

Abstract
The oral bacterial microbiome encompasses ca. 700 commonly occurring phylotypes, approximately half of which can be present at any time in any individual. These bacteria are largely indigenous to the oral cavity; this limited range suggests that interactions between the various phylotypes, and between the phylotypes and their environment, are crucial for their existence. Molecular cataloging has confirmed many basic observations on the composition of the oral microbiome that were formulated well before ribosomal RNA-based systematics, but the power and the scope of molecular taxonomy has resulted in the discovery of new phylotypes and, more importantly, the speed and detail of molecular analyses are impossible to achieve through classical approaches. Community structure varies with location within the mouth, and changes in community structure are related to disease initiation and disease progression. Factors that influence the formation and the evolution of communities include selective adherence to epithelial or tooth surfaces, specific cell-to-cell binding as a driver of early community composition, and interorganismal interaction leading to alteration of the local environment, which represents the first step on the road to oral disease. A comprehensive understanding of how these factors interact to drive changes in the composition of the oral microbial community can lead to new strategies for the inhibition of periodontal diseases and dental caries.
The ease with which the oral flora can be sampled has made oral microorganisms prominent in bacteriology. Dental plaque was one of the first bacterial communities examined using the earliest microscopes. Except for certain yet-to-be-cultured phylotypes (e.g. TM7, discussed later) and a few anaerobic phylotypes associated with the skin or the gut (e.g. Propionibacterium acnes), the bacteria of the oral cavity do not commonly occur elsewhere in the body or in the natural environment. Oral bacteria are inseparably intertwined with diseases that will affect every human at some point in life: gingivitis, periodontal diseases and dental caries. Treatment and prevention of these diseases rests primarily on the mechanical removal of the bacteria. In contrast to most other bacterially mediated diseases, antibiotic treatment alone may not be effective (43), for the following reasons: (i) oral bacterial diseases are polymicrobial (i.e. no single organism is the etiological agent); and (ii) as the pathogens are also naturally occurring members of the oral microflora in healthy individuals, reinfection is inevitable (58). We know more about the oral microbial community at the level of individual phylotypes than we know about the bacterial membership of most other natural systems. However, we know relatively little about the basic physiological interactions that permit (and perhaps require) association of the individual phylotypes with one another in their daily existence.
Members of a selective club
Modern molecular taxonomic approaches, in particular the ability to rapidly obtain large numbers of 16S ribosomal DNA sequences, have paved the way for exhaustive surveys of the oral microflora. The so-called Illumina and 454 technologies provide the highest number of sequences per sample, and thus the most coverage, but comparisons between these approaches in the current literature are difficult to interpret because the samples vary from study to study and the clustering/analysis methods can differ. A pair of recent reports illustrates this point. In 197,000 high-quality reads obtained from samples of supragingival plaque, 10,052 operational taxonomic units (i.e. the molecular equivalent of a species) were defined at 3% genetic difference (38). Of these operational taxonomic units, 95% could be accounted for by ca. 2000 of the most abundant sequences. Genus-level sequences that made up at least 1% of the total operational taxonomic units were: Streptococcus (10.3%), Actinomyces (7.0%), Prevotella (6.3%), Capnocytophaga (6.3%), Fusobacterium (5.7%), Corynebacterium (4.4%), Veillonella (4.3%), Rothia (3.6%), Neisseria (3.5%), the TM7 phylum (3.1%), Leptotrichia (2.7%), Selenomonas (2.0%), Porphyromonas (2.0%), Treponema (1.2%), Campylobacter (1.1%), Haemophilus (1.1%) and Gemella (1.0%). Interestingly, unclassifiable sequences also occurred at > 1% of the total. These sequences were in the order Clostridiales (1.8%), in the family Neisseriaceae (1.4%), in the family Pasteurellaceae (1.2%) and in the class Gammaproteobacteria (1.2%). However, from a more intensive sampling that included mucosal surfaces and interproximal plaque, 452,000 reads yielded only 818 operational taxonomic units at 3% difference (77). Of these, genus-level or higher operational taxonomic units making up ≥1% of the total were: Streptococcus (19.2%), Corynebacterium (6.1%), Neisseria (8.3%), Haemophilus (4.2%), Actinomyces (4.2%), Rothia (3.8%), members of the family Veillonellaceae (3.3%), Granulicatella (2.2%), Prevotella (1.9%), Porphyromonas (1.8%), Capnocytophaga (1.2%) and Actinobaculum (1.0%). One group of unclassified sequences that made up more than 1% of the total were sequences in the Firmicutes (1.7%). It is noteworthy that the number of sequences classifiable only at levels higher than the genus comprised a much smaller fraction of abundant operational taxonomic units and was less diverse in this latter study than in the first example. More importantly, data in the latter study were analyzed according to subject, rather than by pooling – this approach revealed that the number of operational taxonomic units at 3% difference in any one of the three subjects was 540–650, and that approximately 390 operational taxonomic units were shared between all subjects.
The results of high-throughput sequencing studies can be compared with those of more traditional 16S ribosomal DNA investigation based on cloning and sequencing, for which assembly of two recently available oral microbiome databases will be used as comprehensive examples. The Human Oral Microbiome Database (www.homd.org) (18) is actively curated, consists (as of June 2012) of 640 taxa and was created initially though the analysis of full-length human oral taxonomic sequences reported in the literature along with sequences from the creators’ collection. This database is intended to serve as a framework for taxonomic assignment of any sequence obtained from the oral cavity; therefore, it is not static and its power is dependent on exhaustive analysis of full-length cloned sequences obtained from clinical samples. To explore the coverage of the database, 34,753 filtered cloned sequences were examined that originated in 633 libraries and that were representative of a wide variety of healthy and diseased sites throughout the oral cavity. Coverage of the sequence collection was enhanced by using more than one primer set during PCR amplification. The breadth of sample origin, including severe pathologies such as orofacial gangrene, implies that the collection cannot be interpreted as representative of a standard community composition in the oral cavity, if such a composition exists. Furthermore, because the oral cavity is an open system, the authors made the quite defensible assertion that the number of operational taxonomic units required to account for 100% of organisms found in the oral cavity, as measured in different individuals, at different times and in different geographical locations, is that required to account for all microbes in nature.
However, from a data-driven perspective, the 34,753 clones yielded 1179 operational taxonomic units and 99% of the clones can be accounted for by 875 of the operational taxonomic units. In other words, the remaining 304 operational taxonomic units together comprise only 347 clones and therefore represent organisms rarely retrieved from the oral cavity. At the genus level, taxa (defined as operational taxonomic units, which represent organisms currently in pure culture as well as those representative of yet-to-be cultured organisms) that made up ≥ 1% of the clones were: Streptococcus (26.9%), Veillonella (9.8%), Selenomonas (3.5%), Gemella (3.3%), Fusobacterium (3.0%), Prevotella (2.7%), Lactobacillus (2.3%), Neisseria (2.3%), Dialister (2.0%), Actinomyces (1.9%), Capnocytophaga (1.9%), Granulicatella (1.9%), Campylobacter (1.5%), Treponema (1.5%), Enterococcus (1.3%), Eubacterium (1.3%), Atopobium (1.1%), Bacteroides (1.1%) and Propionibacterium (1.0%). Taxa identified at abundances of ≤ 1% in the clone-based study, but at ≥ 1% in the previously discussed pyrosequencing studies, were Rothia (0.7%), Haemophilus (0.7%), Corynebacterium (0.6%) and Actinobaculum (0.06%). Through this extensive clone-based analysis, 400 oral bacterial sequences were discovered that were not present in the Human Oral Microbiome Database but which should probably be included. Curation criteria for inclusion in the Human Oral Microbiome Database (examples of which include that sequences should be near full length and retrieved more than once) will be used to validate newly discovered operational taxonomic units for addition to the 640 now in the database. A second sequence database that is comparable to the Human Oral Microbiome Database is CORE (29) (http://microbiome.osu.edu), which is designed to represent a minimally redundant collection of operational taxonomic units regularly found in the oral cavity. Its primary value is robust identification at the genus and species levels. The database was formulated in a manner similar to the Human Oral Microbiome Database: it started with the cultured and validly named oral bacterial isolates, then the list was supplemented with clinically derived sequences obtained from several published studies. The database contains 636 phylotypes at a 2% difference cut-off, 365 of which presently lack a cultured member and none of which are singleton sequences. A set of 1000 pyrosequencing reads, representing subgingival samples from 24 patients, was obtained from the Human Microbiome Project’s Data Analysis and Coordination Center (www.hmpdacc.org) and was analyzed using the databases CORE, Human Oral Microbiome Database, Ribosome Database Project and GenBank. At the 2% difference cut-off, CORE and the Human Oral Microbiome Database performed similarly and either was better than the Ribosome Database Project or GenBank (29). CORE had a somewhat lower number of distinctly named matches per sequence than did the Human Oral Microbiome Database, thereby delivering slightly less ambiguity.
In a forward-looking clone-based study in which subject-dependent differences were determined (6), 26 sites in each of 10 orally healthy (low clinical attachment loss and lack of bleeding on probing) individuals were sampled. Subgingival and supragingival tooth surfaces were sampled using a curette, and saliva was collected. The samples were pooled to create an overall sample for each individual. Sequences were compared at a much higher level of similarity (99%, or 1% difference cut-off), which yielded a comparatively lower number of operational taxonomic units from the entire population: 247. The phylum-level composition was: Firmicutes, 33.2%; Proteobacteria, 27.5%; Bacteriodetes, 16.6%; Actinobacteria, 14.5%; Fusobacteria, 6.7%; TM7, 1.3%; and Spirochaetes, OD2 and Synergistes, all <1%. The most abundant genera within the population were Streptococcus (19.2%), Haemophilus (11.7%), Neisseria (9.2%), Prevotella (8.6%), Veillonella (8.6%) and Rothia (7.2%). With the exception of the universally high abundance of streptococci, the abundance of other genera showed sometimes striking variation between individuals. Furthermore, the dominant genus varied between the individuals; Streptococcus was the dominant genus in only five individuals. In two individuals, Prevotella dominated. In the remaining individuals, the dominant genus was Veillonella, Neisseria or Haemophilus. The above-listed six most abundant genera were found in all individuals, as were the other common oral genera Actinomyces, Atopobium, Capnocytophaga, Campylobacter, Corynebacterium, Granulicatella and Fusobacterium. However, less-well-known genera (Bergeyella and Cardiobacterium) were also universally detected, as was the yet-to-be cultured TM7 group.
Most data from these studies are similar in two respects: the dominance of streptococci; and the presence of periodontal pathogens in healthy individuals. However, in one respect they are very different: the number of operational taxonomic units. Very large data sets produced by high-throughput methods are subject to inflation through a number of processes that result in spurious sequences; many of these sequences can be removed through careful filtering. However, it has recently been shown that subsequent analysis of these large data sets using multiple sequence alignment and complete-linkage clustering can bias operational taxonomic unit numbers by as much as 60% (32). This finding does not change the perspective that, regardless of habitat, natural microbial populations are much more diverse than previously recognized, and that a large component of the diversity consists of rare sequences. However, the finding does suggest that the number of unique taxa is likely to be inflated in high-throughput studies that use common clustering approaches. A recent comparison of 454-based 16S sequencing with a hybridization chip assay (HOMIM) based on the most prevalent organisms in the Human Oral Microbiome Database showed little difference between the two approaches (2), the exception being the detection of Spirochaetes to a higher degree with HOMIM. Given our understanding of oral diseases as being bacterial-community based, it seems unlikely that deep sequencing will have an impact on oral microbial ecology other than to discover the degree to which sequences of rare and exogenous organisms can be detected in particular individuals.
Functional metagenomic analysis is another way to describe communities from a molecular perspective. A recent analysis (76) yielded results which were substantially different from those of the previously discussed studies that focused solely on 16S ribosomal DNA sequences. Shotgun sequencing on Illumina and 454 sequencing platforms produced, respectively, 15 million and 176,000 high-quality reads from pooled supragingival and interproximal molar plaque obtained from an individual who had not conducted oral hygiene for 24 hours. Approximately one third of the reads were of human origin. Nine analysis strategies (four based on 16S sequences and five based on open reading frames) were used to produce phylum-level taxonomic profiles. In all but one analysis, Firmicutes was not the predominate phylum. Instead, Proteobacteria dominated, in most cases by a factor of two; a major difference from most other studies cited above. This difference can have multiple causes, including 16S primer bias in the earlier PCR-based studies, database bias and the relatively small percentage of 16S sequences (< 1% of all sequences) retrieved using the metagenome approach. However, the most likely causes are individual variation and sample type. No saliva was sampled; and the supragingival and subgingival plaque had developed over a 48-hr period without oral care. The most common Proteobacteria genera in this metagenome study were Haemophilus and Neisseria, the very genera that dominated in two individuals as reported in the study by Bik et al. (6). Independent verification of what might be considered an atypical distribution at the phylum level is exciting and worthy of further investigation. Likewise unexpected was the low number of reads correlated to completely sequenced oral reference strains. Only 4% of the reads could be matched to reference strains at 97% identity, and the top five phylotypes (those with the most frequently encountered sequences) were three Capnocytophaga spp. and two Corynebacterium spp., members, respectively, of the phyla Bacteroidetes and Actinobacteria. The next highest were Streptococcus mitis and Streptococcus sanguinis (common commensal bacteria of the Firmicutes), and in eighth place came a member of the Proteobacteria (Neisseria subflava). Using the BlastX and the SEED databases, 668 bacterial operational taxonomic units were obtained, a result comparable to that reported in the previously discussed high-throughput 16S-based study on single-person samples (77).
One collective interpretation of these and other molecular studies is that the number of operational taxonomic units typical for the oral cavity is 600–800, but also that significantly fewer are normally present in any one individual. The preponderance of the data suggests that Firmicutes comprise the majority of the operational taxonomic units and that Streptococcus is the dominant genus, but several examples of individual microflora that do not fit this paradigm exist. These exceptions are very interesting because they give us clues as to how bacterial community composition is modulated by interorganismal interactions, environmental conditions and host genetics. As specific methodological limitations (freely pointed out by all authors) are overcome, a clearer picture of the importance of these data to oral microbial ecology will materialize. In the meantime, large baseline data sets should be assembled that can be used in comparisons of healthy vs. diseased sites, or in comparisons of healthy individuals with those with nonoral pathologies. When assembling the databases, it is important that data are recorded and analyzed at the level of a single individual and at the site of sample origin, lest we miss the opportunity to delve into the ecological relationships important in establishing these clearly different communities.
Some new(er) members of the club
One of the most important contributions of molecular taxonomic studies has been to highlight organisms that were underappreciated in cultural studies. Certain organisms have been demonstrated to be present at much greater frequencies than in cultural studies, and several taxa known primarily (or solely) through molecular data have been linked with particular disease situations.
Synergistetes
A recently created phylum (37), this group of organisms is often represented by clones in molecular surveys of common oral infections: periodontitis, caries and endodontic pus. Of potentially great importance is that these organisms seem to be absent in subgingival plaque from periodontally healthy patients (74). Organisms from the oral cavity are divided into two clusters; one of which only recently gained a culturable representative (73).
Filifactor
The oral representatives of this genus, formerly placed in the genus Fusobacterium (based on fermentation products and fatty acid composition), were reclassified in 1999 based on their 16S ribosomal RNA sequence (33). The genus’ members are known from periodontitis/gingivitis and from animal soft-tissue infections. The organism displays a clear relationship to periodontal disease, and two studies (28, 62) show it to be at least as prevalent in periodontal disease as are the commonly accepted periodontal pathogens Porphyromonas gingivalis and Tannerella forsythia. One study also used a retrievable substratum to demonstrate that the organism occurs most frequently close to the soft tissue rather than close to the tooth (62).
Scardovia/Parascardovia
Split off from the genus Bifidobacteria on the basis of the sequence of heat shock protein 60 (35), phylotypes of these genera are frequently found in bacteriological and molecular analyses of caries communities (46, 47).
Dialister
This genus is frequently recovered from endodontal infections and deep periodontal pockets. Its closest relatives are in the genus Megasphaera. Dialister pneumosintes has been recognized for some time; it was first placed in the genus Bacteriodes and then was moved to the newly created genus Dialister (50) based on fatty-acid profiling. However, the species Dialister invisus and Dialister microaerophilus have only recently been described (21, 36).
“omics” at a crossroad
The power of molecular analyses in defining bacterial community composition is without question. However, identification of operational taxonomic units can take us only so far, and that point seems to have been reached. One major conclusion is, not surprisingly, that the oral microbial population is more diverse than previously believed. One can substitute any environment for “oral” and the sentence remains true. Perhaps viewing the issue from the opposite direction is more interesting – much was, in fact, already known about the oral microflora before deep sequencing studies became available: more than for any other bacterial community. The yet-to-be cultured operational taxonomic units in the Human Oral Microbiome Database make up 18% of the total clones, yet these represent 68% of the total taxa. Some taxonomic groups consist almost entirely of culturable organisms. For instance, the genus Streptococcus comprises 41 operational taxonomic units, only four of which are uncultured and have a combined recovery of less than 0.02%. If the taxonomic status of an organism reflects its physiology, one might ask why these bacteria remain uncultured despite many decades of oral bacteriological research. In some cases, obscure nutrients or growth factors prevent them from being cultured under conditions that succeed for their close relatives. For example, the genera Abiotrophia and Gemella had their origins in a group of what were initially called “nutritionally variant streptococci” (60, 61). Also, because many oral isolates exist for which no molecular taxonomic data exist, it is possible that certain yet-to-be-cultured bacteria have in fact been cultured and reside in strain collections; many operational taxonomic units in the Human Oral Microbiome Database are cultured but lack the valid taxonomic description required to propose a species name. In other cases, uncultured organisms lack close relatives that have been cultured; TM7 clones fit this description. Sequences of the TM7 group, first seen in a molecular survey of a peat bog, have been recovered from many different habitats outside the oral cavity (31). The wide habitat range of the group suggests that it is very adaptable, but these organisms have thus far proven impossible to bring into pure culture. In the case of lineages very different from those of “typical” oral bacteria, it seems obvious that investigators should attempt to find the keys for culture – entire groups of organisms with unique chemistries would have been passed over without advances in culture technique (68). However, should scientists spend the time and effort necessary to obtain isolates of very close relatives of other well-documented species? Sequences from community analyses have clear utility, but physiology and phenotype are of far greater importance in furthering our mechanistic understanding of the interactions between bacteria, or between bacteria and the host. In polymicrobial situations, the latter interactions are those between the community as a whole (a physiological unit comprising different species) and the host – for this purpose, molecular analyses of bulk community samples (e.g. subgingival scrapings) can correlate community composition with host status. When examined within a host population, periodontally diseased sites, as well as caries sites, have a community composition different from that of healthy sites; molecular analysis has made it much easier to determine the extent of the differences while also revealing bacteria that were previously unnoticed. However, it is also true that differences exist between individuals in these otherwise characteristic communities. Most clinical studies do not analyze data at the level of the individual; rather, all data from individuals are combined. This approach is necessary for assessment of the physiological outcome in the host population because the commonalities are important for measuring treatment efficacy; for example, in answering the question of whether a therapy will reduce a range of periodontal pathogens in most patients. However, the differences, rather than the commonalities, give us clues for understanding the physiological basis of bacterial interactions. Can a particular niche in an oral microbial community be filled by, or more significantly, be defined by, more than one bacterium? This is certainly the case in other bacterial communities (16). Current periodontal treatment regimens are based on brute-force elimination of periodontal pathogens that are also inhabitants of healthy periodontal pockets. By understanding the interactions between bacteria, and between bacteria and the host, we can perhaps design treatments that manipulate conditions within the pocket toward niches that can be preferentially occupied by commensal bacteria. In this approach, as in nature, ecological parameters control community composition, and sophisticated community taxonomic analysis techniques are absolutely required to define that composition.
A sense of community
In the broadest terms, three communities exist within the mouth that correlate with the region sampled: the teeth; saliva, together with the dorsal/lateral surfaces of the tongue; and the remaining epithelial surfaces (45). It is interesting that the flora of the dorsal/lateral surfaces of the tongue is more similar to that of saliva than is the flora of the tongue to that of other epithelial surfaces. It is generally accepted that saliva has no true indigenous flora because salivary turnover (the time from secretion to swallowing) is in the order of a few minutes; therefore, the bacterial growth rate is too slow (in the order of hours) to permit establishment of an indigenous salivary population, especially at the low levels of nutrients present in saliva (13). Instead, the bacteria in saliva are those shed from biofilms on oral tissues. The lateral and the dorsal surfaces of tongue are highly papillate and thus have a topography different from that of other epithelial surfaces, including the tongue’s ventral surface. The papillate surfaces harbor a flora skewed towards anaerobic genera such as Prevotella and Veillonella, whereas the ventral surface bears a flora rich in streptococci and Gemella (45). All epithelial surfaces desquamate and thereby release bacteria into saliva; however, the tongue appears to contribute disproportionately to this process.
The tooth surfaces, either supragingival or subgingival, are the only nonshedding surfaces in the oral cavity and thereby represent a stable location for long-term biofilm development. Furthermore, tooth enamel is a substratum to which salivary proteins adsorb and thereby create the oral conditioning film (salivary pellicle): the basis for selective adherence of bacteria from saliva (44). However, it is striking that the majority of indigenous oral bacterial species are found everywhere in the mouth (e.g. the composition of the microbial community on teeth differs from that on other surfaces, not in the presence/absence of species but rather in the proportions of species).
Founding a hamlet
Given that the majority of oral bacterial species can be found throughout the mouth, how do communities with different characteristic ratios of species develop? One factor is initial adherence. As noted above, tooth enamel is a mineral surface to which host salivary proteins adsorb. Bacteria can adhere to protein-coated surfaces through so-called nonspecific mechanisms (11), but many oral bacteria also have adhesins that specifically interact with components in the pellicle. Streptococcus and Actinomyces strains are the earliest colonizers of freshly cleaned teeth, and these bacteria bind to two major salivary components in the pellicle: mucins and proline-rich proteins. The same molecules that form the pellicle are present in bulk saliva, and therefore adhesins on the bacterial cell surface should be saturated through interacting with saliva. Hypotheses regarding how saturation kinetics can be overcome include multivalent receptor–adhesin interactions, either through different adhesion–receptor types (49) or through the cumulative effect of a single type in which one component is stationary (15). However, two other mechanisms can account for preferential adhesion to substrata, and both involve protein conformation. The cryptitope hypothesis has existed since the pioneering work of Gibbons & Hay (24). It postulates that, upon adsorption, a change in protein conformation exposes a previously inaccessible adhesin-recognition site. Conformational changes in proline-rich proteins upon adsorption to hydroxyapatite have been demonstrated (22), but a consequence for bacterial adhesion has not been reported. A hypothesis developed more recently rests on the counterintuitive observation that binding of Escherichia coli to adsorbed mannose-bearing molecules is increased by shear stress (70). A model of this interaction has the adsorbed receptor molecule acting as an anchor and, upon increased shear, a conformational change takes place in the adhesin to form a so-called catch-bond. Like the finger-trap toy, the catch-bond becomes stronger when pulled. For these bonds, an increase in shear results in no removal of bacterial cells from a substratum. Rather, the interaction between the cell and the substratum becomes stronger. This can be observed by time-resolved examination of cells as they roll along a substratum – during an increase in shear the percentage of stationary cells increases. When shear is returned to a lower value, stationary cells begin rolling again. It has been speculated that this mechanism could be widespread, despite a limited number of examples (66). Recent experiments carried out with oral bacteria demonstrate that the interaction of the streptococcal sialic acid-binding protein, Hsa, with the model sialic acid-presenting glycoprotein, fetuin, is shear-enhanced (20). Shear-enhancement was greater with adsorbed whole saliva than with fetuin; while difficult to assess unambiguously, this observation suggests that the mixture of components in adsorbed saliva augments shear-enhancement. However, no evidence for shear-enhancement was seen in the interaction of Actinomyces spp. fimbriae with proline-rich proteins, with the Galβ1-3GalNAc-presenting protein, asialofetuin, or with saliva. For Actinomyces cells, a very large percentage was already stationary at very low shear and this percentage did not change when shear was increased. These bacteria appear to have a much stronger interaction with the artificial pellicle than do streptococcal cells. Thus, shear-enhanced adhesion is not a universal feature of adhesion–receptor interactions in the oral cavity, despite continuous salivary flow. However, it is possible that shear-enhancement observed in streptococcal sialic-acid binding is also important in bacterial endocarditis, a situation in which the bacterium experiences greater shear than in the oral cavity and for which Hsa is a virulence factor (69).
Another factor important in the earliest steps of biofilm formation is cell-to-cell recognition, known in oral microbiology as coaggregation. The term co-adhesion has been coined to describe the consequences of coaggregation for recruitment of planktonic bacteria into an extant biofilm (7). Decades of research have demonstrated that oral bacterial isolates commonly adhere to one another. When a broth culture of an isolate is mixed with that of an interacting partner, coaggregates (flocs) that contain cells of each isolate become visible and are often large enough to settle out in a matter of seconds. Through coaggregation assays performed on hundreds of isolates, patterns have emerged for certain groups of oral bacteria (40). There is often coaggregation among bacteria of different genera that are present in the initial stages of biofilm formation (Streptococcus, Actinomyces and Veillonella) and, in the case of streptococci, coaggregation also occurs within the genus. The initial colonizers as a group have a restricted degree of coaggregation with periodontal pathogens. However, Fusobacterium spp. were found to interact with initial colonizers as well as with periodontal pathogens and other bacteria found in mature plaque. Fusobacterium spp. were thus postulated to bridge (physically connect) initial colonizers with pathogens, a process that leads to gingivitis and periodontal disease (41). Coaggregation data obtained using bacterial isolates under laboratory conditions strongly support a role for cell-to-cell binding in the assembly of multispecies communities; however, the situation in vivo was not explored until the last decade. Direct evidence of the importance of these interactions in supragingival biofilms has been obtained through confluence of confocal microscopy, highly specific bleach-resistant fluorescent primary antibodies and a retrievable enamel chip model system that yields intact oral biofilm from humans. Not only were bacteria bearing complementary coaggregation-mediating molecules on their cell surfaces found to be in immediate proximity to one another in nascent plaque (52), but also a colony containing only a few cells was captured by micromanipulation and shown to consist of coaggregating bacteria (14). Equally important is the ability to quantify such interactions. An analysis of spatial relationships between bacteria within a plaque sample, as documented by a sophisticated fluorescence in-situ hybridization study, confirmed the relevance in vivo of several coaggregation patterns simultaneously (72). Notably, the in-vivo data did not support earlier in-vitro experiments that showed Fusobacterium spp. to be promiscuous coaggregation partners. This discrepancy is worthy of further examination given the postulated importance of Fusobacterium spp. in the transition from commensal biofilm to pathological community.
Community fitness under difficult circumstances
Bacterial biofilms that develop on retrievable substrata have multispecies colonies from the earliest point at which cells are easily visible when viewed by microscopy (19, 52). Cell-to-cell recognition and binding seems to be very important in the community composition of biofilm colonies. Why? One simple explanation is that bacteria not adhered to a surface are rapidly transferred to less hospitable environments. Therefore, the greater the number of adherence mechanisms, the better the possibility of retention. This explanation should result in a large number of nonspecific cell-to-cell recognition interactions. However, the interactions are not random, and in some cases are highly specific, an observation that suggests an underlying evolutionary pressure. Because bacteria do not exhibit behavior, the only driving force is physiological interaction between organisms. Nutrient supply is a key factor for growth and, for bacteria of supragingival biofilms, nutrient supply is characterized by feast and famine, primarily the latter. Easily fermentable, low-molecular-weight carbohydrates are present for the short period of time between food intake and saliva washout. The paradigmatic Stephan curve sets a short time span of ca. 30 minutes, during which acidogenic saccharolytic oral bacteria can be highly active (23, 67). Experiments on plaque accumulation in monkeys showed that the numbers of various bacteria reached a plateau at approximately 18 hours, and did not vary between animals that were fed, animals that were fasted and animals that were given water with 0.5% glucose (5). Not only do these results raise the question of the importance of simple sugars for bacterial biomass production in vivo, but they also demonstrate that growth must be supported by saliva alone as the substrate.
The recognition that saliva is an important substrate in vivo coincided with early experiments in vitro that used saliva or model salivary proteins as carbon sources. For example, it was demonstrated that an oral bacterial co-culture (a plaque inoculum) could grow to turbidities of approximately 0.35 in stimulated saliva that had been reduced by the addition of dithiothreitol, centrifuged and then filter-sterilized (17). The levels of salivary glycoprotein-associated oligosaccharides, expressed as total hexose, decreased by approximately 85% during bacterial growth. Streptococcal isolates obtained from this enrichment culture could be grown in 50% saliva in a chemostat as monocultures, but the results of those experiments are difficult to interpret because the medium was supplemented with 100 μM glucose. An important consideration in these types of studies is the preparation of saliva: centrifugation and filtration may result in the removal of some of the most abundant glycoprotein – high-molecular-weight mucin. The sialic acid content of mucins is approximately 4% (weight by weight), whereas other sugars (namely galactose, N-acetylglucosamine, N-acetylgalactosamine and fucose) are present at a weight-percent of approximately three- to six-fold higher (64). All these sugars are present at similar weight-percentages in other salivary glycoproteins, such as proline-rich glycoproteins, which are too small to be removed by sterile filtration. In addition to these experiments using sterilized saliva, numerous important chemostat-based (pH-stat) studies, using hog gastric mucin as a culture supplement, were conducted by Marsh, Bradshaw, Keevil and colleagues. The medium for these studies contained tryptone, yeast extract, proteose peptone and, in some experiments, glucose. The concentrations of these components in early studies were similar to those in standard bacteriological medium. One of the earlier studies (25) compared growth of a standardized plaque inoculum on a synthetic saliva (mineral salts with mucin as the sole carbon source) with growth on this rich medium that contained either glucose alone or mucin alone, or glucose plus mucin. Yield (g dry weight/L) was 0.23 for the synthetic saliva, 2.18 for the rich medium with glucose only as well as for the rich medium with mucin only, but was 2.68 on the rich medium that contained glucose plus mucin. The number of streptococci expressed as a percentage of the total number of colony-forming units increased from approximately 4% in synthetic saliva to 12% in the rich medium with mucin alone, to nearly 50% in the medium with glucose regardless of the presence of mucin. While these results showed that glucose was the limiting factor for the growth of streptococci under these conditions, they also showed that the addition of mucin to the glucose-containing medium had a synergistic effect whereby the yield was disproportionately higher than that with mucin alone or with glucose alone. Later experiments employed a five-fold dilution of the rich medium – this modification lowered the concentration of oligopeptide and potential carbon sources. Mucin was used as the sole carbon source (9) and a defined inoculum was employed that contained Streptococcus mutans, Streptococcus sanguis (now S. sanguinis), Streptococcus mitior (now S. mitis), Actinomyces viscosus (now A. naeslundii), Lactobacillus casei, Neisseria subflava, Veillonella dispar, Bacteroides intermedius (now Prevotella intermedia) and Fusobacterium nucleatum. When the number of streptococci present in the chemostat was expressed as a percentage of the total viable count, the value was approximately the same as in the earlier experiments that used an undefined inoculum with glucose-containing medium, but the carbon source for growth was clearly mucin rather than glucose. When glucose was added in daily pulses to the chemostat, while pH was maintained near neutrality, the percentage of S. mutans increased slightly over time (from 0.3 to 1.0), but the percentages of other streptococci and of L. casei did not change appreciably. If, however, streptococcal acid production was not titrated, a pH of 5 was reached after 3 h. After 10 days of pulsing, the time required to reach pH 5 became only 45 min. The community now consisted of 20% S. mutans and 36% L. casei, whereas the percentage of S. sanguis had decreased from 25 to 0.2, and that of S. mitior from 16.9 to 1.3. This experiment demonstrated a key concept of what would come to be called the Ecological Plaque Hypothesis (48), namely that a microbiologically mediated interplay exists between carbon availability and pH, which runs in a feedback loop to control the composition of the oral biofilm flora. The addition of easily fermentable carbon causes an increase in streptococcal biomass, a reduction in pH and an eventual overgrowth by highly acidogenic/aciduric organisms, such as S. mutans and lactobacilli. In the short term this result is transient and can be reversed through removal of carbohydrate and buffering of pH by salivary flow but, if sugar introduction is extensive, the population of less-acidogenic/aciduric commensal streptococci is reduced to the point at which it can no longer compete with S. mutans, particularly in spatially restricted areas on the teeth such as interproximal spaces and coronal fissures where the outcome is caries. However, as will be discussed later, S. mutans is not the sole pathogen. All the above experiments reveal that mixtures of streptococci, and perhaps other early colonizers of the tooth surface, are able to grow using salivary glycoprotein as the sole carbon source.
Despite the fundamental nature of saliva as a nutrient source, little is known about the mechanisms of bacterial growth on this complex mixture of proteins and glycoproteins. However, through experiments that revolve around a single model of glycoprotein, we understand that oligosaccharide catabolism takes place through the sequential action of secreted and cell wall-anchored glycoside hydrolases. Growth of a Streptococcus oralis strain and of the common nasophyarnx inhabitant, Streptococcus pneumoniae, has been examined using α-1 acid glycoprotein (from human serum) as the carbon source (10, 12). A recent review has nicely assembled the results of numerous studies to show the diversity of glycoside hydrolases within S. pneumoniae (39). Most of the bacterium’s seven well-characterized glycoside hydrolase activities are conferred by proteins predicted to be cell wall-bound based on the presence of an LPXTG anchor motif within the gene sequence. The exceptions are neuraminidase and a β-galactosidase (β-galactosidase C). Neuraminidase activity is provided by a set of three enzymes, one of which is cell wall-anchored (neuraminidase A), one of which is secreted (neuraminidase B) and one of which is predicted to be cell wall-anchored (neuraminidase C), but for which an enzymatic activity has yet to be verified. Beta-galactosidase C does not have an LPXTG motif and it does not contain any other recognized cell wall-association or cell wall-secretion motifs, but the protein has been shown, via immunofluorescence microscopy, to reside at the cell surface (34). This observation demonstrates that the absence of cell wall-anchoring motifs does not conclusively identify a protein as secreted into the extracellular milieu rather than being located at the cell surface. With its suite of seven glycoside hydrolase activities, S. pneumoniae is well positioned to make use of oligosaccharides as carbon sources. The genomes of many oral bacteria are now annotated and searchable, so one can compare the presence of glycoside hydrolase sequences across a number of species. A database exists for this comparison: Carbohydrate-Active enZYmes (CAZY; www.cazy.org). The database identifies glycoside hydrolases based on their sequence and organizes them into protein families by shared folding patterns and molecular mechanisms. The families do not necessarily conform to EC activity or naming. In other words, not all β-galactosidases are found in a single family. However, the framework is very useful for rapid comparison of glycoside hydrolase sequences between sequenced organisms. The results of such a comparison for early-colonizing oral bacteria using S. pneumoniae as a template are presented in Table 1. The organism with the most diversity and highest number in glycoside hydrolase families does not belong to the genus Streptococcus, but rather to the genus Prevotella. Ten glycoside hydrolase families, comprising 15 sequences, are present in Prevotella but absent from all other organisms listed; however, Prevotella lacks two families (35 and 101) that are known to be important for oligosaccharide hydrolysis in S. pneumoniae. Members of the phylum Bacteroidetes are major contributors to the anaerobic digestion of complex carbohydrates in the gut. Whether these capabilities contribute to the survival and integration of Prevotella and other members of the Bacteroidetes in early supragingival plaque is unknown. Perhaps Prevotella in particular represents a pioneer organism in the shift from the primarily aerobic/facultative nature of early supragingival plaque to the primarily anaerobic physiology of more mature plaque or of subgingival plaque. The other obligate anaerobic bacterium in Table 1 is V. dispar, an organism that uses short-chain fatty acids as carbon sources and therefore has a predictably small contingent of glycoside hydrolases. The streptococci as a whole have similar suites of enzymes, the notable exception being S. mutans, which has only one of the six families critical for oligosaccharide breakdown. Conversely, S. mutans possesses two families not present in the other streptococci. These observations clearly demonstrate a major difference in basic carbon acquisition and utilization for commensal streptococci vs. the highly acicuric/acidogenic mutans streptococci. It will be interesting to examine the enzyme suite in Streptococcus sobrinus, an acidophilic streptococcus for which sequence data have not yet been analyzed.
Table 1.
Glycoside hydrolases in representative oral bacteria, and the number of sequences associated with that family in each organism’s genome[A35].
| Glycoside hydrolase family number# | Sequenced Organism | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Streptococcus. pneumoniae TIGR4 |
Streptococcus. gordonii DL1 |
Streptococcus. oralis Uo5 |
Streptococcus. oralis* 35037 |
Streptococcus. sanguinis SK36 |
Streptococcus. parasanguinis 15912 |
Streptococcus. mitis B6 |
Streptococcus. mutans UA159 |
Streptococcus. mutans NN2025 |
Actinomyces naeslundii* MG1 |
Veillonella. parvula DSM 2008 |
Haemophilus. parainfluenzae T3T1 |
Rothia. dentocariosa 17931 |
Prevotella. denticola F0289 |
|
| 1 | 6 | 5 | 2 | 2 | 4 | 4 | 2 | 4 | 4 | 1 | 1 | |||
| 2 | BgaA β-galactosidase 1 |
1 | 1 | 1 | 2 | 1 | 3 | |||||||
| 3 | 1 | 2 | 2 | 2 | 1 | 1 | ||||||||
| 8 | 1 | 1 | 1 | 1 | 1 | 8 | ||||||||
| 5 | 1 | |||||||||||||
| 13 | spuA pullanase 7 |
6 | 7 | 4 | 8 | 7 | 7 | 6 | 6 | 13 | 2 | 3 | 8 | |
| 15 | 1 | |||||||||||||
| 16 | 2 | 1 | ||||||||||||
| 18 | 1 | 3 | ||||||||||||
| 20 | strH N-acetyl glucosaminidase 3 |
3 | 4 | 1 | 1 | 3 | 2 | 2 | ||||||
| 23 | 1 | 2 | 2 | 1 | 3 | 1 | 1 | 1 | 1 | 3 | 2 | |||
| 24 | 1 | 1 | 2 | |||||||||||
| 25 | 2 | 2 | 1 | 1 | 2 | 2 | 2 | 2 | 2 | 1 | 3 | |||
| 26 | 2 | |||||||||||||
| 27 | 1 | |||||||||||||
| 29 | 1 | 2 | 2 | 2 | 1 | 3 | ||||||||
| 31 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||
| 32 | 2 | 3 | 1 | 1 | 2 | 2 | 1 | 3 | 3 | 5 | 1 | 1 | 3 | |
| 33 | nanA, B, C neuraminidase 3 |
1 | 1 | 1 | 2 | 1 | ||||||||
| 35 | BgaC β-galactosidase 1 |
1 | 1 | 2 | 1 | 1 | ||||||||
| 36 | 1 | 1 | 2 | 2 | 1 | 1 | 1 | |||||||
| 37 | 1 | |||||||||||||
| 38 | 1 | 1 | 1 | 1 | 1 | |||||||||
| 42 | 1 | |||||||||||||
| 43 | 2 | |||||||||||||
| 53 | 1 | |||||||||||||
| 57 | 1 | |||||||||||||
| 66 | 1 | 1 | ||||||||||||
| 68 | 1 | 1 | 1 | |||||||||||
| 70 | 1 | 1 | 1 | 1 | ||||||||||
| 73 | 1 | 1 | 1 | 1 | 3 | 1 | 1 | 3 | 3 | 2 | 1 | |||
| 76 | 1 | |||||||||||||
| 77 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | ||||
| 78 | 1 | |||||||||||||
| 84 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||
| 85 | endoD endo-β-N-acetyl glucosaminidase 1 |
1 | 1 | 1 | 1 | 1 | ||||||||
| 92 | 1 | 1 | 1 | 1 | 1 | 5 | ||||||||
| 95 | 2 | 1 | 1 | 1 | ||||||||||
| 97 | 2 | |||||||||||||
| 98 | 1 | |||||||||||||
| 101 | Eng O-glycosidase 1 |
1 | 1 | 1 | ||||||||||
| 102 | 1 | |||||||||||||
| 103 | 1 | |||||||||||||
| 109 | 1 | |||||||||||||
| 112 | 1 | |||||||||||||
| 125 | 1 | 1 | 1 | 1 | 1 | 1 | ||||||||
| NC | 1 | 1 | ||||||||||||
The number does not necessarily indicate the number of glycoside hydrolase genes because the sequences are those of molecular domains within a protein; it is possible that a single gene may contain more than one of the sequences. Enzyme families important for the catabolism of high-molecular-weight oligosaccharides in S. pneumoniae (39) are shown in bold.
Data from completed genomes not yet in the Carbohydrate-Active enZYmes (CAZY) database, courtesy of Bernard Henrissat and the CAZY team (www.cazy.org)
An interdependence between coaggregating strains of A. naeslundii and S. oralis has been demonstrated in vitro when these bacteria are grown using saliva as the sole carbon source (53); indeed, it is clear from Table 1 that potentially complementary sets of enzymes occur in these organisms. A. naeslundii (like S. mutans) lacks five key glycoside hydrolase enzyme families present in S. oralis. However, it has six other families not found in S. oralis. This observation provides a genetic and metabolic basis for determining the importance of oligosaccharide catabolism to the mutualistic interaction, an interaction that might drive evolution of cell-to-cell recognition. It is important to recognize that models which employ single bacterial strains, together with mucin or other single glycoproteins as carbon sources, reflect neither the complement of glycoproteins in saliva nor the diversity of bacteria in the oral cavity. Surveys for, and descriptions of, interactions within communities should use bacterial co-cultures together with a relevant complex carbon source, such as saliva or gingival crevicular fluid. While whole saliva is thought of as a well-mixed combination of secretions from the complement of different salivary glands, it may not be the fluid to which supragingival biofilms in various locations in the mouth are exposed. For example, effects of salivary flow stimulation on the protein profile, proximity to a particular gland and saliva throughput should be considered. Furthermore, the penetration of high-molecular-weight components, such as mucins, into deeper regions of a biofilm is likely to be minimal (71). Lastly, much of the above discussion has only limited relevance to the ecology of subgingival biofilms.
Communities as etiological agents
Broad-based molecular community analysis has provided the most comprehensive evidence that neither caries nor periodontal disease results from the presence of a single pathogen. In a series of studies on childhood caries (1, 4), a reverse-capture assay of PCR products (56) for detection of 110 species was used to assess the prevalence of particular bacterial species. Actinomyces spp. and nonmutans streptococci were associated with white spots and thus may be important in the transition from health to early disease. Bifidobacteria spp. and Lactobacillus spp. seem to be as important as S. mutans in carious lesions, and some carious lesions bore no S. mutans.
In the more complex ecology of periodontal disease, a relatively simple early study, in which the presence of only 40 bacterial species was assessed, provided strong evidence for the existence of sets of co-occurring organisms (color-coded “complexes”) associated with clinically defined periodontal disease states (65). The yellow complex, comprising streptococci, was associated with relatively healthy pockets; the orange complex, comprising Fusobacterium, Prevotella and Campylobacter, was identified as a transitional population between health and severe periodontal disease; and the red complex, comprising P. gingivalis, Bacteroides forsythus (now Tannerella forsythia) and Treponema denticola, was associated with the most severely compromised pockets. These complexes were then used as a template for examining periodontal treatment efficacy in 461 patients (30). Reductions in the numbers of bacteria of the red complex and of the orange complex were identified 12 months after starting treatment. These changes corresponded to clinical measures of treatment efficacy. For example, across all locations where attachment loss was greatly reduced through therapy, the numbers of bacteria of the red and orange complexes were also reduced. Conversely, in sites where attachment loss increased despite therapy, no changes were observed in these complexes. Perhaps of greatest interest were the results obtained when the prevalence of the different complexes was assessed at the level of the individual patient. The patients fell into 11 clusters and, when clinical parameters were assessed across these clusters, it became clear that the composition of the microbial community was related to treatment efficacy. In other words, patients with the highest level of red-complex bacteria were those in which treatment was most effective. Another broad-based study examined the percentages of numerous bacteria in individuals whose periodontal status was monitored over time, but who received no treatment (42). Here, while numerous changes in composition of the microbial community took place, none of the changes could be statistically correlated with worsening or improving periodontal health. A recent molecular taxonomic study examined the prevalence of several periodontal pathogens, including red-complex organisms, across a range of clinically defined periodontal disease states (59): periodontitis resistant; chronic periodontitis; and generalized aggressive periodontitis. While the target organisms – P. gingivalis and Treponema lecithinolyticum – were found more often in diseased individuals, they were also present in healthy persons. The presence of T. lecithinolyticum was shown to distinguish the two periodontitis populations, but it could not distinguish the healthy controls from patients with chronic periodontitis. However, the presence of P. gingivalis was strongly correlated with pocket depth across all populations. The results of these latter two studies clearly show the difficulty in establishing a single bacterial species as a marker for periodontitis, but at the same time reinforce the importance of certain bacteria in disease states.
Some “chicken-or-egg” discussion exists for polymicrobial diseases, primarily because it seems impossible to fulfill Koch’s postulates: no single pathogen exists. However, as is true outside the human body, the bacterial community correlates with the environment, and changes in the environment result in changes in the bacterial community. Because most infectious diseases are caused by a single pathogen, an ecological perspective has been missing from much clinical research and, until recently, from much human microbiome research. The presence/absence of the pathogen is the sole significant measure for most infectious diseases, but for polymicrobial diseases this yardstick is not valid. It is tempting to think that human environments, the buccal surface of the third molar, for example, are identical from person to person. This thinking is appropriately reflected in clinical studies. Only by studying a large population of diseased individuals as a group do we appreciate the impact of a medical intervention. If we recognize that water-quality differences between streams can have a profound impact on the composition of the associated microflora, then we must recognize that the differences between, for example, immune effectors or gingival crevicular fluid composition in individuals with periodontal disease may reflect or influence subgingival bacterial profiles. These differences provide keys for understanding the interactions between organisms, and how bacterial communities interact with the host. It is therefore important for data from multiperson studies to be analyzed at the level of the individual. An example of the importance of this perspective, individual-as-ecosystem, is the previously discussed study on plaque from healthy adults by Bik et al. (6); alone, the dominance of Prevotella in one individual is of great interest, a detail not apparent in their pooled sample. Equally important in assessing “atypical” microbial community profiles are the environmental parameters in that individual. For example, in studies of healthy supragingival plaque, salivary chemistry data are desirable. For subgingival communities, gingival crevicular fluid should be characterized. Through such approaches, environmental factors important in establishing and maintaining particular oral biofilm compositions will be identified, and strategies can be tested for creating and maintaining healthy microbial profiles through the physiology of microbial interactions.
For the future
Numerous in-vitro and in-vivo model systems exist for the documentation of, and the experimentation on, interspecies interactions – a few examples are provided in Figs. 1 and 2, and a summary of their characteristics is presented in Table 2. These two approaches can be employed in a complementary manner. The in-vivo approach can document spatial relationships of bacteria in situ, provide clinical isolates through micromanipulation and deliver spatially resolved genomic information (or potentially even physiological information) at the level of the single cell. However, the in-vivo models are primarily descriptive and are difficult to use in an experimental manner. The controlled environment provided by in-vitro systems allows the manipulation of communities in hypothesis-driven experiments. One can identify physiological interactions important to a particular community in vitro by using oral bacterial isolates under conditions that approximate, as closely as possible, those in vivo. When particular communities seem ecologically fitter than others, the importance of the communities, rather than of single bacterial species, can be assessed using an in-vivo system. Both approaches can be applied to hypotheses on the importance of particular ecological conditions (e.g. the salivary protein profile) to community occurrence (in vivo) and to interbacterial physiological interaction (in vitro). The two most important criteria for in-vitro experiments are conditions that mimic as closely as possible the in-vivo ecological parameters and the selection of organisms known to occur together in communities in vivo. The most important criteria for the in-vivo studies are testable hypotheses that drive the choices of which organisms to monitor and the collection of ecological data relevant to establishment or action of the community. At present, good in-vitro models of the complex subgingival biofilm environment are lacking, but reasonable models of the simpler supragingival plaque environment exist. While the suite of organisms used in both models has been limited, we do have solid evidence, provided by sophisticated molecular studies, for the co-occurrence of many species within supragingival or subgingival environments and a potential relationship of these communities to disease. Using these data as a starting point, hypotheses on the physiology of bacterial interactions can be investigated in vitro, and evidence for these interactions (and their consequences) can be sought on a spatially resolved basis in vivo. Oral-biofilm biology remains paradigmatic in all biofilm research, especially in its potential for understanding interorganismal interactions that directly impact humans, regardless of systemic health status. Going forward, the focus of oral-biofilm research should be on the ecological principles underlying the interactions among bacteria in communities and between bacterial communities and the host.
Fig. 1.

In-vitro biofilm model systems. (A) Flowcell for microscopy. (B) Commercial pneumatically driven microcapillary flow system (Fluxion Biosystems, www.fluxionbio.com). (C) Drip-flow reactor. (D) Multiwell plate with pins in lid. (E) Stirred fermenter with coupons held in rods. (F) Constant-depth film fermentor. Images C and E courtesy of Montana State University Center for Biofilm Engineering. Image F courtesy of Dr Jonathan Pratten, UCL Eastman Dental Institute. ePTFE.
Fig. 2.
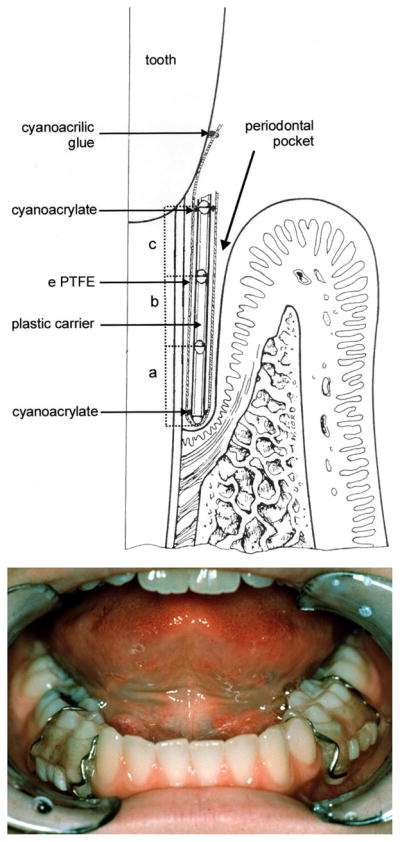
In-vivo oral-biofilm models. (A) foil-jacketed rod for retrieval of subgingival plaque, reprinted by permission from reference (74). (B) Retrievable enamel chip of the model of supragingival plaque.
Table 2.
| Approach | Type of device | Scale (amount of biomass) and biomass accessibilitya | Analysis strategiesb | Variable substratac | Advantages | Disadvantages | Selected references |
|---|---|---|---|---|---|---|---|
| In vitro | Microscopy flowcell | Micro Poor |
Microscopy Isolation of organisms from flowthrough |
No (glass), or through special fabrication | Single-cell resolution Moderately easy to use |
Labor-intensive, large volume of medium, precision pump is expensive | (55), (51) |
| Microcapillary flowcell | Micro Poor |
Microscopy Isolation of organisms from flowthrough |
No (glass) | Single-cell resolution Excellent shear control Small volume of medium (use of valuable reagents) Moderately high number of samples |
Unwieldy for multiple inoculations or medium changes, expensive | (20), www.fluxionbio.com | |
| Drip-flow reactor | Macro Excellent |
Bulk Chemical extraction |
Yes | Gas–liquid interface Headspace can be sampled |
Aseptic handling difficult | (26) | |
| Multiwell plate +/− peg cover | Micro/semimicro Good |
Chemical extraction Isolation of organisms from liquid phase (well contents) Isolation of organisms from biofilm (sonication, scraping) Microscopy (of inserts or of glass- bottomed plates) |
No (polystyrene), but substrata can be inserted as disks within the wells | High throughput Easy to use Gas–liquid interface Single-cell resolution |
Stagnant system (i.e. similar to a batch culture), potential for “bath-tub ring” growth at gas–liquid interface | (3), (71) | |
| Stirred reactor | Semimicro/macro Excellent |
Bulk Chemical extraction |
Yes | Chemostat-like operation High number of replicate samples |
Unwieldy, moving parts | (27), (8) | |
| Constant-depth film fermentor | Semimicro/macro Excellent |
Bulk Chemical extraction |
Yes | High number of replicate samples Gas–liquid interface Controlled biofilm thickness |
Unwieldy, moving parts | (57) | |
| Artificial mouth | Macro Excellent |
Bulk Chemical extraction |
Yes | Replicate samples of very thick plaque Analog Gas–liquid interface |
Custom construction, unwieldy | (63) | |
| In vivo | Jacketed rod | Micro Excellent |
Microscopy Micromanipulation |
Limited to thin foils of biologically inert materials | Natural system | Clinician required, individualized components | (75) |
| Retrievable enamel chip | Micro Excellent |
Microscopy Micromanipulation |
Limited to small pieces of biologically inert materials | Natural system | Clinician required, individualized components | (54), (19) |
Accessibility, ability to harvest or otherwise physically manipulate biomass; macro, easy to see (much biomass); micro, difficult to see; semimicro, can be clearly visible.
Bulk, colony-forming unit counts and isolation of organisms from biofilm; chemical extraction, nucleic acid, protein, polysaccharide.
Ability to integrate different types of substrata for biofilm attachment.
Acknowledgments
Dr Palmer is supported by the Intramural Research Program of the National Institute of Dental and Craniofacial Research at the National Institutes of Health.
References
- 1.Aas JA, Griffen AL, Dardis SR, Lee AM, Olsen I, Dewhirst FE, Leys EJ, Paster BJ. Bacteria of dental caries in primary and permanent teeth in children and young adults. J Clin Microbiol. 2008;46:1407–1417. doi: 10.1128/JCM.01410-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahn J, Yang L, Paster BJ, Ganly I, Morris L, Pei Z, Hayes RB. Oral microbiome profiles: 16S rRNA pyrosequencing and microarray assay comparison. PLoS One. 2011;6:e22788. doi: 10.1371/journal.pone.0022788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahn SJ, Ahn SJ, Wen ZT, Brady LJ, Burne RA. Characteristics of biofilm formation by Streptococcus mutans in the presence of saliva. Infect Immun. 2008;76:4259–4268. doi: 10.1128/IAI.00422-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Becker MR, Paster BJ, Leys EJ, Moeschberger ML, Kenyon SG, Galvin JL, Boches SK, Dewhirst FE, Griffen AL. Molecular analysis of bacterial species associated with childhood caries. J Clin Microbiol. 2002;40:1001–1009. doi: 10.1128/JCM.40.3.1001-1009.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beighton D, Smith K, Hayday H. The growth of bacteria and the production of exoglycosidic enzymes in the dental plaque of macaque monkeys. Arch Oral Biol. 1986;31:829–835. doi: 10.1016/0003-9969(86)90137-8. [DOI] [PubMed] [Google Scholar]
- 6.Bik EM, Long CD, Armitage GC, Loomer P, Emerson J, Mongodin EF, Nelson KE, Gill SR, Fraser-Liggett CM, Relman DA. Bacterial diversity in the oral cavity of 10 healthy individuals. Isme J. 2010;4:962–974. doi: 10.1038/ismej.2010.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bos R, van der Mei HC, Busscher HJ. Co-adhesion of oral microbial pairs under flow in the presence of saliva and lactose. J Dent Res. 1996;75:809–815. doi: 10.1177/00220345960750021201. [DOI] [PubMed] [Google Scholar]
- 8.Bradshaw DJ, Marsh PD, Allison C, Schilling KM. Effect of oxygen, inoculum composition and flow rate on development of mixed-culture oral biofilms. Microbiology. 1996;142(Pt 3):623–629. doi: 10.1099/13500872-142-3-623. [DOI] [PubMed] [Google Scholar]
- 9.Bradshaw DJ, McKee AS, Marsh PD. Effects of carbohydrate pulses and pH on population shifts within oral microbial communities in vitro. J Dent Res. 1989;68:1298–1302. doi: 10.1177/00220345890680090101. [DOI] [PubMed] [Google Scholar]
- 10.Burnaugh AM, Frantz LJ, King SJ. Growth of Streptococcus pneumoniae on human glycoconjugates is dependent upon the sequential activity of bacterial exoglycosidases. J Bacteriol. 2008;190:221–230. doi: 10.1128/JB.01251-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Busscher HJ, Norde W, van der Mei HC. Specific molecular recognition and nonspecific contributions to bacterial interaction forces. Appl Environ Microbiol. 2008;74:2559–2564. doi: 10.1128/AEM.02839-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Byers HL, Tarelli E, Homer KA, Beighton D. Sequential deglycosylation and utilization of the N-linked, complex-type glycans of human alpha1-acid glycoprotein mediates growth of Streptococcus oralis. Glycobiology. 1999;9:469–479. doi: 10.1093/glycob/9.5.469. [DOI] [PubMed] [Google Scholar]
- 13.Carlsson J. Growth and nutrition as ecological factors. In: Kuramitsu HK, Ellen RP, editors. Oral Bacterial Ecology. Horizon Scientific Press; Norfolk, England: 2000. pp. 67–130. [Google Scholar]
- 14.Chalmers NI, Palmer RJ, Jr, Cisar JO, Kolenbrander PE. Characterization of a Streptococcus sp.-Veillonella sp. community micromanipulated from dental plaque. J Bacteriol. 2008;190:8145–8154. doi: 10.1128/JB.00983-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cisar JO, Takahashi Y, Ruhl S, Donkersloot JA, Sandberg AL. Specific inhibitors of bacterial adhesion: observations from the study of gram-positive bacteria that initiate biofilm formation on the tooth surface. Advances in Dental Research. 1997;11:168–175. doi: 10.1177/08959374970110010801. [DOI] [PubMed] [Google Scholar]
- 16.Daims H, Taylor MW, Wagner M. Wastewater treatment: a model system for microbial ecology. Trends in Biotechnology. 2006;24:483–489. doi: 10.1016/j.tibtech.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 17.de Jong MH, van der Hoeven JS. The growth of oral bacteria on saliva. Journal of Dental Research. 1987;66:498–505. doi: 10.1177/00220345870660021901. [DOI] [PubMed] [Google Scholar]
- 18.Dewhirst FE, Chen T, Izard J, Paster BJ, Tanner ACR, Yu WH, Lakshmanan A, Wade WG. The human oral microbiome. J Bacteriol. 2010;192:5002–5017. doi: 10.1128/JB.00542-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dige I, Nilsson H, Kilian M, Nyvad B. In situ identification of streptococci and other bacteria in initial dental biofilm by confocal laser scanning microscopy and fluorescence in situ hybridization. Eur J Oral Sci. 2007;115:459–467. doi: 10.1111/j.1600-0722.2007.00494.x. [DOI] [PubMed] [Google Scholar]
- 20.Ding AM, Palmer RJ, Cisar JO, Kolenbrander PE. Shear-enhanced oral microbial adhesion. Applied and Environmental Microbiology. 2010;76:1294–1297. doi: 10.1128/AEM.02083-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Downes J, Munson M, Wade WG. Dialister invisus sp. nov., isolated from the human oral cavity. Int J Syst Evol Microbiol. 2003;53:1937–1940. doi: 10.1099/ijs.0.02640-0. [DOI] [PubMed] [Google Scholar]
- 22.Elangovan S, Margolis HC, Oppenheim FG, Beniash E. Conformational changes in salivary proline-rich protein 1 upon adsorption to calcium phosphate crystals. Langmuir. 2007;23:11200–11205. doi: 10.1021/la7013978. [DOI] [PubMed] [Google Scholar]
- 23.Fejerskov O, Scheie AA, Manji F. The effect of sucrose on plaque pH in the primary and permanent dentition of caries-inactive and -active Kenyan children. J Dent Res. 1992;71:25–31. doi: 10.1177/00220345920710010401. [DOI] [PubMed] [Google Scholar]
- 24.Gibbons RJ, Hay DI, Childs WC, 3rd, Davis G. Role of cryptic receptors (cryptitopes) in bacterial adhesion to oral surfaces. Arch Oral Biol. 1990;35(Suppl):107S–114S. doi: 10.1016/0003-9969(90)90139-2. [DOI] [PubMed] [Google Scholar]
- 25.Glenister DA, Salmamon KE, Smith K, Beighton D, Keevil CW. Enhanced growth of complex communities of dental palque bacteria in mucin-limited continuous culture. Microbial Ecology in Health and Disease. 1988;1:31–38. [Google Scholar]
- 26.Goeres DM, Hamilton MA, Beck NA, Buckingham-Meyer K, Hilyard JD, Loetterle LR, Lorenz LA, Walker DK, Stewart PS. A method for growing a biofilm under low shear at the air-liquid interface using the drip-flow biofilm reactor. Nat Protoc. 2009;4:783–788. doi: 10.1038/nprot.2009.59. [DOI] [PubMed] [Google Scholar]
- 27.Goeres DM, Loetterle LR, Hamilton MA, Murga R, Kirby DW, Donlan RM. Statistical assessment of a laboratory method for growing biofilms. Microbiology. 2005;151:757–762. doi: 10.1099/mic.0.27709-0. [DOI] [PubMed] [Google Scholar]
- 28.Griffen AL, Beall CJ, Campbell JH, Firestone ND, Kumar PS, Yang ZK, Podar M, Leys EJ. Distinct and complex bacterial profiles in human periodontitis and health revealed by 16S pyrosequencing. ISME Journal. 2012;6:1176–1185. doi: 10.1038/ismej.2011.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Griffen AL, Beall CJ, Firestone ND, Gross EL, Difranco JM, Hardman JH, Vriesendorp B, Faust RA, Janies DA, Leys EJ. CORE: a phylogenetically-curated 16S rDNA database of the core oral microbiome. PLoS One. 2011;6:e19051. doi: 10.1371/journal.pone.0019051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haffajee AD, Teles RP, Socransky SS. The effect of periodontal therapy on the composition of the subgingival microbiota. Periodontology 2000. 2006;42:219–258. doi: 10.1111/j.1600-0757.2006.00191.x. [DOI] [PubMed] [Google Scholar]
- 31.Hugenholtz P, Tyson GW, Webb RI, Wagner AM, Blackall LL. Investigation of candidate division TM7, a recently recognized major lineage of the domain Bacteria with no known pure-culture representatives. Appl Environ Microbiol. 2001;67:411–419. doi: 10.1128/AEM.67.1.411-419.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huse SM, Welch DM, Morrison HG, Sogin ML. Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol. 2010;12:1889–1898. doi: 10.1111/j.1462-2920.2010.02193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jalava J, Eerola E. Phylogenetic analysis of Fusobacterium alocis and Fusobacterium sulci based on 16S rRNA gene sequences: proposal of Filifactor alocis (Cato, Moore and Moore) comb. nov. and Eubacterium sulci (Cato, Moore and Moore) comb. nov. Int J Syst Bacteriol. 1999;49(Pt 4):1375–1379. doi: 10.1099/00207713-49-4-1375. [DOI] [PubMed] [Google Scholar]
- 34.Jeong JK, Kwon O, Lee YM, Oh DB, Lee JM, Kim S, Kim EH, Le TN, Rhee DK, Kang HA. Characterization of the Streptococcus pneumoniae BgaC protein as a novel surface beta-galactosidase with specific hydrolysis activity for the Galβ1-3GlcNAc moiety of oligosaccharides. J Bacteriol. 2009;191:3011–3023. doi: 10.1128/JB.01601-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jian W, Dong X. Transfer of Bifidobacterium inopinatum and Bifidobacterium denticolens to Scardovia inopinata gen. nov., comb. nov., and Parascardovia denticolens gen. nov., comb. nov., respectively. Int J Syst Evol Microbiol. 2002;52:809–812. doi: 10.1099/00207713-52-3-809. [DOI] [PubMed] [Google Scholar]
- 36.Jumas-Bilak E, Jean-Pierre H, Carlier JP, Teyssier C, Bernard K, Gay B, Campos J, Morio F, Marchandin H. Dialister micraerophilus sp. nov. and Dialister propionicifaciens sp. nov., isolated from human clinical samples. Int J Syst Evol Microbiol. 2005;55:2471–2478. doi: 10.1099/ijs.0.63715-0. [DOI] [PubMed] [Google Scholar]
- 37.Jumas-Bilak E, Roudiere L, Marchandin H. Description of ‘Synergistetes’ phyl. nov. and emended description of the phylum ‘Deferribacteres’ and of the family Syntrophomonadaceae, phylum ‘Firmicutes’. Int J Syst Evol Microbiol. 2009;59:1028–1035. doi: 10.1099/ijs.0.006718-0. [DOI] [PubMed] [Google Scholar]
- 38.Keijser BJ, Zaura E, Huse SM, van der Vossen JM, Schuren FH, Montijn RC, ten Cate JM, Crielaard W. Pyrosequencing analysis of the oral microflora of healthy adults. J Dent Res. 2008;87:1016–q020. doi: 10.1177/154405910808701104. [DOI] [PubMed] [Google Scholar]
- 39.King SJ. Pneumococcal modification of host sugars: a major contributor to colonization of the human airway? Mol Oral Microbiol. 2010;25:15–24. doi: 10.1111/j.2041-1014.2009.00564.x. [DOI] [PubMed] [Google Scholar]
- 40.Kolenbrander PE. Surface recognition among oral bacteria - multigeneric coaggregations and their mediators. Critical Reviews in Microbiology. 1989;17:137–159. doi: 10.3109/10408418909105746. [DOI] [PubMed] [Google Scholar]
- 41.Kolenbrander PE, London J. Adhere today, here tomorrow: oral bacterial adherence. Journal of Bacteriology. 1993;175:3247–3252. doi: 10.1128/jb.175.11.3247-3252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kumar PS, Leys EJ, Bryk JM, Martinez FJ, Moeschberger ML, Griffen AL. Changes in periodontal health status are associated with bacterial community shifts as assessed by quantitative 16S cloning and sequencing. J Clin Microbiol. 2006;44:3665–3673. doi: 10.1128/JCM.00317-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.LeBlanc DJ, Flynn TR, Simos C, Lantz MS. Antibiotics and treatment of infectious diseases. In: Lamont RJ, Burne RA, Lantz MS, LeBlanc DJ, editors. Oral Microbiology and Immunology. American Society for Microbiology Press; Washington, D.C: 2006. pp. 379–422. [Google Scholar]
- 44.Liljemark WF, Bloomquist C. Human oral microbial ecology and dental caries and periodontal diseases. Crit Rev Oral Biol Med. 1996;7:180–98. doi: 10.1177/10454411960070020601. [DOI] [PubMed] [Google Scholar]
- 45.Mager DL, Ximenez-Fyvie LA, Haffajee AD, Socransky SS. Distribution of selected bacterial species on intraoral surfaces. J Clin Periodontol. 2003;30:644–654. doi: 10.1034/j.1600-051x.2003.00376.x. [DOI] [PubMed] [Google Scholar]
- 46.Mantzourani M, Fenlon M, Beighton D. Association between Bifidobacteriaceae and the clinical severity of root caries lesions. Oral Microbiol Immunol. 2009;24:32–37. doi: 10.1111/j.1399-302X.2008.00470.x. [DOI] [PubMed] [Google Scholar]
- 47.Mantzourani M, Gilbert SC, Sulong HN, Sheehy EC, Tank S, Fenlon M, Beighton D. The isolation of bifidobacteria from occlusal carious lesions in children and adults. Caries Res. 2009;43:308–313. doi: 10.1159/000222659. [DOI] [PubMed] [Google Scholar]
- 48.Marsh PD. Are dental diseases examples of ecological catastrophes? Microbiology. 2003;149:279–294. doi: 10.1099/mic.0.26082-0. [DOI] [PubMed] [Google Scholar]
- 49.Mishra A, Wu C, Yang J, Cisar JO, Das A, Ton-That H. The Actinomyces oris type 2 fimbrial shaft FimA mediates co-aggregation with oral streptococci, adherence to red blood cells and biofilm development. Mol Microbiol. 2010 doi: 10.1111/j.1365-2958.2010.07252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moore LV, Moore WE. Oribaculum catoniae gen. nov., sp. nov.; Catonella morbi gen. nov., sp. nov.; Hallella seregens gen. nov., sp. nov.; Johnsonella ignava gen. nov., sp. nov.; and Dialister pneumosintes gen. nov., comb. nov., nom. rev., anaerobic gram-negative bacilli from the human gingival crevice. Int J Syst Bacteriol. 1994;44:187–192. doi: 10.1099/00207713-44-2-187. [DOI] [PubMed] [Google Scholar]
- 51.Palmer RJ., Jr Microscopy flowcells: perfusion chambers for real-time study of biofilms. Methods in Enzymology. 1999;310:160–166. doi: 10.1016/s0076-6879(99)10014-4. [DOI] [PubMed] [Google Scholar]
- 52.Palmer RJ, Jr, Gordon SM, Cisar JO, Kolenbrander PE. Coaggregation-mediated interactions of streptococci and actinomyces detected in initial human dental plaque. J Bacteriol. 2003;185:3400–3409. doi: 10.1128/JB.185.11.3400-3409.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Palmer RJ, Jr, Kazmerzak K, Hansen MC, Kolenbrander PE. Mutualism vs. independence: strategies of mixed-species oral biofilms in vitro using saliva as the sole nutrient source. Journal of Bacteriology. 2001;69:5794–5804. doi: 10.1128/IAI.69.9.5794-5804.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Palmer RJ, Jr, Wu R, Gordon S, Bloomquist C, Liljemark WF, Kilian M, Kolenbrander PE. Retrieval of biofilms from the oral cavity. Methods in Enzymology. 2001;337:393–403. doi: 10.1016/s0076-6879(01)37028-3. [DOI] [PubMed] [Google Scholar]
- 55.Palmer RJJ, Haagensen AJ, Neu TR, Sternberg C. Confocal microscopy of biofilms - spatiotemporal approaches. In: Pawley JB, editor. Handbook of Biological Confocal Microscopy. 3. Springer; New York: 2006. pp. 870–888. [Google Scholar]
- 56.Paster BJ, I, Bartoszyk M, Dewhirst FE. Identification of oral streptococci using PCR-based, reverse-capture, checkerboard hybridization. Methods in Cell Science. 1998;20:223–231. [Google Scholar]
- 57.Pratten J. Growing oral biofilms in a constant depth film fermentor (CDFF) Curr Protoc Microbiol. 2007;Chapter 1(Unit 1B):5. doi: 10.1002/9780471729259.mc01b05s6. [DOI] [PubMed] [Google Scholar]
- 58.Quirynen M, Vogels R, Pauwels M, Haffajee AD, Socransky SS, Uzel NG, van Steenberghe D. Initial subgingival colonization of ‘pristine’ pockets. Journal of Dental Research. 2005;84:340–344. doi: 10.1177/154405910508400409. [DOI] [PubMed] [Google Scholar]
- 59.Riep B, Edesi-Neuss L, Claessen F, Skarabis H, Ehmke B, Flemmig TF, Bernimoulin JP, Gobel UB, Moter A. Are putative periodontal pathogens reliable diagnostic markers? J Clin Microbiol. 2009;47:1705–1711. doi: 10.1128/JCM.01387-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ruoff KL. Nutritionally variant streptococci. Clin Microbiol Rev. 1991;4:184–90. doi: 10.1128/cmr.4.2.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sato S, Kanamoto T, Inoue M. Abiotrophia elegans strains comprise 8% of the nutritionally variant streptococci isolated from the human mouth. J Clin Microbiol. 1999;37:2553–2556. doi: 10.1128/jcm.37.8.2553-2556.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schlafer S, Riep B, Griffen AL, Petrich A, Hubner J, Berning M, Friedmann A, Gobel UB, Moter A. Filifactor alocis--involvement in periodontal biofilms. BMC Microbiol. 2010;10:66. doi: 10.1186/1471-2180-10-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sissons CH, Cutress TW, Hoffman MP, Wakefield JS. A multi-station dental plaque microcosm (artificial mouth) for the study of plaque growth, metabolism, pH, and mineralization. J Dent Res. 1991;70:1409–1416. doi: 10.1177/00220345910700110301. [DOI] [PubMed] [Google Scholar]
- 64.Slomiany BL, V, Murty L, Slomiany A. Structural features of carbohydrate chains in human salivary mucins. Int J Biochem. 1993;25:259–265. doi: 10.1016/0020-711x(93)90015-7. [DOI] [PubMed] [Google Scholar]
- 65.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 66.Sokurenko EV, Vogel V, Thomas WE. Catch-bond mechanism of force-enhanced adhesion: counterintuitive, elusive, but…. widespread? Cell Host & Microbe. 2008;4:314–323. doi: 10.1016/j.chom.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stephan R. Changes in hydrogen-ion concentration on tooth surfaces and in carious lesions. Journal of the American Dental Association. 1940;27:718–723. [Google Scholar]
- 68.Stetter KO, Fiala G, Huber R, Segerer A. Life above the boiling point of water? Experientia. 1986;42:1187–1191. [Google Scholar]
- 69.Takahashi Y, Takashima E, Shimazu K, Yagishita H, Aoba T, Konishi K. Contribution of sialic acid-binding adhesin to pathogenesis of experimental endocarditis caused by Streptococcus gordonii DL1. Infect Immun. 2006;74:740–743. doi: 10.1128/IAI.74.1.740-743.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Thomas WE, Nilsson LM, Forero M, Sokurenko EV, Vogel V. Shear-dependent ‘stick-and-roll’ adhesion of type 1 fimbriated Escherichia coli. Molecular Microbiology. 2004;53:1545–1557. doi: 10.1111/j.1365-2958.2004.04226.x. [DOI] [PubMed] [Google Scholar]
- 71.Thurnheer T, Gmur R, Shapiro S, Guggenheim B. Mass transport of macromolecules within an in vitro model of supragingival plaque. Appl Environ Microbiol. 2003;69:1702–1709. doi: 10.1128/AEM.69.3.1702-1709.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Valm AM, Welch JL, Rieken CW, Hasegawa Y, Sogin ML, Oldenbourg R, Dewhirst FE, Borisy GG. Systems-level analysis of microbial community organization through combinatorial labeling and spectral imaging. Proc Natl Acad Sci U S A. 2011;108:4152–4157. doi: 10.1073/pnas.1101134108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vartoukian SR, Palmer RM, Wade WG. Cultivation of a Synergistetes strain representing a previously uncultivated lineage. Environ Microbiol. 2010;12:916–928. doi: 10.1111/j.1462-2920.2009.02135.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vartoukian SR, Palmer RM, Wade WG. Diversity and morphology of members of the phylum “Synergistetes” in periodontal health and disease. Appl Environ Microbiol. 2009;75:3777–3786. doi: 10.1128/AEM.02763-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wecke J, Kersten T, Madela K, Moter A, Göbel UB, Friedmann A, Bernimoulin JP. A novel technique for monitoring the development of bacterial biofilms in human periodontal pockets. Fems Microbiology Letters. 2000;191:95–101. doi: 10.1111/j.1574-6968.2000.tb09324.x. [DOI] [PubMed] [Google Scholar]
- 76.Xie G, Chain PS, Lo CC, Liu KL, Gans J, Merritt J, Qi F. Community and gene composition of a human dental plaque microbiota obtained by metagenomic sequencing. Mol Oral Microbiol. 2010;25:391–405. doi: 10.1111/j.2041-1014.2010.00587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009;9:259. doi: 10.1186/1471-2180-9-259. [DOI] [PMC free article] [PubMed] [Google Scholar]