

Abstract
Background
Glucose-sensor-induced tissue reactions (e.g., inflammation and wound healing) are known to negatively impact sensor function in vivo. The roles of cytokine networks in controlling these tissue reactions (i.e., sensor biofouling) is not understood. In the present study, we investigated the role of interleukin-1 receptor antagonist (IL-1Ra), a key anti-inflammatory antagonist of the proinflammatory interleukin-1 cytokines [i.e. interleukin-1 (IL-1) alpha and IL-1 beta] in controlling continuous glucose monitoring (CGM).
Methods
To investigate the role of IL-1Ra in long-term CGM in vivo, we compared CGM in transgenic mice that overexpress IL-1Ra [interleukin-1 receptor antagonist overexpresser (IL-1Ra~OE), B6.Cg-Tg(IL1rn)1Dih/J] or are deficient in IL-1Ra [interleukin-1 receptor antagonist knockout (IL-1Ra~KO), B6.129S-IL1rntm1Dih/J] with mice that have normal levels of IL-1Ra (C57BL/6) over a 28-day time period.
Results
Mean absolute relative difference (MARD) analysis of CGM results among the mice of varying IL-1Ra levels demonstrated that during the first 21 days, IL-1~KO mice had the greatest tissue inflammation and the poorest sensor performance (i.e., higher MARD values) when compared with normal or IL-1Ra~OE mice. By 28 days post-sensor implantation, the inflammatory reactions had subsided and were replaced by varying degrees of fibrosis.
Conclusions
These data support our hypothesis on the importance of the IL-1 family of agonists and antagonists in controlling tissue reactions and sensor function in vivo. These data also suggest that local delivery of IL-1Ra genes or recombinant proteins (anakinra) or other IL-1 antagonists such as antibodies or soluble IL-1 receptors would suppress sensor-induced tissue reactions and likely enhance glucose sensor function by inhibiting inflammation and wound healing at sensor implantation sites.
Keywords: angiogenesis, biosensor, continuous glucose monitoring, diabetes, fibrosis, inflammation
Introduction
It is generally believed that loss of glucose sensor function in vivo is caused by sensor-induced tissue reactions, including inflammation and wound healing (i.e., fibrosis), which is also accepted to induce sensor biofouling. It is known that cytokine and growth factor networks are central to controlling inflammation and wound healing (e.g., fibrosis and neovascularization) in a wide variety of diseases and tissue injuries.1,2 Consequently, it is likely that cytokine/ growth factor networks play a major role in controlling inflammation, wound healing, and therefore sensor biofouling at sites of in vivo glucose sensor implantation. One of the major cytokine families involved in controlling inflammation and wound healing is the interleukin-1 (IL-1) family. The major IL-1 proinflammatory cytokines interleukin-1 alpha (IL-1a) and interleukin-1 beta (IL-1B) are powerful proinflammatory cytokines. Effective regulation of IL-1a and IL-1B in vivo is critical to preventing uncontrolled inflammation, tissue destruction, and fibrosis associated with acute and chronic inflammatory processes, including foreign body reactions. Crucial to controlling IL-1a and IL-1B mediated inflammation is the naturally occurring IL-1 antagonist, interleukin-1 receptor antagonist (IL-1Ra),3,4 a competitive antagonist that competes with IL-1a and IL-1B for binding to the IL-1 receptors and prevents IL-1 activation of both leukocytes and tissue cells.5–11 A recombinant version of IL-1Ra, designated anakinra, has been used to treat inflammation associated with rheumatoid arthritis.4 Additionally, there are a number of ongoing clinical trials that are utilizing not only anakinra, but also a soluble IL-1 decoy receptor (rilonacept) as well as neutralizing antibodies to IL-1B (canakinumab), IL-1a, and IL-1 receptors to treat variety of other diseases, including diabetes.4 We hypothesized that proinflammatory cytokines such as IL-1a and IL-1B and their antagonist IL-1Ra play central roles in controlling inflammation and wound healing at sites of glucose sensor implantation and thereby control glucose sensor biofouling. We further hypothesize that at sites of glucose sensor implantation, both tissue-cell- and leukocyte-derived IL-1a and IL-1B are key sources for IL-1 expression both acutely and chronically. Specifically, we hypothesize that the initial sensor implantation trauma triggers release of IL-1a from dead and dying tissue cells, which initiates acute inflammation, including leukocyte recruitment (Figure 1). These recruited leukocytes induce additional cell (i.e., tissue cells and leukocytes) and tissue destruction, resulting in further release of IL-1a and IL-B. This additional release of IL-1 proinflammatory cytokines not only amplifes acute inflammation, but also drives chronic inflammation (Figure 1). Alternatively IL-1 antagonists such as IL-Ra can inhibit the IL-1-induced tissue reaction at several steps in the process (Figure 1). To begin to probe this hypothesis, we have utilized transgenic mice that are interleukin-1 receptor antagonist overexpressers (IL-1Ra~OEs) or interleukin-1 receptor antagonist knockouts (IL-1Ra~KOs). For these initial studies, we compared sensor function in transgenic mice that (1) overexpress IL-1Ra expression (B6.Cg-Tg(II1rn)1Dih/J) and (2) are deficient in IL-1Ra expression (B6.129S-Il1rntm1Dih/J) with mice that have normal levels of IL-1Ra expression (C57BL/6) for a 7-day period of continuous glucose monitoring (CGM).12 These short-term sensor studies indicated that (1) the IL-1 family of cytokines, likely IL-1 agonists IL-1a and IL-1B, play a critical role in controlling tissue reactions and sensor function in vivo and (2) IL-1Ra is critical in controlling tissue reactions and sensor function in vivo. For the present study, we extended our initial short-term sensor studies from 7 to 28 days, using these same transgenic mouse models. These long-term studies support our hypothesis on the importance of the IL-1 family of cytokines in sensor function in vivo. It demonstrated that although IL-1Ra plays a major role in controlling inflammation in the earlier stages (weeks 1–3) of tissue reactions to glucose sensor implantation (i.e., inflammation and wound healing), it is less important in the later stages of tissue response to the implanted sensor (e.g., fibrosis). Future studies directed to determine the impact of local-delivery IL-1Ra gene or recombinant protein (anakinra) or other IL-1 antagonists, such as neutralizing antibodies and soluble IL-1 receptors,4 would confirm our transgenic mouse studies and would suggest possible future therapeutic strategies to target sensor biofouling.
Figure 1.
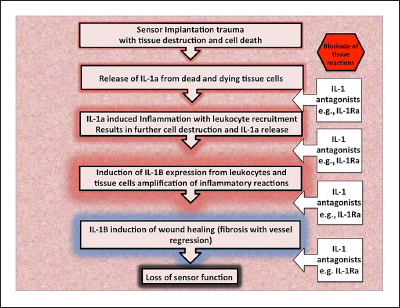
General model of the role of IL-1 (IL-1a and IL-1B) and IL-1Ra in glucose sensor induced tissue reactions. This model illustrates the cascade of IL-1-related events from the release of IL-1 from damaged tissues to loss of sensor function. At each step following the release of internal cellular IL-1a due to sensor implantation trauma, IL-1Ra can inhibit the cascade. Interleukin-1a release from damaged tissues induces further IL-1 alpha inflammation and cell destruction with leukocyte recruitment. Interleukin-1 beta is expressed by the leukocytes, leading to other inflammatory reactions and ultimately wound healing processes, including fibrosis with vessel regression, a severe inhibitor of accurate and timely sensor function.
Methods
Interleukin-1 Receptor Antagonist Knockout and Interleukin-1 Receptor Antagonist Overexpression Mouse Models
In the present studies, female IL-1Ra~KO mice and IL-1Ra~OE mice were utilized. The IL-1Ra~KO (B6.129S-Il1rntm1Dih/J) and IL-1Ra~OE mice (B6.Cg-Tg(II1rn)1Dih/J) were obtained from Jackson Laboratory (Bar Harbor, ME). Additionally, female C57BL/6 mice were used as normal controls for these studies and were also obtained from Jackson Laboratory.
Glucose Sensors, Implantation, and Murine Continuous Glucose Sensor System
The modified Navigator glucose sensors used in these in vivo studies were obtained from Abbott Diabetes Care. Glucose sensors were implanted into IL-1Ra~KO, IL-1Ra~OE, or C57BL/6j mice, and CGM was undertaken for a period up to 28 days as described previously12–14 Blood glucose reference measurements were obtained periodically over the 28-day implantation period, using blood obtained from the tail vein and a FreeStyle blood glucose monitor. The Institutional Animal Care and Use Committee of the University of Connecticut Health Center (Farmington, CT) approved all mice studies.
Continuous Glucose Monitoring Data Analysis
Reference blood measurements were used to calculate the mean absolute relative difference (MARD) over a 4-week experiment for the three groups of mice with distinct IL-1Ra genetic backgrounds.15 Equations (1)–(3) describe the MARD calculation in detail. Sensitivity (S; mg/dl/nA) is calculated for each mouse experiment based on the reference blood glucose and the sensor output (I; nA) measurements in an initial reference stage of the experiment, i.e., k in Equation (2) is approximately 5, for the first initial five measurements across 2 days.
| (1) |
| (2) |
| (3) |
Statistical Analysis
Since the mean MARD values were non-normal in distribution with the exception of the IL-1Ra~KO mice, Kruskal-Wallis tests were used to conduct statistical comparisons among the three groups of MARD values, as a nonparametric equivalent to analysis of variance. Mann-Whitney U tests were then conducted to determine the statistical differences between pairs of average mean MARD values, as non-parametric equivalents to Student’s -tests.
Histopathologic Analysis of Tissue Reactions at Glucose Sensor Implantation Sites
In order to evaluate tissue responses to glucose sensor implantation at various time points, individual mice were euthanized and the full thickness of the skin and sensors were removed en bloc in approximately 3 × 3 cm2 sections and immediately placed in tissue fixative. Tissue was fixed in zinc buffer for 24 h followed by standard processing, embedded in paraffin, and sectioned. The resulting 4–6 μm sections were then stained using standard protocols for hematoxylin eosin stain and Masson trichrome to evaluate fibrosis. Histopathologic evaluation of tissue reactions at sites of sensor implantation was performed on mouse specimens obtained at 1–28 days post-sensor implantation. The tissue samples were generally examined for signs of necrosis and inflammation, including leukocyte influx, fibrosis, angiogenesis, and vessel regression. To provide an initial evaluation of the inflammatory reactions at the sensor–tissue interface, we utilized a semiquantitative evaluation scoring system from 0–4. For this system, the tissue reactions were scored as follows: 0, no inflammation (no leukocyte infiltration present near the implanted sensor); 1, trace inflammation (occasional leukocyte infiltration present near the implanted sensor); 2, mild inflammation (scattered and consistent leukocyte infiltration present near the implanted sensor); 3, moderate inflammation (significant leukocyte infiltration near the implanted sensor); 4, severe inflammation (dense leukocyte infiltration near the implanted sensor).
The individual histologic sections were evaluated in a double-blind fashion, and the mean inflammation index was determined. Since the average inflammation index values were non-normal in distribution, with the exception of the day 14 IL-1Ra~KO and IL-1Ra~OE inflammation index values, Kruskal–Wallis tests were used to conduct statistical comparisons among the three groups of inflammation index values of differing genetic background. Mann–Whitney U tests were then conducted to determine the statistical differences between pairs of average inflammation index values. Microsoft Excel for Mac 2011 (version 14.1.4) and IBM SPSS Statistics 20 (release 20.0.0) were the software packages used for the calculations and statistical analyses, respectively, for both the MARDs and inflammation indices.
Results
Continuous Glucose Monitoring in the Normal and Interleukin-1 Receptor Antagonist Transgenic Mice
To begin our mice studies, we first determined the general CGM profile for the normal C57BL/6J and the two IL-1Ra transgenic mice. Figure 2 represents data of CGM over the 28-day time period for these mice. As expected and consistent with our previously published work,12 glucose sensing closely followed blood glucose levels during the first 7 days post-sensor implantation for the normal C57BL/6J mice (Figure 2A) as well as the transgenic IL-1Ra~OE mice (Figure 2B) but not for the IL-1Ra~KO mice (Figure 2C). Sensor function beyond 7–10 days post-sensor implantation displayed very heterogeneous patterns of CGM for all three mice strains. It appears that the IL-1Ra~KO mice consistently performed the worst and the IL-1Ra~OE mice performed the best when compared with C57BL/6J and IL-1Ra~KO mice.
Figure 2.
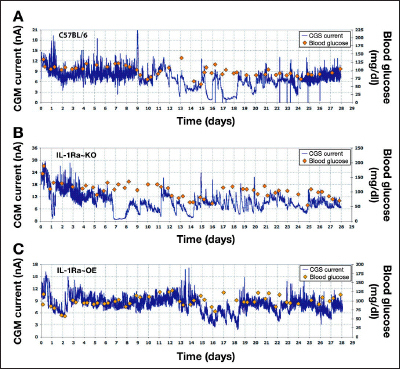
Continuous glucose monitoring in normal C57BL/6, IL-1Ra~KO, and IL-1Ra~OE mice over a 28-day time period. Representative examples of CGM in (A) normal C57BL/6, (B) IL-1Ra~KO, and (C) IL-1Ra~OE mice over a 28-day time period. Sensor function (blue line) is expressed as CGM current and externally monitored blood glucose (red diamonds) is expressed as blood glucose.
Quantitative Accuracy Assessment Using the Mean Absolute Relative Difference
To measure the role of IL-1Ra in the inflammatory and wound healing processes around glucose sensor implantation, we evaluated the effect of IL-1Ra on glucose sensor function by calculating the MARD over a 4-week experiment for the mice with three distinct IL-1Ra expression backgrounds. The impact of IL-1Ra expression on CGM is presented in Figure 3, in which IL-1Ra~KO mice have statistically higher MARD values when compared with the normal or IL-1Ra~OE mice in weeks 1 to 3 post-sensor implantation. The IL-1Ra~OE mice generally had lower MARD values when compared with C57BL/6J and IL-1Ra~KO during weeks 1–3 post-sensor implantation (Figure 3). By week 4 post-sensor implantation, there was no longer any statistically significant differences between the three groups of mice (Table 1). The IL-1Ra~KO mice had the worst total mean MARD of 29.75% ± 10.98%, whereas the C57BL/6 mice had an intermediate MARD of 23.50% ± 9.83%, and IL-1Ra~OE had the best overall MARD of 21.49% ± 13.84% for the 28-day study. Table 2 demonstrates that each comparison between the various MARD for IL-1Ra~KO mice to the other IL-1Ra genetic backgrounds was statistically significant (p < .05). The difference among the mice of the three different IL-1Ra genetic backgrounds, for the total MARD values (all 4 weeks together), was statistically significant, as per the Kruskal–Wallis tests (p = .006). Additionally, when observing the difference among the three groups of mice over time, we find a statistically significant difference in weeks 1 to 3 but not in week 4 (p = .013, .011, .039, and .283 for weeks 1–4, respectively; Kruskal–Wallis tests). We also observed that the difference in MARD between the various genetic IL-1Ra backgrounds completely disappears by week 4 or between 21 and 28 days, as shown in Table 1 and Figure 3.
Figure 3.
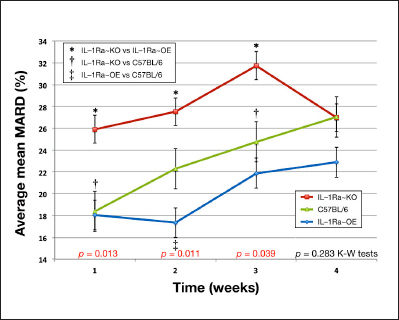
Impact of IL-1Ra expression on cumulative average mean MARD values from weeks 1 to 4 by IL-1Ra genetic background in a murine model of CGM. Cumulative average mean MARD from weeks 1 to 4 by IL-1Ra genetic background. * represents the statistical significance (p < .05) of the difference between IL-1Ra~KO and IL-1Ra~OE mean MARD values for that week. † represents the statistical significance (p < .05) of the difference between IL-1Ra~KO and C57BL/6 control mean MARD values for that week. ‡ represents the statistical significance (p < .05) of the difference between IL-1Ra~OE and C57BL/6 control mean MARD values for that week. Error bars present standard errors of the mean MARD for each individual week and genetic background. To test for statistical differences among all three genetic groups at once, Kruskal–Wallis (K–W) tests were conducted on the MARD values as a nonparametric equivalent to analysis of variance for each week.
Table 1.
Total Mean Absolute Relative Difference Values of Mice with Various Interleukin-1 Receptor Antagonist Genetic Backgrounds for All Four Weeksa
| Total mean MARD data | C57BL/6 average mean MARD = 23.50 ± 9.83% n = 26 | IL-1Ra~KO average mean MARD = 29.75 ± 10.98% n = 22 | IL-1Ra~OE average mean MARD = 21.49 ± 13.84% n = 20 |
| C57BL/6 average mean MARD = 23.50 ± 9.83% n = 26 | — | 0.230 | 0.1160 |
| IL-1Ra~KO average mean MARD = 29.75 ± 10.98% n = 22 | — | 0.0050 | |
| IL-1Ra~OE average mean MARD = 21.49 ± 13.84% n = 20 | — |
Error values following “±” are standard deviations from the average of the individual genetic background group’s MARD values. P-values within the boxes represent the statistical significance of the comparisons of the two treatment groups indicated in the axes, calculated by Mann–Whitney U tests, as nonparametric equivalents to Student’s t-tests. Mann–Whitney U tests were conducted because only the IL-1Ra~KO group had normally distributed mean MARD values.
Table 2.
Average Mean Absolute Relative Difference Values for Week 4 of Surviving Mice with Various Interleukin-1 Receptor Antagonist Genetic Backgroundsa
| Cumulative 4 weeks mean MARD data | C57BL/6 average mean MARD = 27.02 ± 10.95% n = 15 | IL-1Ra~KO average mean MARD = 22.51 ± 7.22% n = 6 | IL-1Ra~OE average mean MARD = 19.86 ± 8.07% n = 8 |
| C57BL/6 average mean MARD = 27.02 ± 10.95% n = 15 | — | 0.3809 | 0.2132 |
| IL-1Ra~KO average mean MARD = 22.51 ± 7.22% n = 6 | — | 0.2824 | |
| IL-1Ra~OE average mean MARD = 19.86 ± 8.07% n = 8 | — |
Error values following “±” are standard deviations from the average of the individual genetic background group’s MARD values. P-values within the boxes represent the statistical significance of the comparisons of the two treatment groups indicated in the axes, calculated by Mann–Whitney U tests, as nonparametric equivalents to Student’s t-tests. Mann–Whitney U tests were conducted because only the IL-1Ra~KO group had normally distributed mean MARD values.
Inflammation and Fibrosis at the Sites of Glucose Sensor Implantation
After demonstrating the key role of IL-1/IL-1Ra in controlling sensor function in vivo, the next obvious question is, do alterations in IL-1Ra expression influence sensor function in vivo? Since we hypothesized that IL-1 drives inflammation and fibrosis, we would predict that by removing IL-1Ra control of the IL-1 activity (i.e., IL-1Ra deficiency/knockout), there would be an increase in inflammation and fibrosis at sites of sensor implantation. To investigate this possibility, we evaluated sensor tissue sites at 7–28 DPI using hematoxylin eosin histological evaluation of inflammation.
As can be seen in Figure 4, IL-1Ra deficiency (IL-1Ra~KO mice) had significant increased tissue reactions of inflammation when compared with normal C57BL/6 or IL-1Ra~OE mice. A comparison of the degree of inflammation (inflammation index) at sites of sensor implantation demonstrated statistically greater inflammatory tissue reactions when compared with IL-1Ra~KO mice over the 4-week period and for 3 weeks when compared with normal mice ( C 5 7 B L / 6 ; Figure 4 and Table 3 ). Comparison of the sensor-induced inflammatory reactions in IL-1Ra~OE versus C57BL/6 mice only demonstrated statistical significance related to the inflammation index for the 14-day histology samples (Figure 4 and Table 3). These studies clearly demonstrate that sensor-induced inflammatory reactions are more intense and prolonged in the IL-1Ra~KO mice when compared with IL-1Ra~OE or C57BL/6 mice. Further analysis of tissue reactions at sensor implantation sites using trichrome staining for evaluation of fibrosis in tissues also demonstrated that there was generally more fibrosis at sensor implantation sites in the normal C57BL/6J and IL-1Ra~KO mice when compared with the IL-1Ra~OE mice (data not shown).
Figure 4.
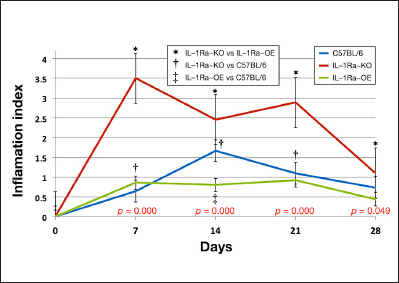
Quantitation of inflammation reactions at sensor implantation sites in normal and IL-1Ra transgenic mice. Average inflammation index values from weeks 1 to 4 by IL-1Ra genetic background. * represents the statistical significance (p < .05) of the difference between IL-1Ra~KO and IL-1Ra~OE inflammation index values for that week. † represents the statistical significance (p < .05) of the difference between IL-1Ra~KO and C57BL/6 control inflammation index values for that week. ‡ represents the statistical significance (p < .05) of the difference between IL-1Ra~OE and C57BL/6 control inflammation index values for that week. Error bars present standard errors of the inflammation index values for each individual week and genetic background. To test for statistical differences among all three genetic groups at once, Kruskal–Wallis tests were conducted on the inflammation index values as a nonparametric equivalent to analysis of variance for each week, with p values indicated at the bottom of the graph.
Table 3.
Statistical Analysis of the Inflammation Index at Glucose Sensor Implantation Sites of Normal and Interleukin-1 Receptor Antagonist Transgenic Micea
| Average inflammation index Mann-Whitney U-test comparisons | Day 7 | Day 14 | Day 21 | Day 28 |
| IL-1Ra~KO vs IL-1Ra~OE | 0.0016 | 0.0000 | 0.0000 | 0.0147 |
| IL-1Ra~KO vs C57BL/6 | 0.0000 | 0.0192 | 0.0006 | 0.3562 |
| IL-1Ra~OE vs C57BL/6 | 0.5622 | 0.0003 | 0.6452 | 0.2093 |
| K-W test for all 3 groups | 0.000 | 0.000 | 0.000 | 0.049 |
P-values within the boxes represent the statistical significance of the paired comparisons of the inflammation index values of the mice with different genetic backgrounds indicated in the horizontal axis, calculated by Mann–Whitney U tests, as nonparametric equivalents to Student’s t-tests. Mann–Whitney U tests were conducted because only the day 14 IL-1Ra~KO and IL-1Ra~OE groups had normally distributed inflammation values. For that single box, the p-value was calculated with the traditional Student’s t-test. At the bottom of the table, Kruskal– Wallis tests were used to conduct statistical comparisons among the three groups of inflammation index values as a nonparametric equivalent to analysis of variance. K–W, Kruskal-Wallis.
Discussion
Inflammation and Continuous Glucose Monitoring
The loss of sensor function in vivo is a result of tissue reactions that include biofouling of the sensor, sensor encapsulation (fibrosis), formation of metabolic barriers by inflammatory cells, and loss of vasculature (vessel regression).13,16 Unfortunately, the specific mediators and mechanisms involved in the loss of sensor function in vivo remains unclear. However, the importance of inflammation in this loss of sensor function is well established. For example, our laboratory demonstrated that anti-inflammatory drugs such as corticosteroids (dexamethasone) not only suppress inflammation at sites of sensor implantation, but also extend sensor lifespan in vivo.17 Unfortunately, steroids have many drawbacks. For example, although corticosteroids are effective anti-inflammatory agents for short-term suppression of inflammation, steroids can have significant negative side effects when used long term.18,19 Additionally, corticosteroids are broad-spectrum anti-inflammatory agents that affect a wide range of inflammatory and wound-healing pathways, thus the specific mechanism(s) and mediators that are affected by corticosteroids are not completely cataloged or understood. Interestingly, part of the mechanism by which steroids produce anti-inflammatory effects appears to be related to suppression of cytokine expression within various cells.20–22 For example, steroids are known to suppress IL-1B expression in various cell types.23–25
Interleukin-1 Cytokine Family of Agonists and Antagonists
The IL-1 family consists of two agonists (IL-1a and IL-1B), a competitive antagonist (IL-1Ra/IL-1Ra), and two receptors [interleukin-1 receptor I (IL-1RI) and interleukin-1 receptor II (IL-1RII)]. Interleukin-1 alpha is the acidic form while IL-1B is the neutral form. Interestingly, these cytokines lack classical signal peptides (for secretion), yet IL-1a and IL-1B exert their physiological effects by binding to specific receptors. While IL-1a remains intracellular and is released upon cell death, IL-1B is secreted out of the cell. Crucial to controlling an inflammatory event is the concentration of the IL-1Ra, and the ratio of IL-1Ra/IL-1 within the tissue microenvironment. The IL-1Ra competes for binding to IL-1RI and IL-1RII and thereby prevents IL-1 from activating the receptor. Isoforms of IL-1Ra have been identified and include one secreted form and three intracellular forms (1, 2, and 3).4 While secreted IL-1Ra competitively inhibits IL-1 receptor binding, intracellular IL-1Ra may inhibit not only IL-1 binding, but also regulate IL-1 responses beyond the receptor level. Interleukin-1RI and IL-1RII are both receptor members of the immunoglobulin superfamily. Where IL-1RI has a 213 amino acid cytoplasmic domain, IL-1RII contains only 29 amino acids in this region. Interleukin-1RI is the signal-transducing receptor, and IL-1RII does not transduce a signal when IL-1 is bound to it and is considered an IL-1 “sink.” Additionally, IL-1RII not only exists as a membrane-bound form, but can also be found as a soluble form in the circulation of healthy adults. Therefore, IL-1RI mediates IL-1 signal transduction, and IL-1RII is involved in downregulation or inhibition of IL-1 activation. Lastly, IL-1 activation requires that IL-1/IL-1RI complex associate with IL-1 receptor accessory protein to mediate signal transduction.11 The mechanism by which IL-1 mediates its activity is via activation of the inhibitor of |B/nuclear factor-|B (I|B/NF|B) and AP-1 transcription factor pathways.26Nuclear factor|B has been shown or implicated in the regulation of a number of protumorigenic activities, including regulation of invasiveness/metastasis factors such as metalloproteinase,27 urokinase plasminogen activator,28 endothelial cell adhesion molecules (selectins) critical for angiogenesis,29 and a number angiogenic/mitogenic cytokines such as growth-regulated oncogene protein,30 interleukin-8, vascular endothelial growth factor, basic fibroblast growth factor, and tumor necrosis factor as well as the motility factor, interleukin-6.31
Interleukin-1 Receptor Antagonist and Continuous Glucose Monitoring
Based on these results, we have also developed a hypothetical model to explain the results seen in both transgenic and normal mice (see Figure 5). In our model, the key factor is the balance between expression of proinflammatory factors such as IL-1 (derived from tissue cells and inflammatory cells) and anti-inflammatory factors such as IL-1Ra [derived from plasma (vasopermeability) as well as tissue and inflammatory cells]. In this model, initial implantation of the sensors in normal mice (C57BL/6J) triggers the release of local inflammatory mediators (e.g. IL-1B, vasopermeability factors, and leukocyte chemotactic factors) from tissue cells, including mast cells. The locally expressed leukocyte chemotactic factors, in turn, recruit both polymorphonuclear leukocytes and monocytes/macrophages, and both cell types are known to express IL-1 and IL-1Ra. Mast-cell-derived vasopermeability factor such as histamine and serotonin induce vasopermeability, resulting in an influx of plasma components, including IL-1Ra. Ultimately, the balance between cell-derived IL-1 and IL-1Ra as well as plasma-derived IL-1Ra determines whether inflammation and fibrosis or wound healing (angiogenesis) will dominate the tissue surrounding the implanted sensor. We believe that in the IL-1Ra~KO mice, the lack of plasma and cell-derived IL-1Ra allow locally expressed IL-1B to dominate the tissue microenvironment at the sensor implantation site and thereby promote inflammation, tissue destruction, and fibrosis, which is particularly noticeable in the first few weeks following sensor implantation (Figure 3 and Table 1). Alternatively, we hypothesize that in the case of IL-1Ra~OE mice, both the enhanced levels of IL-1Ra seen in the plasma, the recruited cells (polymorphonuclear leukocytes and macrophages), as well as tissue cells create an anti-inflammatory tissue microenvironment at the site of sensor implantation.
Figure 5.
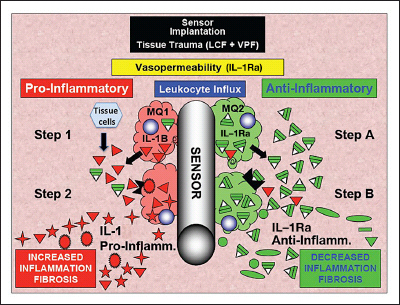
Hypothetical model of IL-1 and IL-1Ra tissue and sensor interactions at sites of glucose sensor implantation in normal tissue. Hypothetical model of role of IL-1(IL-1a and IL-1B) and IL-1Ra at sensor implantation sites of glucose sensors. This hypothetical model outlines the various possible IL-1-related pathways /interactions that are involved in controlling tissue reactions at sites of glucose sensor–tissue reactions as well as glucose sensor function in vivo. M1, (proinfammatory) macrophage subpopulations, red cells; M2, (anti-infammatory) macrophage subpopulations, green cells; red triangles, IL-1 (IL-1a and IL-1B); green triangles, IL-1Ra; red stars, IL-1-induced proinflammatory and profbrotic factors; green circles and ovals, IL-1Ra-induced/related anti-inflammatory and antifibrosis factors; VPF, vasopermeability factors; LCF leukocyte chemotactic factors.
Conclusions
To investigate the role of the IL-1 family of cytokines in glucose sensor function and tissue reactions in vivo using our murine model of CGM,14 we compared sensor function in transgenic mice that (1) overexpress IL-1Ra [B6.Cg-Tg(IL1rn)1Dih/J] and (2) are deficient in IL-1Ra (B6.129S-Il1rntm1Dih/J) with mice that have normal levels of IL-1Ra (C57BL/6). These studies indicate that (1) the IL-1 family of cytokines, especially IL-1, play a critical role in controlling tissue reactions and sensor function in vivo in the first few weeks sensor implantation and (2) the IL-1 antagonist IL-1Ra is critical in controlling tissue reactions and sensor function in vivo, particularly in the early phases of tissue injury and inflammation. These studies suggest that targeting the IL-1 family of cytokines for the first 2–3 weeks following sensor implantation, e.g., local delivery of IL-1 antagonists at sites of sensor implantation, will likely enhance both short-term and long-term sensor function in vivo.
Acknowledgments
We thank Abbott Diabetes Care, Alameda, CA, for providing us with the modified Navigator sensors.
Glossary
- (CGM)
continuous glucose monitoring
- (IL-1)
interleukin-1
- (IL-1a)
interleukin 1 alpha
- (IL-1B)
interleukin-1 beta
- (IL-1RI)
interleukin-1 receptor I
- (IL-1RII)
interleukin-1 receptor II
- (IL-1Ra)
interleukin-1 receptor antagonist
- (IL-1Ra~KO)
interleukin-1 receptor antagonist knockout
- (IL-1Ra~OE)
interleukin-1 receptor antagonist overexpresser
- (MARD)
mean absolute relative difference
Funding
These studies were supported by grants from the National Institutes of Health (NIDDK DK081171) and the American Diabetes Association.
References
- 1.Li J, Chen J, Kirsner R. Pathophysiology of acute wound healing. Clin Dermatol. 2007;25(1):9–18. doi: 10.1016/j.clindermatol.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 2.Barrientos S, Stojadinovic O, Golinko MS, Brem H, Tomic-Canic M. Growth factors and cytokines in wound healing. Wound Repair Regen. 2008;16(5):585–601. doi: 10.1111/j.1524-475X.2008.00410.x. [DOI] [PubMed] [Google Scholar]
- 3.Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S. Reduced Inflammation in the Tumor Microenvironment Delays the Accumulation of Myeloid-Derived Suppressor Cells and Limits Tumor Progression. Cancer Res. 2007;67(20):10019–10026. doi: 10.1158/0008-5472.CAN-07-2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11(8):633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arend WP, Gabay C. Physiologic role of interleukin-1 receptor antagonist. Arthritis Res. 2000;2(4):245–248. doi: 10.1186/ar94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arend WP, Guthridge CJ. Biological role of interleukin 1 receptor antagonist isoforms. Ann Rheum Dis. 2000;(59 Suppl 1):i60–i64. doi: 10.1136/ard.59.suppl_1.i60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greenfeder SA, Nunes P, Kwee L, Labow M, Chizzonite RA, Ju G. Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. J Biol Chem. 1995;270(23):13757–13765. doi: 10.1074/jbc.270.23.13757. [DOI] [PubMed] [Google Scholar]
- 8.Martin MU, Falk W. The interleukin-1 receptor complex and interleukin-1 signal transduction. Eur Cytokine Netw. 1997;8(1):5–17. [PubMed] [Google Scholar]
- 9.Muzio M, Polentarutti N, Facchetti F, Peri G, Doni A, Sironi M, Transidico P, Salmona M, Introna M, Mantovani A. Characterization of type II intracellular IL-1 receptor antagonist (IL-1ra3): a depot IL-1ra. Eur J Immunol. 1999;29(3):781–788. doi: 10.1002/(SICI)1521-4141(199903)29:03<781::AID-IMMU781>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 10.Muzio M, Polentarutti N, Sironi M, Poli G, De Gioia L, Introna M, Mantovani A, Colotta F. Cloning and characterization of a new isoform of the interleukin 1 receptor antagonist. J Exp Med. 1995;182(2):623–628. doi: 10.1084/jem.182.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wesche H, Korherr C, Kracht M, Falk W, Resch K, Martin MU. The interleukin-1 receptor accessory protein (IL-1RAcP) is essential for IL-1-induced activation of interleukin-1 receptor-associated kinase (IRAK) and stress-activated protein kinases (SAP kinases) J Biol Chem. 1997;272(12):7727–7731. doi: 10.1074/jbc.272.12.7727. [DOI] [PubMed] [Google Scholar]
- 12.Klueh U, Liu Z, Feldman B, Kreutzer D. Importance of interleukin-1 and interleukin-1 receptor antagonist in short-term glucose sensor function in vivo. J Diabetes Sci Technol. 2010;4(5):1073–1086. doi: 10.1177/193229681000400506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klueh U, Kaur M, Qiao Y, Kreutzer DL. Critical role of tissue mast cells in controlling long-term glucose sensor function in vivo. Biomaterials. 2010;31(16):4540–4551. doi: 10.1016/j.biomaterials.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Klueh U, Liu Z, Cho B, Ouyang T, Feldman B, Henning TP, Kaur M, Kreutzer D. Continuous glucose monitoring in normal mice and mice with prediabetes and diabetes. Diabetes Technol Ther. 2006;8(3):402–412. doi: 10.1089/dia.2006.8.402. [DOI] [PubMed] [Google Scholar]
- 15.Cunningham DD, Stenken JA. In vivo glucose sensing. In: Winefordner JD, editor. Chemical analysis: a series of monographs on analytical chemistry and its applications. Vol. 454. Hoboken: John Wiley and Sons; 2009. [Google Scholar]
- 16.Klueh U, Liu Z, Ouyang T, Cho B, Feldman B, Henning TP, Kreutzer D. Blood-induced interference of glucose sensor function in vitro: implications for in vivo sensor function. J Diabetes Sci Technol. 2007;1(6):842–849. doi: 10.1177/193229680700100607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Klueh U, Kaur M, Montrose DC, Kreutzer DL. Inflammation and glucose sensors: use of dexamethasone to extend glucose sensor function and life span in vivo. J Diabetes Sci Technol. 2007;1(4):496–504. doi: 10.1177/193229680700100407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Friedl KE. Corticosteroid modulation of tissue responses to implanted sensors. Diabetes Technol Ther. 2004;6(6):898–901. doi: 10.1089/dia.2004.6.898. [DOI] [PubMed] [Google Scholar]
- 19.McGregor VP, Banarer S, Cryer PE. Elevated endogenous cortisol reduces autonomic neuroendocrine and symptom responses to subsequent hypoglycemia. Am J Physiol Endocrinol Metab. 2002;282(4):E770–E777. doi: 10.1152/ajpendo.00447.2001. [DOI] [PubMed] [Google Scholar]
- 20.Herbert C, Hettiaratchi A, Webb DC, Thomas PS, Foster PS, Kumar RK. Suppression of cytokine expression by roflumilast and dexamethasone in a model of chronic asthma. Clin Exp Allergy. 2008;38(5):847–856. doi: 10.1111/j.1365-2222.2008.02950.x. [DOI] [PubMed] [Google Scholar]
- 21.Smith SJ, Piliponsky AM, Rosenhead F, Elchalal U, Nagler A, Levi-Schaffer F. Dexamethasone inhibits maturation, cytokine production and Fc epsilon RI expression of human cord blood-derived mast cells. Clin Exp Allergy. 2002;32(6):906–913. doi: 10.1046/j.1365-2745.2002.01418.x. [DOI] [PubMed] [Google Scholar]
- 22.Yi ES, Remick DG, Lim Y, Tang W, Nadzienko CE, Bedoya A, Yin S, Ulich TR. The intratracheal administration of endotoxin: X. Dexamethasone downregulates neutrophil emigration and cytokine expression in vivo. Inflammation. 1996;20(2):165–175. doi: 10.1007/BF01487403. [DOI] [PubMed] [Google Scholar]
- 23.Langereis JD, Oudijk EJ, Schweizer RC, Lammers JW, Koenderman L, Ulfman LH. Steroids induce a disequilibrium of secreted interleukin-1 receptor antagonist and interleukin-1β synthesis by human neutrophils. Eur Respir J. 2011;37(2):406–415. doi: 10.1183/09031936.00170409. [DOI] [PubMed] [Google Scholar]
- 24.Newman SP, Flower RJ, Croxtall JD. Dexamethasone suppression of IL-1β-induced cyclooxygenase 2 expression is not mediated by lipocortin-1 in A549 Cells. Biochem Biophys Res Commun. 1994;202(2):931–939. doi: 10.1006/bbrc.1994.2019. [DOI] [PubMed] [Google Scholar]
- 25.Tong Z, Dai H, Chen B, Abdoh Z, Guzman J, Costabel U. Inhibition of cytokine release from alveolar macrophages in pulmonary sarcoidosis by pentoxifylline: comparison with dexamethasone. Chest. 2003;124(4):1526–1532. doi: 10.1378/chest.124.4.1526. [DOI] [PubMed] [Google Scholar]
- 26.O’Neill LA. Molecular mechanisms underlying the actions of the pro-inflammatory cytokine interleukin 1. Royal Irish Academy Medal Lecture. Biochem Soc Trans. 1997;25(1):295–302. doi: 10.1042/bst0250295. [DOI] [PubMed] [Google Scholar]
- 27.Rutter JL, Benbow U, Coon CI, Brinckerhof CE. Cell-type specific regulation of human interstitial collagenase-1 gene expression by interleukin-1 beta (IL-1 beta) in human fibroblasts and BC-8701 breast cancer cells. J Cell Biochem. 1997;66(3):322–336. [PubMed] [Google Scholar]
- 28.Hansen SK, Nerlov C, Zabel U, Verde P, Johnsen M, Baeuerle PA, Blasi F. A novel complex between the p65 subunit of NF-kappa B and c-Rel binds to a DNA element involved in the phorbol ester induction of the human urokinase gene. EMBO J. 1992;11(1):205–213. doi: 10.1002/j.1460-2075.1992.tb05043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nguyen M, Corless CL, Kräling BM, Tran C, Atha T, Bischof J, Barsky SH. Vascular expression of E-selectin is increased in estrogen-receptor-negative breast cancer: a role for tumor-cell-secreted interleukin-1 alpha. Am J Pathol. 1997;150(4):1307–1314. [PMC free article] [PubMed] [Google Scholar]
- 30.Rangnekar VV, Waheed S, Davies TJ, Toback FG, Rangnekar VM. Antimitogenic and mitogenic actions of interleukin-1 in diverse cell types are associated with induction of gro gene expression. J Biol Chem. 1991;266(4):2415–2422. [PubMed] [Google Scholar]
- 31.Sica A, Matsushima K, Van Damme J, Wang JM, Polentarutti N, Dejana E, Colotta F, Mantovani A. IL-1 transcriptionally activates the neutrophil chemotactic factor/IL-8 gene in endothelial cells. Immunology. 1990;69(4):548–553. [PMC free article] [PubMed] [Google Scholar]