in punctate sub-nuclear structures (Figure 1B). Runx2 and CoAA were found co-localized in some nuclear speckles; however, the degree of co-localization varied and was more extensive in cells expressing higher levels of Runx2. Together these data demonstrate that Runx2 and CoAA can be components of the same protein complex, but that they are not obligate partners.
Figure 1. CoAA Interacts with Runx2 In Vivo.
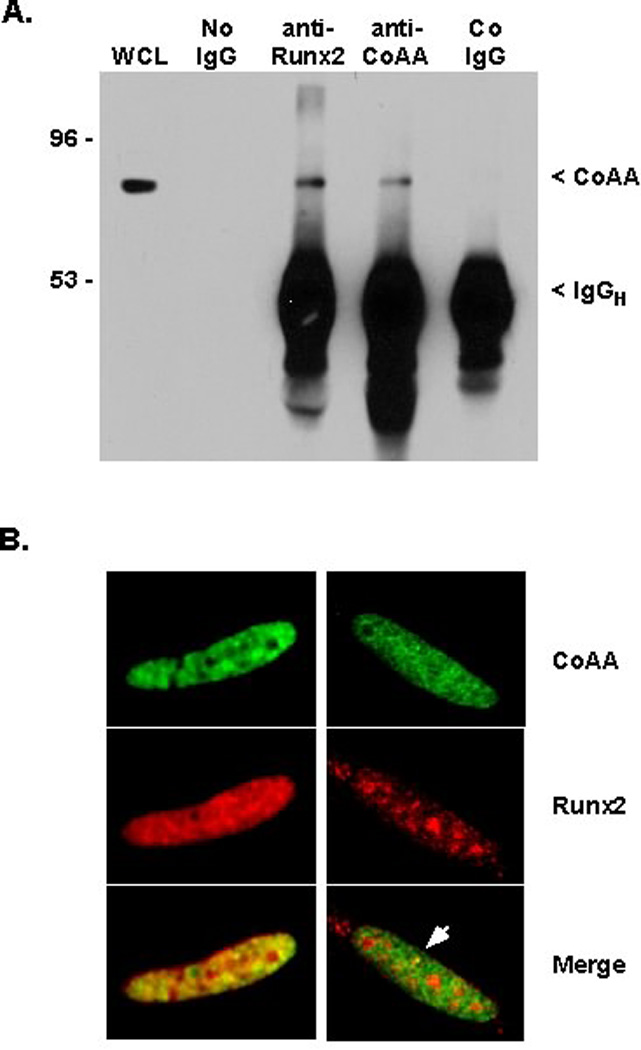
A. CoAA Immunoprecipitates with Runx2. Whole cell lysates (WCL) of the rat osteosarcoma cell line, UMR-106-1 were immunoprecipitated with Protein G Dynabeads alone (no IgG), or antibodies to CoAA, Runx2, or FHOD1 (control IgG). Proteins were transferred to membranes and detected by immunoblotting with CoAA antibodies. IgGH denotes the immunoglobulin heavy chain. B. The co-localization (yellow) of Runx2 (red) and CoAA (green) was confirmed in U2-OS cells transfected with HA-Runx2 by immunofluorescence.
The Carboxy-Terminal Portion of CoAA Associates with Runt Domain of Runx2
In vitro pulldown assays were performed to identify the region(s) of Runx2 that associates with CoAA. For these experiments, GST-Runx2 fusion proteins (Figure 2A) were incubated with in vitro transcribed and translated CoAA. CoAA bound to Runx2 proteins containing amino acids 1–227, 1–250, 1–327 and 1–383 (numbering is from the MRIPV isoform) (Figure 2B). CoAA did not bind to GST or to GST-Runx2 fusion proteins containing amino acids 1–117 and 383–513 of Runx2. These data demonstrate that amino acids 118–227 of Runx2 are necessary for binding CoAA. These residues include the highly conserved Runt domain, which is a well-characterized DNA and protein-binding region. To determine if a Runt domain was sufficient to bind CoAA, we tested a GST-Runx1 fusion protein containing residues 50–179 of isoform AML-1B [Meyers et al., 1995]. CoAA associated strongly with the Runt domain from Runx1. Together, these data demonstrate that the Runt domain is sufficient and necessary for CoAA associations with Runx factors.
Figure 2. CoAA Binds the Runt Domain of Runx Factors.

A. This diagram illustrates the GST-Runx2 and GST–Runx1 fusion proteins used in this assay. RuntD is the Runt domain, which is 93% identical in Runx1 and Runx2. B. GST or the indicated GST-Runx fusion proteins were isolated from E. coli lysates with glutathione beads. These beads were incubated with in vitro transcribed and translated, 35S-labeled CoAA. Following extensive washing, the proteins were eluted from the beads with heat, resolved by SDS-PAGE and visualized by autoradiography.
CoAA contains two RRMs in its amino-terminus and 27 copies of a tyrosine- and glutamine-rich (YxxQ) sequence in it carboxy-terminus, where×is a small amino acid (e.g. glycine, alanine, serine or proline) (Figure 3A). The carboxy-terminus is necessary for binding the TRBP co-activator [Iwasaki et al., 2001]. The amino-terminal RRMs are maintained in the naturally occurring co-activator modulator (CoAM) protein, which is generated by alternative splicing of the CoAA gene. To identify the regions of CoAA that bound Runx proteins and determine if CoAM interacts with Runx2, in vitro transcribed and translated CoAA or CoAM were incubated with GST-Runx2 (1–383). CoAA, but not CoAM, bound to the amino-terminus of Runx2 (Figure 3B). A CoAA protein in which all the tyrosines in the 27 YxxQ sequences were mutated to alanines also failed to associate with the Runx2 amino terminus. These data indicate that the RRMs of CoAA do not associate directly with Runx2 and that the tyrosines in the carboxy-terminal half of CoAA are essential for interacting with Runx2.
Figure 3. The Carboxy-terminus of CoAA is Necessary for Binding Runx2.
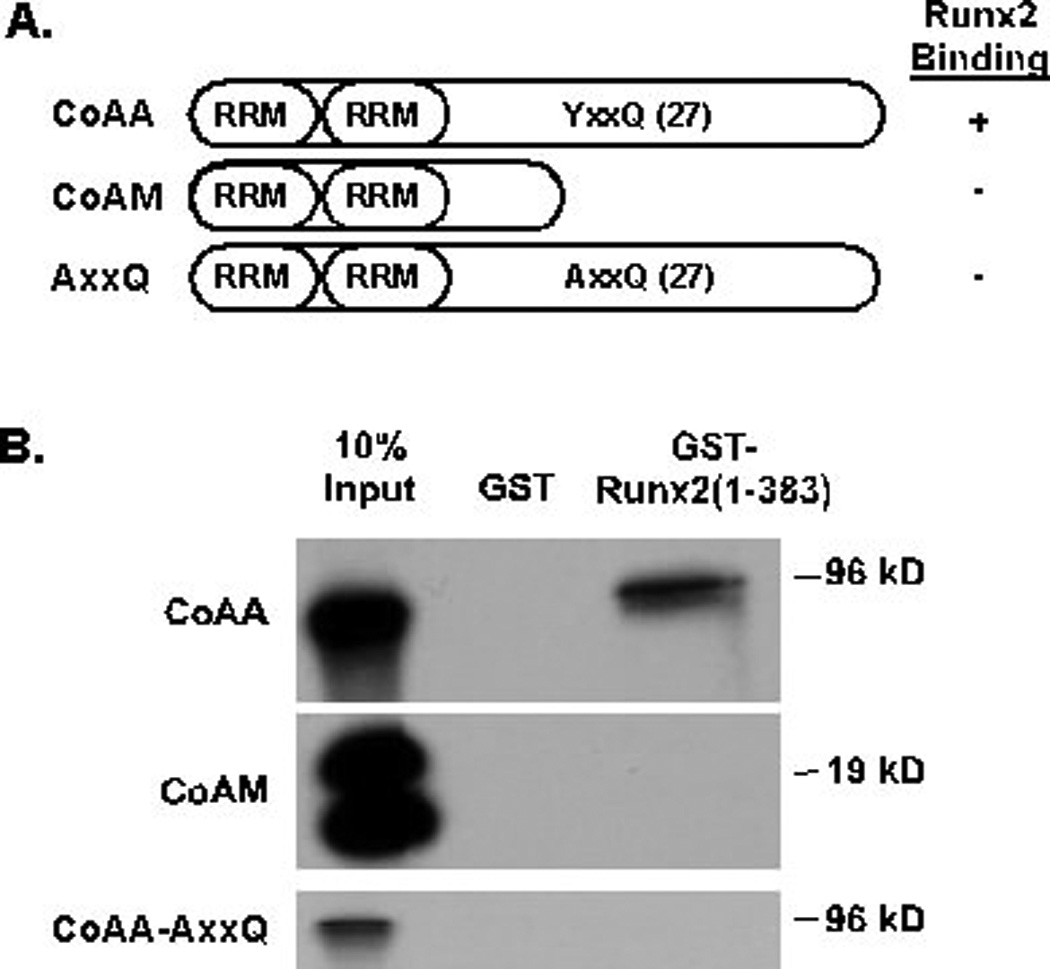
A. This diagram illustrates the CoAA and CoAM proteins used in this assay. The YxxQ motif is repeated 27 times in the carboxy-terminus of CoAA. In the AxxQ protein, all the tyrosines (Y) were changed to alanine (A). RRMs are RNA recognition motifs. B. Glutathione beads bound to GST or GST-Runx2(1–383) proteins were incubated with in vitro transcribed and translated, 35S-labeled CoAA, CoAM or CoAA-AxxQ. Proteins were detected as described in Figure 2.
CoAA Represses Runx Factor Transcriptional Activity in a Histone Deacetylase Independent Manner
Because CoAA was identified as a transcriptional co-factor of steroid hormone receptors, we assessed the effects of CoAA on Runx2 transcriptional activity. CoAA suppressed the basal activity of p6OSE2-luciferase, a Runx responsive reporter, by 40% in C2C12 cells. Runx2 isoforms I and II activated this reporter by 12 to 60 fold, but CoAA completely blocked the Runx2-dependent stimulation (Figure 4A and B). CoAA also blocked the transcriptional activation of p6OSE-luciferase by Runx1 and Runx3 (Figure 4B) and the basal activity of the osteocalcin promoter (Figure 4C), which contains three essential Runx2 binding sites. The naturally occurring CoAA isoform, CoAM, which contains the two RRMs, did not block Runx2-dependent transcription, but CoAA-AxxQ partially inhibited Runx2 (Figure 4D). Neither CoAM nor the AxxQ mutant competitively inhibited CoAA repression of Runx2 (Figure 4E), as was expected since neither bound to Runx2 (Figure 3). Co-transfection of CoAA expression plasmids did not affect the stability or alter the levels of Runx2 proteins (data not shown); thus, alternative mechanisms of repression were examined.
Figure 4. CoAA Represses Runx Factor Transcriptional Activity in a Histone Deacetylase Independent Manner.

The effects of CoAA on Runx factor transcriptional activity were performed in C2C12 cells co-transfected with p6OSE2-luciferase or mOG2-luciferase, pRenilla luciferase and, unless otherwise noted, 300 ng of the indicated expression plasmids. Empty pcDNA3 was added to equalize DNA amounts transfected into cells. Firefly luciferase levels were normalized to renilla luciferase levels. Results represent the mean of triplicate samples. A. CoAA blocks Runx2-I (begins with amino acids MRIPV)-dependent activation of p6OSE2. B. CoAA blocks Runx1, Runx2-II (begins with residues MASNSL), and Runx3-induced activation of p6OSE2. C. CoAA blocks the basal activity of the mouse osteocalcin promoter (mOG2)-luciferase. D. Runx2-II-dependent activation of p6OSE2-luciferase is completely blocked by CoAA, but not by CoAM. CoAA-AxxQ mutant constructs partially inhibit Runx2-II-dependent activation. E. Neither CoAM nor AxxQ inhibits CoAA-mediated repression of Runx2-II-driven p6OSE2-luciferase. F. Hdac3 and CoAA repress Runx2-II activity, but do not synergize to repress Runx2-II. E. The Hdac inhibitor, trichostatin A (TSA) was added to cell culture during the last 18 hours of incubation. TSA increases Runx2-II-dependent activation of p6OSE2-luciferase but did not relieve CoAA repression of Runx2-II.
Although it was originally identified as a co-activator, CoAA was recently shown to interact with Hdac3 and repress the expression of the c-myc proto-oncogene [Kang et al., 2008]. As we previously reported [Schroeder et al., 2004], Hdac3 repressed Runx2 transcriptional activity (Figure 4F); however, co-transfection of Hdac3 did not augment CoAA-mediated repression of Runx2 activity on p6OSE2-Luc. Moreover, the Hdac inhibitor, trichostatin A (TSA), did not block CoAA-directed repression (Figure 4E), although it enhanced the basal activity of the reporter as we previously showed [Schroeder et al., 2004]. These data indicate that CoAA represses Runx factor transcriptional activity in a deacetylase-independent manner.
CoAA Interferes with Runx2-DNA Interactions
CoAA bound to GST-Runx2 proteins that contained the highly conserved Runt domain (Figure 2) and repressed transcriptional activation by Runx1, Runx2 and Runx3 (Figure 4). Therefore, it was hypothesized that CoAA interferes with the binding of Runx factors to DNA. To test this hypothesis, Runx2-expressing lysates were incubated with a radiolabeled double-stranded oligonucleotide probe in the presence of increasing concentrations of CoAA-programmed lysates. As more CoAA protein was added to the mixture, approximately 40% fewer Runx2-probe complexes were detected (Figure 5A). The shorter isoform, CoAM, did not affect Runx2-DNA probe complexes (Figure 5B).
Figure 5. CoAA Prevents Runx2 from Binding DNA.
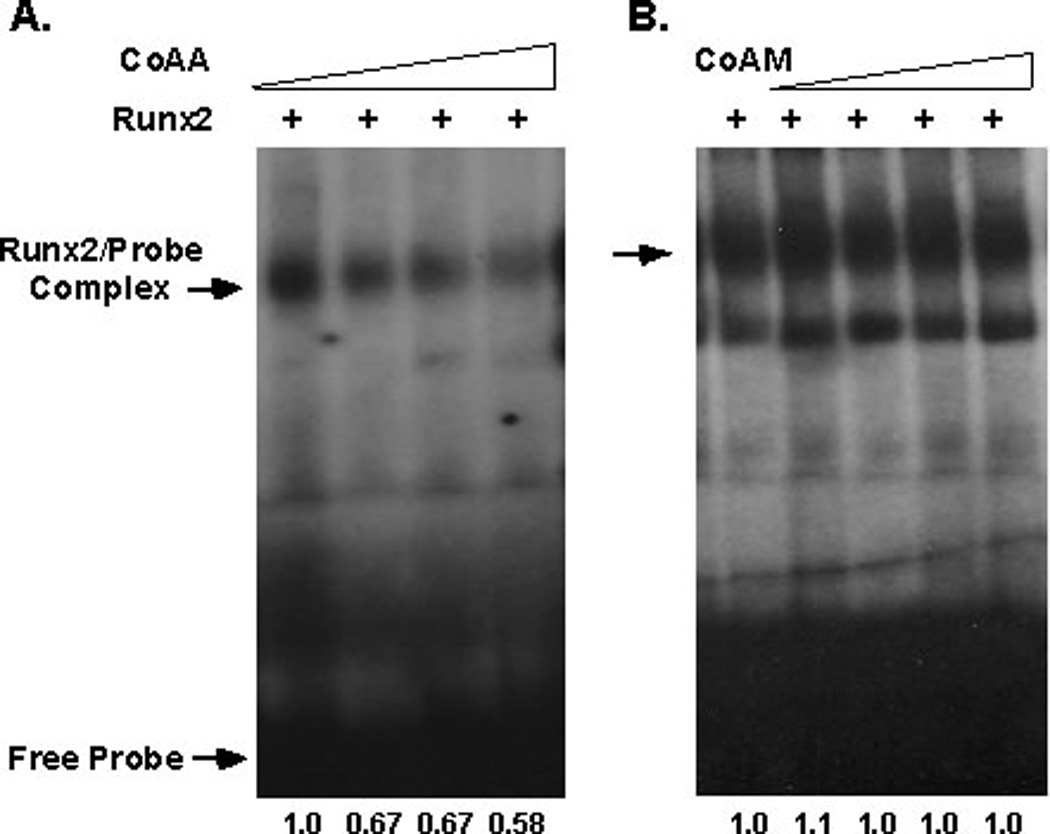
EMSAs were performed with radiolabeled double-stranded oligonucleotides containing a Runx binding site. Runx2 expressing lysates (2 µg) were mixed with the probe and increasing amounts (0, 1, 2, 4 or 8 µl) of CoAA- (A) or CoAM- (B) programmed in vitro transcribed and translated reactions. Relative quantities of Runx2-probe complexes were determined by densitometry with Image J software.
Runx2 is an activator as well as a repressor of gene expression. Data presented in Figure 2 show that CoAA blocks Runx2-dependent activation of reporter constructs. If CoAA prevents Runx2 DNA binding, then it should also relieve the repressive effects of Runx2 on specific target genes. We found that Axin2 mRNA levels are elevated 5.5-fold in mesenchymal cells derived from calvaria of Runx2−/− mice as compared to cells isolated from calvaria of wildtype mice (data not shown). Axin2 protein levels were also higher in the Runx2-deficient cells (Figure 6A). Four consensus Runx binding sites were identified in the Axin2 promoter (data not shown). Runx2 repressed the Axin2 promoter (Figure 6B). CoAA prevented Runx2 repression of the Axin2 promoter construct in a concentration-dependent manner (Figure 6B and 6C). The short isoform, CoAM, had no effect on Runx2 repression (Figure 6C). These data support the conclusion that CoAA inhibits Runx factor transcriptional activity by blocking DNA binding.
Figure 6. CoAA Prevents Runx2-Directed Repression of the Axin2 Promoter.
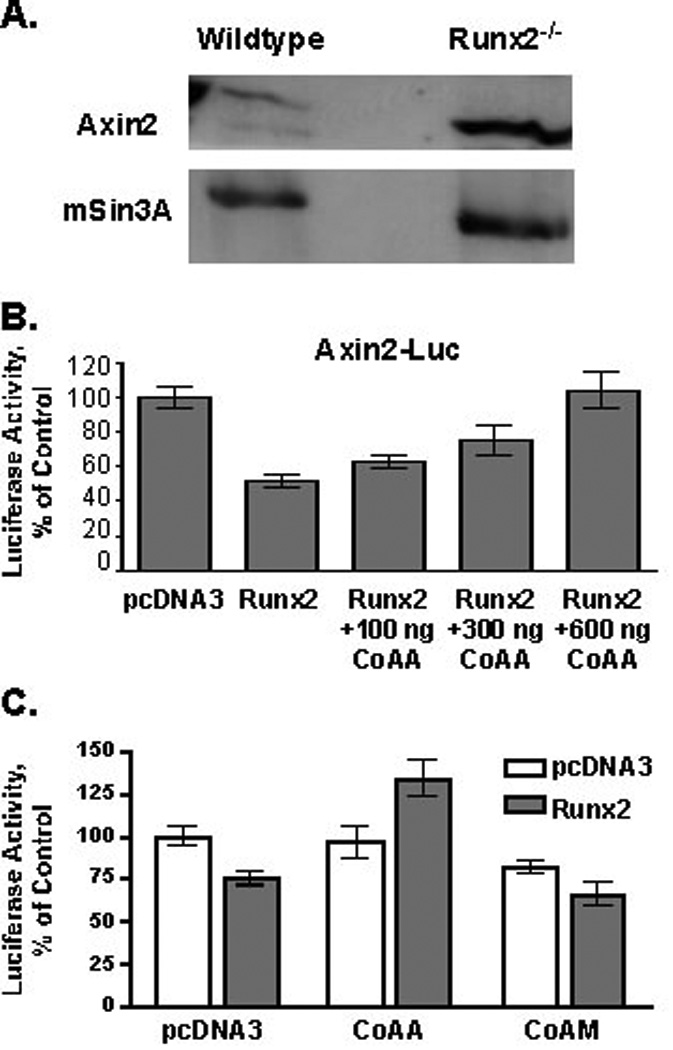
A. Axin2 is elevated in Runx2-deficient cells. Immunoblot analysis of Axin2 and mSin3a (loading control) in wildtype and Runx2−/− calvarial cells. B. CoAA blocks Runx2-mediated repression of the Axin2-promoter. C2C12 cells were transiently transfected with Axin2-firefly luciferase (luc) reporter, pRenilla-luciferase (pRL), pcDNA3-Runx2 (300 ng) and increasing concentrations of CoAA expression plasmids. Empty pcDNA3 was added to equalize DNA amounts transfected into cells. Firefly luciferase levels were normalized to renilla luciferase levels. Results represent the mean of triplicate samples. C. CoAA, but not CoAM, blocks Runx2-mediated repression of the Axin2-promoter. C2C12 cells were transiently transfected as described above using 300 ng of the CoAA and CoAM expression plasmids.
CoAA is Necessary for Osteosarcoma Survival In Vitro
CoAA is amplified in some human cancers [Auboeuf et al., 2002; Iwasaki et al., 2001; Sui et al., 2007]. In vitro assays showed that CoAA overexpression transforms fibroblasts and that CoAA suppression by RNA interference blocks DNA synthesis in human lung cancer cells [Sui et al., 2007]. However, CoAA siRNAs stimulate the proliferation of human kidney cells and CoAA levels are low in human renal cell carcinomas [Kang et al., 2008]. To assess CoAA levels in human osteoblast lineage cells, immunoblots were performed on lysates from human fetal osteoblasts (FOB) and several osteosarcoma cell lines (U2-OS, 143B, and MG63). CoAA was abundantly expressed in all cell lines (Figure 7A). CoAA expression levels were reduced in U2-OS cells by siRNAs (Figure 7B). CoAA suppression progressively inhibited U2-OS viability and suppressed proliferation by 43% after three days (Figure 7C). These data suggest that CoAA contributes to the proliferation and/or survival of osteosarcomas.
Figure 7. CoAA Suppression Blocks Osteosarcoma Cell Growth In Vitro.
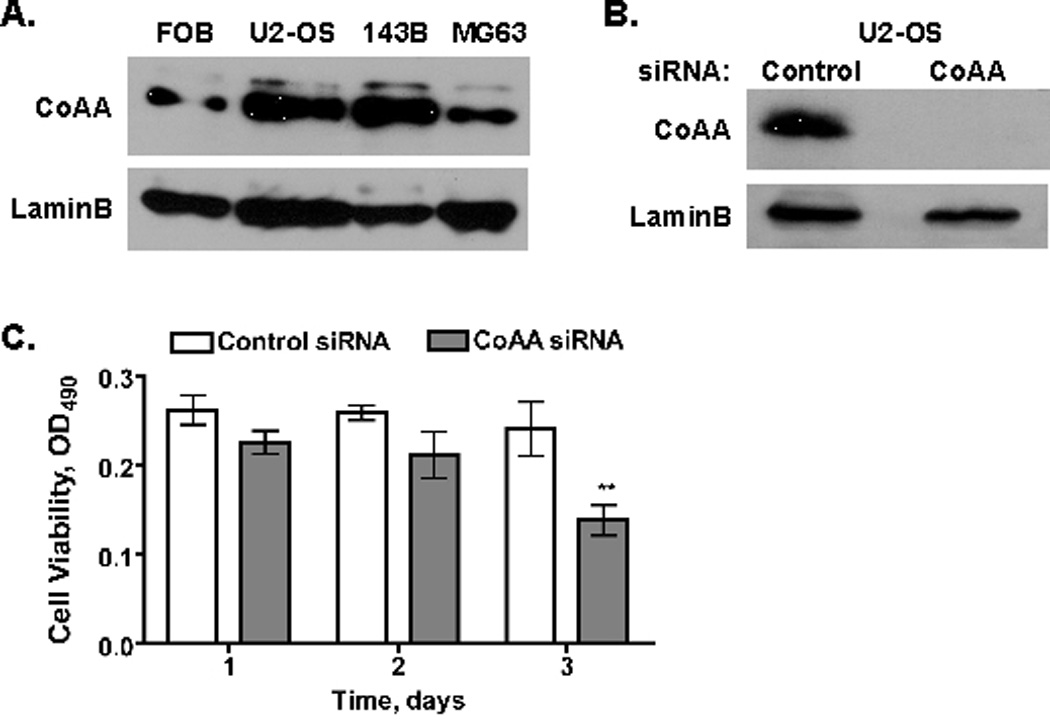
A. CoAA is expressed at high levels in human osteoblast-derived cell lines. Cell extracts from the indicated cell lines were resolved by SDS-PAGE and immunoblotted with antibodies to CoAA or LaminB. B. siRNAs decrease CoAA expression. U2-OS cells were incubated for 2 days with control or CoAA siRNAs. Cell extracts from the indicated cell lines were resolved by SDS-PAGE and immunoblotted with antibodies to CoAA or LaminB. C. CoAA suppression reduces U2-OS cell viability. U2-OS cells were transfected with control or CoAA siRNAs for 1, 2, or 3 days. Cell viability was measured with the MTT assay. Results represent the mean of triplicate samples ± SD; **p<0.01.
DISCUSSION
Runx2 is essential for osteoblast development from mesenchymal progenitors and is necessary for chondrocyte hypertrophy [Komori et al., 1997; Otto et al., 1997]. Although required for proper skeletal development, overexpression of Runx2 is harmful for bone health as it increases osteoclast differentiation and bone resorption [Geoffroy et al., 2002; Liu et al., 2001]. Others have shown that Runx2 has both oncogenic and tumor suppressor activities [Cameron and Neil, 2004; Kilbey et al., 2008; Pratap et al., 2003; Zaidi et al., 2007a]. Together, these seemingly contradictory results indicate that Runx2 activity must be tightly regulated and is context dependent. In fact, Runx2 levels are regulated in numerous ways, including by transcriptional, translational and post-transcriptional mechanisms [Schroeder et al., 2005]. To better understand Runx2’s functions in cells we have focused on identifying proteins that interact with Runx2 and regulate its activity. We previously described a proteomic experiment wherein Runx2 complexes were purified from rat UMR-106 cells stably expressing tandem affinity protein (TAP)-Runx2 fusion proteins [Jensen et al., 2008]. Runx2 interacted strongly with the DEAD box protein, Ddx5 (p68), as well as to actinin, gelsolin and heat shock protein 70 [Jensen et al., 2008]. The proteomic analysis of the gel slice containing Ddx5 also indicated that CoAA was co-precipitated with TAP-Runx2. In this manuscript, we confirm the presence of native Runx2-CoAA complexes in human osteosarcoma cells (U2-OS, Figure 1A). CoAA bound the Runt domains of Runx2 and Runx1 (Figure 2) and prevented Runx2 from binding DNA (Figure 5). This physical interaction between CoAA and the DNA binding domain of Runx proteins blocked the transcriptional activation of a reporter by all Runx factors (Figure 4) and reversed the repression of the Axin2 promoter by Runx2 (Figure 6). Thus, CoAA inhibits direct Runx factor transcriptional activity by preventing DNA binding. Together, these results identify CoAA as a suppressor of Runx2 transcriptional activity.
Confocal microscopy studies indicate that the frequency of the CoAA and Runx2 interactions is variable within cells, but more frequent in cells expressing high levels of Runx2 (Figure 1B). Thus CoAA might provide a mechanism to control excess Runx factor levels. However, since CoAA blocks Runx factor DNA binding, complete co-localization would negate Runx factor transcriptional activity. Thus limited colocalization would be beneficial to cells that require Runx factor activity.
CoAA and Runx factors are both known to localize to highly organized but dynamic domains within the nucleus. Runx factors associate with the nuclear matrix, which is structural network of ribonuclear proteins that excludes chromatin and is resistant to detergents and high salt concentrations in extraction buffers [Zaidi et al., 2007b]. Runx proteins are especially enriched near ribosomal RNA genes in nucleoli during interphase and with nucleolar organizing regions on metaphase chromosomes [Bakshi et al., 2008; Pande et al., 2009; Young et al., 2007]. Structural properties of CoAA predict that it might also associate with the nuclear matrix. CoAA has two RRMs and the YxxQ motif; the latter of which is present in several other transcription-coupled splicing factors and is reminiscent of residue repeats in heterogeneous nuclear ribonucleoproteins that are important for protein-protein interactions [Iwasaki et al., 2001; Perani et al., 2005; Sui et al., 2007]. In fact, CoAA localizes to distinct domains in the nucleus called paraspeckles and was independently described as paraspeckle protein 2 (PSP2) [Fox et al., 2002]. Paraspeckles are interchromatin bodies that are adjacent to splicing speckles. CoAA appears to shuttle between these sub-nuclear compartments and nucleoli [Fox et al., 2002]. This dynamic movement could explain the variability we observed in Runx2 and CoAA co-localization in transiently transfected cells. We observed co-localization of Runx2 and CoAA in some punctate nuclear domains as well as in less organized nuclear regions. Because Runx2 interacts with the nuclear matrix via a carboxy-terminal sequence that is distinct from the DNA- and CoAA-binding Runt domain [Zaidi et al., 2007b], it is possible that CoAA and Runx2 complexes have non-transcriptional functions in nuclei. It is also likely that the CoAA and Runx2 complexes are highly dynamic. Additional immunofluorescence studies are required to understand the activities associated with the CoAA and Runx2 double-positive structures and the environmental cues that influence this partnership.
We have verified two proteins (e.g. Ddx5 and CoAA) identified in our proteomic screen as novel Runx2 binding proteins [Jensen et al., 2008]. It is interesting that both Ddx5 (p68) and CoAA are steroid receptor co-activators and modulators of mini-gene splicing in vivo. There are notable differences in these complexes. First, Ddx5 is a co-activator of Runx2 [Jensen et al., 2008], but CoAA is a competitive inhibitor of its DNA binding. Second, CoAA increases exon skipping induced by ligand-bound steroid receptors [Iwasaki et al., 2001], but Ddx5 prevents androgen receptor mediated splicing [Clark et al., 2008]. Runx2 also interacts with steroid receptors [Ning and Robins, 1999], but it is unknown if Runx2 affects RNA splicing on its own or in a complex with CoAA, Ddx5, or steroid receptors and/or other nuclear matrix associated proteins. There is increasing evidence that pre-mRNA processing, including splicing, is intimately tied to transcription and translation [Moore and Proudfoot, 2009]. Our data suggest that interactions with multi-functional proteins like Ddx5 and CoAA in subnuclear structures might implicate Runx2 in multiple facets of gene expression.
Recently it was found that CoAA contributes to transcriptional repression of the c-myc proto-oncogene as a part of Hdac3 co-repressor complexes [Kang et al., 2008]. We previously showed that Hdac3 interacts with and represses Runx2-dependent transcription in a deacetylase-dependent fashion [Schroeder et al., 2005]. Hdac3 did not augment CoAA repression of Runx2 and in fact, might have partially relieved it (Figure 4F). Moreover, the Hdac inhibitor, TSA, did not relieve CoAA repression of Runx2 (Figure 4G). These data indicate Hdac3 and CoAA are likely to be mutually exclusive binding partners of Runx2.
We demonstrate that Axin2 is a novel Runx2-target gene. Axin2 (Conductin, Axil) and its homolog Axin1 are scaffolding proteins that are essential for Wnt signaling because they assemble β-catenin, the serine/threonine kinases Gsk3β and casein kinase-1, Apc, Wtx, and other proteins into a complex that regulates β-catenin stability [Huang and He, 2008]. Beta-catenin is a crucial for the development and maintenance of numerous tissues, including skeletal, neuronal, hematopoietic and intestinal tissues [Clevers, 2006]. Moreover, β-catenin is often upregulated through expression or mutatagenesis in human cancers [Polakis, 2007]. Thus, the identification of Axin2 as a Runx target gene has wide-ranging implications for Runx-expressing tissues. Axin2 negatively regulates the proliferation and differentiation of osteoblast-lineage cells by augmenting Wnt/ β-catenin and Bmp signaling pathways and Axin2-deficient mice develop postnatal craniosynostosis and craniofacial defects [Liu et al., 2007; Yu et al., 2005]. Axin2 is upregulated in Runx2-deficient mesenchymal cells and Runx2 represses its promoter (Figure 6). There are several consensus Runx binding sites in the promoter and studies are ongoing to test the hypothesis that Runx2 binds the Axin2 promoter to directly repress transcription.
CoAA was easily detected in human osteoblast lineage cells, including immortalized human fetal osteoblasts and several human osteosarcoma cell lines, but it did not seem to be overexpressed in the sarcoma lines (Figure 7). Sui and colleagues showed that CoAA is amplified in some human cancers, including lung and squamous skin carcinomas, pancreatic cancers, and lymphomas [Sui et al., 2007]. CoAA was not amplified in any of the six sarcomas tested in this multiple tumor array. Although it is not clear that how many if any of these samples were osteosarcomas, these data are consistent with our western blots demonstrating relatively equal CoAA expression in a small panel of human osseous cells. In vitro assays showed that CoAA overexpression transforms fibroblasts and that CoAA suppression by RNA interference blocks DNA synthesis in human lung cancer cells [Sui et al., 2007]. However, Kang and colleagues showed that CoAA siRNAs stimulate the proliferation of human kidney cells and CoAA levels are low in human renal cell carcinomas [Kang et al., 2008]. Our data demonstrate that osteosarcoma cells are more similar to lung cancer cells with regards to requiring CoAA for cell viability.
In summary, CoAA was identified as a novel inhibitor of Runx factor activity. CoAA blocks transcriptional activation and repression of Runx target genes and is essential for human osteosarcoma survival in vitro. CoAA interacts with Runx2 in vivo and in vitro. By interacting with the Runt domain of Runx factors, CoAA prevents Runx factors from binding to DNA sequences. CoAA and Runx2 co-localize in a subset of sub-nuclear compartments. These data reveal new mechanisms controlling Runx factor function in nuclear activities and gene expression.
ACKNOWLEDGMENTS
We thank Drs. Lan Ko and Frank Costantini for generously sharing the CoAA expression vectors and the Axin2-luciferase reporter, respectively. We are also grateful to Dr. André VanWijnen for providing the Runx2-deficient cells and Dr. Maran Avudaiappan for the osteosarcoma cell lines. The National Institutes of Health (RO1-AR048147, S10-RR16851) and the Mayo Foundation supported this project.
REFERENCES
- Auboeuf D, Dowhan DH, Li X, Larkin K, Ko L, Berget SM, O'Malley BW. CoAA, a nuclear receptor coactivator protein at the interface of transcriptional coactivation and RNA splicing. Mol Cell Biol. 2004;24:442–453. doi: 10.1128/MCB.24.1.442-453.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auboeuf D, Honig A, Berget SM, O'Malley BW. Coordinate regulation of transcription and splicing by steroid receptor coregulators. Science. 2002;298:416–419. doi: 10.1126/science.1073734. [DOI] [PubMed] [Google Scholar]
- Bakshi R, Zaidi SK, Pande S, Hassan MQ, Young DW, Montecino M, Lian JB, van Wijnen AJ, Stein JL, Stein GS. The leukemogenic t(8;21) fusion protein AML1-ETO controls rRNA genes and associates with nucleolar-organizing regions at mitotic chromosomes. J Cell Sci. 2008;121:3981–3990. doi: 10.1242/jcs.033431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blyth K, Cameron ER, Neil JC. The RUNX genes: gain or loss of function in cancer. Nat Rev Cancer. 2005;5:376–387. doi: 10.1038/nrc1607. [DOI] [PubMed] [Google Scholar]
- Cameron ER, Neil JC. The Runx genes: lineage-specific oncogenes and tumor suppressors. Oncogene. 2004;23:4308–4314. doi: 10.1038/sj.onc.1207130. [DOI] [PubMed] [Google Scholar]
- Clark EL, Coulson A, Dalgliesh C, Rajan P, Nicol SM, Fleming S, Heer R, Gaughan L, Leung HY, Elliott DJ, Fuller-Pace FV, Robson CN. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res. 2008;68:7938–7946. doi: 10.1158/0008-5472.CAN-08-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469–480. doi: 10.1016/j.cell.2006.10.018. [DOI] [PubMed] [Google Scholar]
- Daga A, Tighe JE, Calabi F. Leukaemia/Drosophila homology. Nature. 1992;356:484. doi: 10.1038/356484b0. [DOI] [PubMed] [Google Scholar]
- Ducy P, Karsenty G. Two distinct osteoblast-specific cis-acting elements control expression of a mouse osteocalcin gene. Mol Cell Biol. 1995;15:1858–1869. doi: 10.1128/mcb.15.4.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durst KL, Hiebert SW. Role of RUNX family members in transcriptional repression and gene silencing. Oncogene. 2004;23:4220–4224. doi: 10.1038/sj.onc.1207122. [DOI] [PubMed] [Google Scholar]
- Fox AH, Lam YW, Leung AK, Lyon CE, Andersen J, Mann M, Lamond AI. Paraspeckles: a novel nuclear domain. Curr Biol. 2002;12:13–25. doi: 10.1016/s0960-9822(01)00632-7. [DOI] [PubMed] [Google Scholar]
- Geoffroy V, Kneissel M, Fournier B, Boyde A, Matthias P. High bone resorption in adult aging transgenic mice overexpressing cbfa1/runx2 in cells of the osteoblastic lineage. Mol Cell Biol. 2002;22:6222–6233. doi: 10.1128/MCB.22.17.6222-6233.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, He X. Wnt/beta-catenin signaling: new (and old) players and new insights. Curr Opin Cell Biol. 2008;20:119–125. doi: 10.1016/j.ceb.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y. Oncogenic potential of the RUNX gene family: 'overview'. Oncogene. 2004;23:4198–4208. doi: 10.1038/sj.onc.1207755. [DOI] [PubMed] [Google Scholar]
- Iwasaki T, Chin WW, Ko L. Identification and characterization of RRM-containing coactivator activator (CoAA) as TRBP-interacting protein, and its splice variant as a coactivator modulator (CoAM) J Biol Chem. 2001;276:33375–33383. doi: 10.1074/jbc.M101517200. [DOI] [PubMed] [Google Scholar]
- Jensen ED, Niu L, Caretti G, Nicol SM, Teplyuk N, Stein GS, Sartorelli V, van Wijnen AJ, Fuller-Pace FV, Westendorf JJ. p68 (Ddx5) interacts with Runx2 and regulates osteoblast differentiation. J Cell Biochem. 2008;103:1438–1451. doi: 10.1002/jcb.21526. [DOI] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F. Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol. 2002;22:1172–1183. doi: 10.1128/MCB.22.4.1172-1183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahler RA, Galindo M, Lian J, Stein GS, van Wijnen AJ, Westendorf JJ. Lymphocyte enhancer-binding factor 1 (Lef1) inhibits terminal differentiation of osteoblasts. J Cell Biochem. 2006;97:969–983. doi: 10.1002/jcb.20702. [DOI] [PubMed] [Google Scholar]
- Kamachi Y, Ogawa E, Asano M, Ishida S, Murakami Y, Satake M, Ito Y, Shigesada K. Purification of a mouse nuclear factor that binds to both the A and B cores of the polyomavirus enhancer. J Virol. 1990;64:4808–4819. doi: 10.1128/jvi.64.10.4808-4819.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang YK, Schiff R, Ko L, Wang T, Tsai SY, Tsai MJ, O'Malley BW. Dual roles for coactivator activator and its counterbalancing isoform coactivator modulator in human kidney cell tumorigenesis. Cancer Res. 2008;68:7887–7896. doi: 10.1158/0008-5472.CAN-08-1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilbey A, Terry A, Cameron ER, Neil JC. Oncogene-induced senescence: an essential role for Runx. Cell Cycle. 2008;7:2333–2340. doi: 10.4161/cc.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–764. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- Lamour V, Detry C, Sanchez C, Henrotin Y, Castronovo V, Bellahcene A. Runx2- and histone deacetylase 3-mediated repression is relieved in differentiating human osteoblast cells to allow high bone sialoprotein expression. J Biol Chem. 2007;282:36240–36249. doi: 10.1074/jbc.M705833200. [DOI] [PubMed] [Google Scholar]
- Levanon D, Bettoun D, Harris-Cerruti C, Woolf E, Negreanu V, Eilam R, Bernstein Y, Goldenberg D, Xiao C, Fliegauf M, Kremer E, Otto F, Brenner O, Lev-Tov A, Groner Y. The Runx3 transcription factor regulates development and survival of TrkC dorsal root ganglia neurons. Embo J. 2002;21:3454–3463. doi: 10.1093/emboj/cdf370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QL, Ito K, Sakakura C, Fukamachi H, Inoue K, Chi XZ, Lee KY, Nomura S, Lee CW, Han SB, Kim HM, Kim WJ, Yamamoto H, Yamashita N, Yano T, Ikeda T, Itohara S, Inazawa J, Abe T, Hagiwara A, Yamagishi H, Ooe A, Kaneda A, Sugimura T, Ushijima T, Bae SC, Ito Y. Causal relationship between the loss of RUNX3 expression and gastric cancer. Cell. 2002;109:113–124. doi: 10.1016/s0092-8674(02)00690-6. [DOI] [PubMed] [Google Scholar]
- Lian JB, Stein GS, Javed A, van Wijnen AJ, Stein JL, Montecino M, Hassan MQ, Gaur T, Lengner CJ, Young DW. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev Endocr Metab Disord. doi: 10.1007/s11154-006-9001-5. [DOI] [PubMed] [Google Scholar]
- Liu B, Yu HM, Hsu W. Craniosynostosis caused by Axin2 deficiency is mediated through distinct functions of beta-catenin in proliferation and differentiation. Dev Biol. 2006;2007;301:298–308. doi: 10.1016/j.physletb.2003.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Toyosawa S, Furuichi T, Kanatani N, Yoshida C, Liu Y, Himeno M, Narai S, Yamaguchi A, Komori T. Overexpression of Cbfa1 in osteoblasts inhibits osteoblast maturation and causes osteopenia with multiple fractures. J Cell Biol. 2001;155:157–166. doi: 10.1083/jcb.200105052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makita N, Suzuki M, Asami S, Takahata R, Kohzaki D, Kobayashi S, Hakamazuka T, Hozumi N. Two of four alternatively spliced isoforms of RUNX2 control osteocalcin gene expression in human osteoblast cells. Gene. 2008;413:8–17. doi: 10.1016/j.gene.2007.12.025. [DOI] [PubMed] [Google Scholar]
- Manoukian AS, Krause HM. Control of segmental asymmetry in Drosophila embryos. Development. 1993;118:785–796. doi: 10.1242/dev.118.3.785. [DOI] [PubMed] [Google Scholar]
- Meyers S, Downing JR, Hiebert SW. Identification of AML- 1 and the (8;21) translocation protein (AML- 1/ETO) as sequence-specific DNA-binding proteins: the runt homology domain is required for DNA binding and protein-protein interactions. Mol Cell Biol. 1993;13:6336–6345. doi: 10.1128/mcb.13.10.6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers S, Lenny N, Hiebert SW. The t(8;21) fusion protein interferes with AML-1B-dependent transcriptional activation. Molecular & Cellular Biology. 1995;15:1974–1982. doi: 10.1128/mcb.15.4.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montecino M, Frenkel B, Lian J, Stein J, Stein G. Requirement of distal and proximal promoter sequences for chromatin organization of the osteocalcin gene in bone-derived cells. J Cell Biochem. 1996;63:221–228. doi: 10.1002/(SICI)1097-4644(19961101)63:2%3C221::AID-JCB9%3E3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Moore MJ, Proudfoot NJ. Pre-mRNA processing reaches back to transcription and ahead to translation. Cell. 2009;136:688–700. doi: 10.1016/j.cell.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Niki M, Okada H, Takano H, Kuno J, Tani K, Hibino H, Asano S, Ito Y, Satake M, Noda T. Hematopoiesis in the fetal liver is impaired by targeted mutagenesis of a gene encoding a non-DNA binding subunit of the transcription factor, polyomavirus enhancer binding protein 2/core binding factor. Proc Natl Acad Sci U S A. 1997;94:5697–5702. doi: 10.1073/pnas.94.11.5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ning YM, Robins DM. AML3/CBFalpha1 is required for androgen-specific activation of the enhancer of the mouse sex-limited protein (Slp) gene. J Biol Chem. 1999;274:30624–30630. doi: 10.1074/jbc.274.43.30624. [DOI] [PubMed] [Google Scholar]
- Otto F, Thornell AP, Crompton T, Denzel A, Gilmour KC, Rosewell IR, Stamp GW, Beddington RS, Mundlos S, Olsen BR, Selby PB, Owen MJ. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89:765–771. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- Pande S, Ali SA, Dowdy C, Zaidi SK, Ito K, Ito Y, Montecino MA, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Subnuclear targeting of the Runx3 tumor suppressor and its epigenetic association with mitotic chromosomes. J Cell Physiol. 2009;218:473–479. doi: 10.1002/jcp.21630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perani M, Antonson P, Hamoudi R, Ingram CJ, Cooper CS, Garrett MD, Goodwin GH. The proto-oncoprotein SYT interacts with SYT-interacting protein/co-activator activator (SIP/CoAA), a human nuclear receptor co-activator with similarity to EWS and TLS/FUS family of proteins. J Biol Chem. 2005;280:42863–42876. doi: 10.1074/jbc.M502963200. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Pratap J, Galindo M, Zaidi SK, Vradii D, Bhat BM, Robinson JA, Choi JY, Komori T, Stein JL, Lian JB, Stein GS, van Wijnen AJ. Cell growth regulatory role of Runx2 during proliferative expansion of preosteoblasts. Cancer Res. 2003;63:5357–5362. [PubMed] [Google Scholar]
- Redondo JM, Pfohl JL, Hernandez-Munain C, Wang S, Speck NA, Krangel MS. Indistinguishable nuclear factor binding to functional core sites of the T-cell receptor delta and murine leukemia virus enhancers. Mol Cell Biol. 1992;12:4817–4823. doi: 10.1128/mcb.12.11.4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redondo JM, Pfohl JL, Krangel MS. Identification of an essential site for transcriptional activation within the human T-cell receptor delta enhancer. Mol. Cell. Biol. 1991;11:5671–5680. doi: 10.1128/mcb.11.11.5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder TM, Jensen ED, Westendorf JJ. Runx2: a master organizer of gene transcription in developing and maturing osteoblasts. Birth Defects Res C Embryo Today. 2005;75:213–225. doi: 10.1002/bdrc.20043. [DOI] [PubMed] [Google Scholar]
- Schroeder TM, Kahler RA, Li X, Westendorf JJ. Histone deacetylase 3 interacts with runx2 to repress the osteocalcin promoter and regulate osteoblast differentiation. J Biol Chem. 2004;279:41998–42007. doi: 10.1074/jbc.M403702200. [DOI] [PubMed] [Google Scholar]
- Schroeder TM, Westendorf JJ. Histone deacetylase inhibitors promote osteoblast maturation. J Bone Miner Res. 2005;20:2254–2263. doi: 10.1359/JBMR.050813. [DOI] [PubMed] [Google Scholar]
- Schug J. Using TESS to predict transcription factor binding sites in DNA sequence. Chapter 2. Curr Protoc Bioinformatics. 2008 doi: 10.1002/0471250953.bi0206s21. Unit 2 6. [DOI] [PubMed] [Google Scholar]
- Sui Y, Yang Z, Xiong S, Zhang L, Blanchard KL, Peiper SC, Dynan WS, Tuan D, Ko L. Gene amplification and associated loss of 5' regulatory sequences of CoAA in human cancers. Oncogene. 2007;26:822–835. doi: 10.1038/sj.onc.1209847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniuchi I, Osato M, Egawa T, Sunshine MJ, Bae SC, Komori T, Ito Y, Littman DR. Differential requirements for Runx proteins in CD4 repression and epigenetic silencing during T lymphocyte development. Cell. 2002;111:621–633. doi: 10.1016/s0092-8674(02)01111-x. [DOI] [PubMed] [Google Scholar]
- Westendorf JJ, Mernaugh R, Hiebert SW. Identification and characterization of a protein containing formin homology (FH1/FH2) domains. Gene. 1999;232:173–182. doi: 10.1016/s0378-1119(99)00127-4. [DOI] [PubMed] [Google Scholar]
- Woolf E, Xiao C, Fainaru O, Lotem J, Rosen D, Negreanu V, Bernstein Y, Goldenberg D, Brenner O, Berke G, Levanon D, Groner Y. Runx3 and Runx1 are required for CD8 T cell development during thymopoiesis. Proc Natl Acad Sci U S A. 2003;100:7731–7736. doi: 10.1073/pnas.1232420100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DW, Hassan MQ, Pratap J, Galindo M, Zaidi SK, Lee SH, Yang X, Xie R, Javed A, Underwood JM, Furcinitti P, Imbalzano AN, Penman S, Nickerson JA, Montecino MA, Lian JB, Stein JL, van Wijnen AJ, Stein GS. Mitotic occupancy and lineage-specific transcriptional control of rRNA genes by Runx2. Nature. 2007;445:442–446. doi: 10.1038/nature05473. [DOI] [PubMed] [Google Scholar]
- Yu HM, Jerchow B, Sheu TJ, Liu B, Costantini F, Puzas JE, Birchmeier W, Hsu W. The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development. 2005;132:1995–2005. doi: 10.1242/dev.01786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Pande S, Pratap J, Gaur T, Grigoriu S, Ali SA, Stein JL, Lian JB, van Wijnen AJ, Stein GS. Runx2 deficiency and defective subnuclear targeting bypass senescence to promote immortalization and tumorigenic potential. Proc Natl Acad Sci U S A. 2007a;104:19861–19866. doi: 10.1073/pnas.0709650104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi SK, Young DW, Javed A, Pratap J, Montecino M, van Wijnen A, Lian JB, Stein JL, Stein GS. Nuclear microenvironments in biological control and cancer. Nat Rev Cancer. 2007b;7:454–463. doi: 10.1038/nrc2149. [DOI] [PubMed] [Google Scholar]