

Abstract
Lung inflammation has many etiologies, including diseases of Th2-type immunity, such as asthma and anti-parasitic responses. Inflammatory diseases of the lung involve complex interactions among structural cells (airway epithelium, smooth muscle, and fibroblasts) and immune cells (B and T cells, macrophages, dendritic cells, and innate lymphoid cells). Signal transducer and activator of transcription 6 (STAT6) has been demonstrated to regulate many pathologic features of lung inflammatory responses in animal models including airway eosinophilia, epithelial mucus production, smooth muscle changes, Th2 cell differentiation, and IgE production from B cells. Cytokines IL-4 and IL-13 that are upstream of STAT6 are found elevated in human asthma and clinical trials are underway to therapeutically target the IL-4/IL-13/STAT6 pathway. Additionally, recent work suggests that STAT6 may also regulate lung anti-viral responses and contribute to pulmonary fibrosis. This review will focus on the role of STAT6 in lung diseases and mechanisms by which STAT6 controls immune and structural lung cell function.
Keywords: IL-13, IL-4, STAT6, Th2, asthma
Introduction
The signal transducers and activators of transcription (STATs) including STAT6 are latent cytoplasmic proteins that undergo tyrosine phosphorylation by Janus kinases (JAKs) in response to cytokine exposure in the extracellular milieu. Ligation of cytokines interleukin-4 (IL-4) and interleukin-13 (IL-13) with their receptors that contain the α subunit of the IL-4 receptor (IL-4Rα) result in a common STAT6-mediated signaling pathway critical to the development of Th2 inflammation characteristic of asthma and anti-parasitic responses.1,2 Once phosphorylated, STAT6 is transported to the nucleus where it regulates gene expression in various cell types critical to the balance between host immune defense and allergic inflammatory responses.3 The principle lung cells that are profoundly altered by STAT6 signaling during inflammatory responses include T and B lymphocytes, macrophages, and structural cells including airway epithelial and smooth muscle cells.4 Interestingly, STAT6 contributes to a wide array of distinct IL-4 and IL-13-induced effector functions in different cell types resulting in Th2 helper T-cell differentiation, epithelial mucus production, smooth muscle contractility and chemokine release, alternative activation of macrophages and immunoglobulin class switching to IgE by B cells, which are also reviewed elsewhere.1,3-6
The generation of mice lacking STAT6 (STAT6−/−) has allowed for intensive investigation into the role of STAT6 in numerous lung disease models. In allergen sensitization and challenge asthma models, STAT6−/− mice lack most of the features of the disease including Th2 cell accumulation, chemokine production, airway eosinophilia, epithelial mucus metaplasia, and airway hyperresponsiveness (AHR).7-9 While STAT6 has been show to play a number of important immune regulatory roles including host resistance against helminthes,10 the main focus of this article will be to discuss the role of STAT6 in the pathogenesis of allergen-induced airway inflammation in asthma, as well as other forms of inflammatory lung disease such as pulmonary fibrosis and respiratory viral infections.
STAT6 Signaling Downstream of IL-4/IL-13
IL-4 and IL-13 expressed by Th2 cells, mast cells, basophils, and innate lymphoid cells (ILC2) are key cytokines in the pathogenesis of allergic asthma and atopic disease. Numerous investigations into the mechanisms of IL-4 and IL-13 signaling have provided insight into how these cytokines regulate immune responses. IL-4 and IL-13 each bind to two receptor complexes and have one shared receptor subunit (Fig. 1). IL-4 binds to its cognate receptor complex consisting of the IL-4 receptor α chain (IL-4Rα) and the common gamma chain (γc) to form the type I receptor.11 IL-4 first binds to IL-4Rα subunit with high affinity followed by dimerization with γc receptor subunit and subsequent JAK-STAT6 activation.12,13 Both IL-4 and IL-13 bind to the shared type II receptor complex made up of IL-4Rα and IL-13Rα1. In contrast to the type I and II receptors that transmit intracellular signals through STAT6, the IL-13Rα2 subunit was initially reported to be a decoy receptor because of its short cytoplasmic tail.14 However, some reports have demonstrated that cell membrane-bound IL-13Rα2 may have some signaling capabilities related to tissue remodeling responses thought to be STAT6-independent.15,16

Figure 1. STAT6 signaling results in distinct gene profiles in different lung cell types. IL-4 binds to the type-I IL-4R consisting of the IL-4 receptor α chain (IL-4Rα) and the common gamma chain (γc) to form the type I receptor. Additionally, both IL-4 and IL-13 bind to the type II receptor made up of IL-4Rα/IL-13Rα1. Stimulation of either receptor activates IL-4Rα and associated JAK1 to phosphorylate STAT6 monomers, which then homodimerize and translocate to the nucleus, resulting induction of gene expression. Alternatively, viral infection with dsDNA or dsRNA viruses leads to STING recruitment of TBK1 and STAT6. STAT6 is phosphorylated by TBK1 and an unidentified tyrosine kinase leading to homodimerization and translocation to the nucleus for anti-viral chemokine production (not shown). IL-4/IL-13-induced STAT6 activation leads to distinct gene expression profiles in different cell types involved in lung inflammation including epithelial cells, Th2 cells, B cells, macrophages, and smooth muscle cells.
The IL-4Rα subunit contains three tyrosine residues (Y575, Y603, and Y631) that are phosphorylated after receptor stimulation and can lead to STAT6 activation.17-20 STAT6 monomers bind via Src homology 2 (SH2) domains to the intracellular portion of the IL-4Rα subunit and in turn become phosphorylated by IL-4Rα associated JAK kinases including JAK1. The γc subunit also activates JAK3, whereas IL-13Rα1 activates two other kinases, tyrosine kinase 2 (TYK2) and JAK2.1 STAT6 phosphorylation leads to dimerization followed by translocation to the nucleus where STAT6 regulates gene expression.
Depending on the cell type, STAT6 regulates completely different gene expression profiles after IL-4/IL-13 stimulation (Fig. 1).21 In B cells, STAT6 induces Ig epsilon chain and CD23 gene expression, whereas Th2 differentiation genes gata3 and crth2 are induced in T cells.4,22-24 IL-4/IL-13 stimulation of macrophages induces STAT6-dependent “alternative activation” and transcription of arginase 1, Retnla, and Chi3L3 in mouse and indolamine 2,3-dioxygenase (IDO) in humans.25 STAT6 signaling further induces airway epithelial mucin genes including muc5ac, gob5, as well as eotaxins that regulate eosinophil chemotaxis to the lung.26-28 Smooth muscle contractility genes including Rho as well as eotaxins are also targets of STAT6.29-31 In depth cell-specific regulation by STAT6 will be discussed further below.
In addition to the role of STAT6 as a transcriptional activator promoting Th2 development, STAT6 also functions as a repressor.32 STAT6 exerts inhibition of gene expression through steric hindrance of binding by other transcription factors.33,34 This suppression has a number of diverse effects and likely plays a greater role in lineage commitment of other T helper subsets than previously thought.35 Further, suppressor of cytokine signaling-1 (SOCS-1) is a repressor of STAT6 activation through the inhibition of JAKs.36,37 SOCS-1 is induced by IL-4/IL-13 and has been shown to inhibit Th2 responses in vitro and in vivo.36,38,39 Recent evidence also suggests that SOCS-2 regulates Th2 cell differentiation and STAT6 phosphorylation.40 Additionally, one report demonstrated that the mTORC2-signaling complex (mammalian target of rapamycin complex 2) supports IL-4/STAT6 signaling and contributes to Th2 differentiation by inhibiting SOCS-5.41 Disruption of mTORC2 in T cells lead to impaired differentiation into Th2 cells including inability to upregulate GATA3 and IL-4 expression, whereas Th1 differentiation was nearly unaffected. In contrast, disruption of mTORC1 resulted in a shift toward a Th2 phenotype.41
The STAT6 pathway has also been shown to be negatively regulated by IFN-γ and Bcl-6.42,43 IFN-γ antagonizes many immune responses mediated by IL-4. For example, IFN-γ secreted by T cells of the Th1 lineage represses the development of the Th2 lineage, and inhibits IgE class-switching in B cells mediated by IL-4.44 Using a human B-cell line, stimulation with IFN-γ was shown to result in a loss of IL-4-induced STAT6 tyrosine phosphorylation, nuclear translocation and DNA binding.42 In this same study, treatment with IFN-γ specifically upregulated SOCS-1 (suppressor of cytokine signaling), an inhibitor of cytokine pathways that interacts with JAK kinases to downregulate their activity.37,45 Meanwhile, overexpression of SOCS-1 also effectively blocked STAT6 phosphorylation and transcription induced by IL-4. This suggests that the inhibitory effect of IFN-γ could be mediated by SOCS-1 that interferes with the IL-4 signaling pathway.42
Bcl-6 is a transcriptional repressor normally expressed in lymphocytes that binds to the STAT6 DNA binding site and inhibits IL-4 induced transcription.43 T cells from Bcl-6-deficient mice express predominantly Th2 cytokines and Bcl-6 knockout mice exhibit a Th2 inflammatory disease involving the heart and lungs.43 These findings suggest that Bcl-6 likely has an important role in regulating STAT6 and its absence results in an overactive Th2 response.3 Interestingly, STAT6−/− Bcl6−/− double knockout mice were later found to develop Th2 inflammatory responses, suggesting that Bcl6 suppresses STAT6-independent mechanisms of Th2 differentiation.45
STAT6 in Lung Disease Models
Asthma
Extensive investigation has demonstrated that STAT6 plays a crucial role in the pathogenesis of allergen-induced airway inflammation, mucus production, and airway hyperresponsiveness (AHR). In murine models of allergen-induced airway inflammation, repeated exposure to ovalbumin (OVA) in immunized wild-type mice results in the pathogenic features reminiscent of human asthma, including increased serum IgE, airway eosinophilia, epithelial mucus production, and AHR. In contrast to wild-type mice, STAT6 knockout mice challenged with OVA have significantly reduced lung eosinophilia, peribronchial inflammation, mucus-producing cells, and AHR.8,9
CD4+ T helper Type 2 (Th2) cells that produce cytokines IL-4, IL-5, and IL-13 are thought to play a pivotal role in the induction of allergic asthma. In addition to the role of STAT6 in Th2 differentiation, Th2 cell trafficking and chemokine production from resident parenchymal cells of the lung have been demonstrated to be STAT6-dependent.7 To show this, in vitro differentiated wild-type antigen-specific Th2 cells were adoptively transferred into STAT6−/− sensitized mice and failed to induce pulmonary eosinophilia, mucus production, or AHR.7 More recently, Chapoval et al. performed allergen challenges on wild-type and STAT6−/− mouse bone marrow chimeric mice and found an absence or paucity of eosinophilia in OVA-treated STAT6−/− mice even when reconstituted with wild-type BM or when Th2 cells were adoptively transferred.46 These reports highlight the importance of STAT6 signaling in structural, non-hematopoietic, lung cells in the development of lung inflammation. STAT6 regulation of different cell types involved in asthma pathogenesis is summarized in Figure 2.
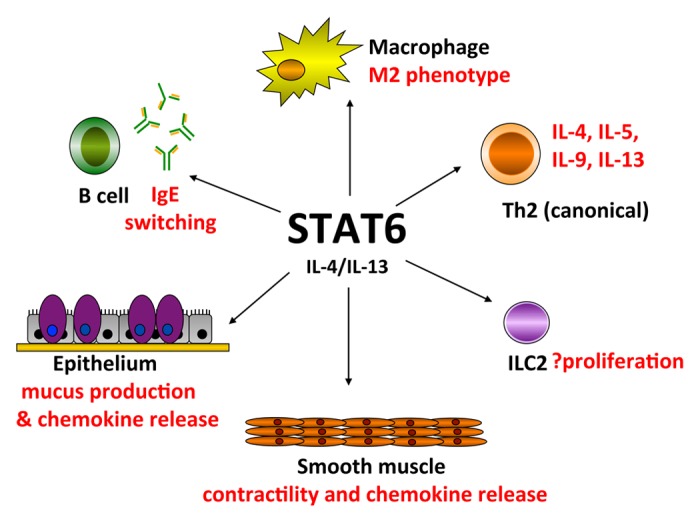
Figure 2. Effector functions mediated by STAT6 in multiple cell types in lung inflammation. IL-4/IL-13 activation of STAT6 leads to polarization of macrophages to the M2 “alternatively activated” phenotype that regulate tissue repair and anti-inflammatory responses. Naïve CD4+ T cells are differentiated into Th2 cells that produce Th2 cytokines (IL-4, IL-5, IL-9, and IL-13) by IL-4 in a STAT6-dependent manner (canonical differentiation). B cell isotype switching to IgE is mediated by STAT6. Lineage-negative type 2 innate lymphoid cells (ILC2) produce Th2 cytokines independent of STAT6 in vivo, but STAT6 may regulate ILC2 proliferation. Further, STAT6 controls IL-4/IL-13 induced epithelial mucus metaplasia that leads to airway mucus secretion found in many lung inflammatory diseases. IL-13 induces STAT6-dependent smooth muscle contractility that may contribute to airway hyperresponsiveness. Both airway epithelial cells and smooth muscle cells produce STAT6-dependent chemokines that result in lung eosinophilia.
Further studies with IL-4 and IL-13 transgenic mice have shed light on the potential roles of these cytokines in the lung. One report demonstrated that lung-specific overexpression of IL-4 led to an inflammatory response characterized by epithelial cell hypertrophy, accumulation of macrophages, lymphocytes, eosinophils, and neutrophils.47 A similar phenotype was detected in transgenic mice overexpressing pulmonary IL-13 but also included profound eosinophilia, mucus metaplasia, subepithelial fibrosis, and AHR.48 IL-4 and IL-13 signaling, as mediated by STAT6, therefore contributes to the multiple pathologic features of asthma including eosinophilic inflammation, airway hyperresponsiveness, subepithelial fibrosis, and excessive mucus production.49
Many of the early mouse models of asthma utilized OVA challenges after systemic immunization with OVA with an adjuvant and thus may be less representative of allergen mucosal sensitization in humans. Reports utilizing innate and adaptive asthma models with fungal allergens have further confirmed the important role of STAT6 in lung inflammation. Sensitization to the fungal allergen Alternaria alternata has been increasingly recognized as a risk factor for persistent or near fatal asthma in humans.50 An innate mouse model of Alternaria-induced asthma revealed that the induction of early eosinophilia and type 2 innate lymphoid cell proliferation occurred in a STAT6-dependent manner.51,52 Further, eosinophil chemokines eotaxin-1 and eotaxin-2 were significantly reduced in the airway after one challenge with Alternaria in STAT6 knockout mice suggesting that STAT6 regulates very early eosinophil influx through chemokines. Separately, another study utilizing a chronic fungal allergen-induced model with Aspergillus fumigatus demonstrated that STAT6 was required for goblet cell hyperplasia, peribronchial inflammation and AHR after conidia intratracheal challenge.53 These reports suggest, that similar to OVA models, natural aeroallergens also use STAT6 pathways to generate the asthma phenotype.
Parasites and Th2 immunity
Given the striking phenotype in Th2 asthma models in STAT6 knockout mice, it is not surprising that STAT6 contributes to anti-parasitic Th2 responses. Indeed, STAT6 has been shown to be important for immunity to helminth parasites including Nippostrongylus brasilensis, Heligmosomoides polygyrus, Trichinella spiralis, and Shistosoma mansoni.54,55 Similar to allergic asthma, STAT6 functions in Th2 differentiation and induction of IgE and chemokine production in resident tissue cells. Recently, innate lymphoid cells type 2 that rapidly produce IL-5 and IL-13 have demonstrated a critical early role in the expulsion of parasites.56,57 Interestingly, ILC2 GATA3 expression was no different in parasite-infected STAT6 knockout mice though total numbers of ILC2 were reduced, suggesting that STAT6 regulates ILC2 development, proliferation, or maintenance independent of GATA3 and cytokine production.58
Adaptive immunity and T helper type 2 (Th2) cells direct immune responses against helminthes through IL-4 and IL-13 production. Kopf et al. observed that mice defective for IL-4 had impaired Th2 responses, characterized by reduced Th2 effector cytokine production, and reduced eosinophilia upon infection with Nippostrongylus brasilensis.59 Further studies showed that mice deficient in IL-4/IL-13, IL-4Rα, or STAT6 signaling develop impaired type 2 immune response against GI parasites.60,61
Interestingly, IL-13 possesses unique functions in vivo independent of IL-4 that have been elucidated by comparing IL-4−/−, IL-13−/−, STAT6−/−, and IL-4Rα−/− mice in models of helminth infection.55 STAT6−/− and IL-4Rα−/− mice are impaired in both IL-4- and IL-13-mediated responses (likely because STAT6 + IL-4Rα share components of both the IL-4 and IL-13 receptor signaling pathways). While IL-4−/− mice display normal expulsion of N. brasiliensis, IL-4Rα and STAT6 knockout mice are unable to expel N. brasiliensis. Therefore, in response to natural helminth infection there may be a greater availability and importance of IL-13 compared with IL-4, as parasite expulsion requires STAT6 signaling in an IL-4-independent manner.
Viral immunity
Stimulator of interferon genes (STING) is an important mediator in innate viral immunity. STING-deficient mice are highly sensitive to both RNA (e.g., vesicular stomatitis virus, sendai virus) and DNA viruses (herpes simplex virus-1).62 Other names for STING include mediator of IRF3 activation (MITA), endoplasmic reticulum interferon stimulator (ERIS), and membrane tetraspanner associated with MHC class II (MPYS).63 Very recently, STAT6 was demonstrated to have an important role in innate immune signaling via STING in response to viral infections.64 Surprisingly, viral infections appear to trigger a cell intrinsic pathway leading to activation of STAT6 that is distinct from the canonical pathway induced by IL-4/IL-13. The authors found that upon recognition of viral nucleic acids, STING is activated and recruits STAT6 to the endoplasmic reticulum along with TBK1 that then phosphorylates STAT6 independent of JAKs.64 TBK1 was also found to activate another unidentified tyrosine kinase (at Tyr641) that phosphorylates STAT6. Virus-induced STAT6 activation was detected in fibroblasts, bone marrow-derived macrophages, and peritoneal macrophages. This is in contrast to the cell type-specific roles of STAT6 in IL-4/IL-13 signaling. Importantly, STAT6−/− mice were shown to be more susceptible to viral infection than wild-type mice. Interestingly, this supports the hypothesis that STAT6 induction by respiratory viruses might contribute to asthma pathogenesis given the strong association between viral illness and asthma.65
STAT6 in lung fibrosis
IL-13 contributes to fibrosis in a number of chronic infectious and autoimmune diseases, and is likely involved in airway fibrosis and smooth muscle increase in asthma as well as interstitial lung disease.6 In addition to the Th2 immune response, IL-13 transgene overexpression in the lung has been shown to induce persistent subepithelial fibrosis and smooth muscle hypertrophy.66,67 Some of the effect of IL-13 induced fibrosis may be STAT6-independent as IL-13 signaling has been shown to mediate TGF-β1 induced fibrosis through IL-13Rα2.16 IL-4 and IL-13 are capable of stimulating fibroblast differentiation, as well as α-SMA and collagen expression suggesting both cytokines have tissue remodeling capabilities.68 Fibroblasts stimulated with IL-4 or IL-13 triggers increased expression of β1-integrin and vascular cell adhesion molecule-1 (VCAM-1) as well as increased production of inflammatory cytokines IL-6 and MCP-1/CCL2.68 Importantly, mice subjected to bleomycin-induced pulmonary fibrosis displayed elevated IL-4 and IL-13 and therapeutic blockade was shown to reduce the pulmonary interstitial fibrosis phenotype.69
Found in inflammatory zone (FIZZ1), also known as resistin-like molecule α (RELMα), or hypoxia induced mitogenic factor (HIMF), is induced in alveolar type II epithelial cells (AECs) in models of bleomycin-induced lung fibrosis.70 FIZZ1 has been shown to induce myofibroblast differentiation in lung fibroblast in vitro as measures by increased expression of α-SMA and type 1 collagen.71 Further, pulmonary microvascular endothelial cells stimulated with FIZZ1 were shown to have increased cell proliferation as well as the expression of vascular endothelial growth factor (VEGF) and MCP-1 production that was dependent on IL-4/IL-4Rα signaling.72 In a mouse model of bleomycin-induced lung fibrosis, mice deficient in IL-4, IL-13, or STAT6 showed marked reduction in FIZZ1 expression in AECs as well as decreased lung fibrosis.73 Together these findings suggest a mechanism by which IL-4/IL-13/STAT6 could play a role in the pathogenesis of pulmonary fibrosis and lung remodeling processes.
STAT6 in Human Lung Disease
Asthma
Increased levels of IL-4 and IL-13 expression have been detected in sputum and bronchial biopsies of severe asthmatics.74,75 Further, mast cell-derived IL-4 and IL-13 was shown to be elevated in airway smooth muscle of individuals with asthma.76 Asthmatics also have a greater number of cells expressing IL-4Rα in their bronchial mucosa compared with controls.77 Further strengthening the role of the IL-4/IL-13 signaling pathways are studies demonstrating that expression of GATA3 and STAT6 are upregulated in bronchial biopsy tissue from patients with atopic and non-atopic asthma, as compared with healthy control subjects.75,78,79 Additionally, non-atopic asthmatics, as determined by lack of skin test positivity to common aeroallergens or elevated IgE, were shown to have fewer STAT6-expressing cells than asthmatics who demonstrated atopic features.79 Samples from allergic asthmatics have demonstrated STAT6 expression in T cells, as well as STAT6-inducible genes in alveolar macrophages and airway epithelial cells.80 Thus, the IL-4/IL-13/STAT6 pathway is strongly upregulated in asthma.
Several linkage association studies have provided evidence that variations in single nucleotide polymorphisms (SNPs) in the genes encoding IL-4, IL-13, and STAT6 may be linked to asthma and allergic disease.81,82 Two separate groups have independently reported that variations in the dinucleotide (GT) repeat sequence in exon 1 of the STAT6 gene were associated with atopic asthma and increased total serum IgE in Japanese and British populations, respectively.83,84 Orosomucoid-like 3 (ORMDL3) has been strongly liked with asthma in genetic association studies.85 A report of allergen-challenged mice first demonstrated that induction of ORMDL3 mRNA expression was dependent on STAT6.86 It has since been shown that STAT6 regulates the expression of human ORMDL3 by directly binding to the promoter region.87 In addition, IL-4/IL-13 treatment increases ORMDL3 promoter activity as well as endogenous ORMDL3 expression in humans.
Currently several promising therapeutics are under investigation that target the IL-4/IL-13/STAT6 pathway in the treatment of asthma, including anti-IL-13 monoclonal antibodies and IL-4 receptor antagonists, reviewed elsewhere.5,14 A phase 2 clinical trial in asthmatic patients showed that the use of pitrakinra, an IL-4Rα antagonist, was associated with fewer adverse asthma related events and a decreased need for rescue inhaler use as compared with placebo group.88 More recently in a double blind, placebo-controlled trial, lebrikizumab, a monoclonal antibody to IL-13, was associated with improved lung function in patients with asthma, and the greatest improvement was seen in individuals with high pretreatment levels of the IL-13-induced protein periostin.89 Other experimental approaches directed at inhibiting STAT6 include small-molecule inhibitors, antisense therapy, as well as RNA interference and dominant-negative peptides.14
Pulmonary fibrosis
Several studies have shown elevated expression of IL-4 and IL-13 in human subjects with idiopathic pulmonary fibrosis (IPF).90,91 Further, a Th2 pattern (characterized by IL-4 and IL-5) predominates in the pulmonary interstitium in patients with cryptogenic fibrosing alveolitis (CFA), a fatal inflammatory lung condition marked by excessive fibroblast activation, deposition of collagen, and scar formation.91 Increased expression of receptor subunits IL-4Rα and IL-13Rα2 have also been detected in fibroblasts from surgical lung biopsies of patients with idiopathic interstitial pneumonia.92,93 Pulmonary fibroblast lines cultured from patients with the most severe form of idiopathic interstitial pneumonia (IIP), usual interstitial pneumonia (UIP), exhibited increased gene and protein expression of IL-4Rα, IL-13Rα1, and IL-13Rα2.92 Thus, the IL-4/IL-13/STAT6 pathway may be a target for intervention in these severe lung diseases.
Cell-Specific Regulation by STAT6
Th2 cell differentiation and recruitment
Naïve T cells differentiate into different effector T-helper subsets that produce characteristic cytokines depending on the cytokine milieu and co-stimulation.94 Th2 cells are thought to be central in the pathogenesis of allergic asthma through induction of key cytokines, including IL-4, IL-5, and IL-13 that result in tissue eosinophilia, mucus metaplasia, IgE production, AHR, and remodeling.95 Transfer of effector allergen-specific Th2 cells into naïve mice followed by allergen challenges is sufficient to induce pathophysiologic features of asthma, including airway eosinophilia, mucus production, and hyperresponsiveness thus demonstrating their importance.7,96,97 STAT6 regulates effector Th2 responses in lung inflammation through multiple mechanisms including canonical Th2 cell differentiation and recruitment, also reviewed elsewhere.32,49
The IL-4 signaling cascade through STAT6 activation is considered the canonical pathway of Th2 differentiation.4,95 While Th2 cells themselves are a source of IL-4, Kopf et al. observed that CD4 cells from IL-4 knockout mice infected with Nippostrongylus brasilensis were impaired in Th2 cytokine production suggesting that IL-4 was critical for their differentiation.59 Subsequently, STAT6−/− mice were also found to have a similar defect in IL-4-mediated Th2 cell differentiation.22-24
Upon activation by IL-4, STAT6 regulates expression of the “master regulator” of Th2 differentiation, GATA3.98 After STAT6 forms dimers that translocate into the nucleus, STAT6 regulates expression of GATA3, a transcription factor that belongs to the GATA family of zinc finger proteins. GATA3 is translated from two distinct transcripts termed GATA3-1a and GATA3-1b that derive from two different promoters. Activated STAT6 induces expression of GATA3 from both promoters and controls the onset and maintenance of its expression. GATA3 in turn binds to and modifies the IL-4, IL-5, and IL-13 locus which results in enhanced expression of Th2-related cytokines.32
GATA3 was initially shown to be selectively expressed in Th2 cells and required for Th2 development.99 Importantly, IL-4-stimulated STAT6−/− T cells demonstrated significantly impaired GATA3 induction.100 In contrast, GATA3 was shown to inhibit Th1 cell development in an IL-4-independent manner providing early insight into IL-4/STAT6-independent roles of GATA3.100 Consistent with this, a follow-up study demonstrated that GATA3 expression and auto-activation can occur in STAT6−/− Th2 cells that produce IL-4.101 Finally, analysis of conditional GATA3 knockout mice confirmed the critical role of GATA3 in Th2 cell differentiation (both IL-4 dependent and IL-4 independent).102 Taken together, these reports have demonstrated that GATA3 expression is critical to Th2 cytokine production regulated by STAT6/IL-4-dependent and -independent mechanisms.
In addition to a role in Th2 differentiation, STAT6 also has a role in Th2 effector cell recruitment into areas of allergic inflammation. An early report demonstrated that transfer of wild-type OVA-specific Th2 cells into STAT6−/− mice were impaired in trafficking to the lung after OVA challenge.7 Subsequently, IL-4 was shown to induce expression of thymus and activation regulated-chemokine (TARC/CCL-17), and its murine homolog (mTARC/ABCD-2), which binds the G protein-coupled chemokine receptor CCR4 to direct Th2 cell recruitment.103 STAT6 has multiple binding sites in the TARC promoter region that are induced by IL-4 in the presence of the PI3K pathway, and these promoter elements have been shown to drive mTARC/STAT6 transgene expression in sites of Th2 inflammation in vivo.104
Though there is strong evidence for a role of STAT6 in canonical Th2 differentiation, several other pathways facilitate Th2 differentiation independent of STAT6 including STAT5 and IL-33.32 In addition to STAT6, STAT5 is also involved in Th2 polarization independent of IL-4Rα signaling.105,106 Constitutive expression of STAT5 has been reported to result in production of IL-4 from Th2 cells even in the absence of IL-4R and STAT6.106 Further, IL-2 contributes to Th2 differentiation by activating STAT5A, which facilitates transcription at the IL-4 gene locus.105 In mouse models, double knockout STAT5A−/− STAT6−/− mice displayed a significant reduction in Th2 cell development and significant decreases in lung eosinophilia after antigen challenge, as compared with STAT6-deficient mice.107 IL-33, an IL-1 family cytokine, is strongly linked to asthma development and has also been shown to induce non-canonical Th2 cells that produce IL-5 and IL-13, but not IL-4, independent of STAT6.108 These reports highlight the complexities of Th2 cell development, and overall, suggest that the role of STAT6 in Th2 cell development is condition-dependent.
Th9 cell differentiation
The ever-expanding T helper cell subsets now include Th9 cells that produce predominately IL-9, but very little IL-4, IL-5, and IL-13.109 Th9 cells express high levels the transcription factor PU.1 that is required for Th9 differentiation.110 Recently, STAT6 has been shown to be critical for the development of Th9 cells.111 Similar to Th2 cells, Th9-cell differentiation also depends on IL-4, STAT6, and GATA3, but in addition requires TGF-β1 for polarization. Though IL-9 has been shown to induce features of type 2 responses in the lung including mucus production, accumulation of mast cells, and airway remodeling, it is unclear whether the Th9 subset or other cells including Th2 cells contribute more to IL-9 induced lung inflammation.109
B-cell class switching and activation
During allergic and parasitic lung responses, B cells differentiate into plasma cells and produce large amounts of IgE. Nearly 25 years ago, IL-4 was shown to be a switch factor for IgE synthesis in B cells.112 Since then, STAT6 has been shown to be critical for IL-4 induced class switching to IgE as well as the expression of cell surface molecules including CD23 and MHCII responsible for antigen presentation by B cells.23,24 STAT6-deficient B cells fail to produce immunoglobulin (IgE) following immunization in vivo with anti-IgD.22 Additionally, levels of IgE are dramatically reduced in STAT6-deficient mice after mice sensitization with antigen or infection with N. brasiliensis.23 Current therapeutic strategies to target IgE in allergic diseases such as asthma have shown some efficacy and highlight the importance of the STAT6/IgE pathway in humans.
Macrophage alternative activation
STAT6 has been shown both in vitro and in vivo to promote IL-4 and IL-13 induced differentiation of “alternatively activated macrophages” or M2 macrophages, which are present during parasitic infections and Th2 immune responses in the lung.25,113 M2 macrophages have been described to have anti-inflammatory and homeostatic functions linked to wound healing, fibrosis, and tissue repair.25 Many of the genes associated with mouse M2 macrophages are regulated by STAT6, including resistin-like-α (Retnla, also known as FIZZ1), arginase 1 (Arg1), chitinase 3-like 3 (Chi3l3, also known as YM1), and macrophage mannose receptor 1 (Mrc1, also known as Cd206).25 Mice deficient in neutrophil/macrophage-specific IL-4Rα lack M2 polarization during mouse models of helminth infection and Th2-mediated inflammation.114 The M2-specific arginase 1 that modulates inducible nitric oxide synthase (iNOS) in asthma is regulated by both STAT6 and C/EBP-β.115 Further, specific macrophage functions in response to IL-13 are greatly impaired in STAT6-deficient mice and lack the ability to mediate IL-13-induced expression of genes such as MHC class II.116
In addition to STAT6, M2 macrophage polarization is mediated by interferon-regulatory factor 4 (IRF4) and peroxisome proliferator-activated receptor-γ (PPAR-γ).117,118 PPAR-γ regulates lipid metabolism in M2 macrophages and is constitutively expressed in adipose tissue where it has an anti-inflammatory role through repression of NFκB.119 PPAR-γ expression may also be induced by IL-4 and IL-13, which suggests that M2 polarization in a Th2-mediated response might also involve PPAR-γ.118 A study by Szanto et al. has now demonstrated that STAT6 facilitates gene transcription mediated by PPAR-γ in vitro, linking the two factors functionally.117 Despite these advances, the precise role of M2 macrophages during inflammatory and remodeling diseases of the lung is considered an ongoing investigation.
Type 2 innate lymphoid cell responses
Type 2 innate lymphoid cells (ILC2s) are non-B non-T lineage-negative lymphocytes that produce large amount of Th2 cytokines and are emerging as an important effector cell type in type-2 responses. ILC2 have been shown to contribute to airway hyperresponsiveness and type-2 lung inflammatory responses in mice infected with influenza virus and after challenge with multiple allergens.51,120-124 GATA3, which regulates Th2 transcription, is expressed in ILC2s in response to TSLP and in a STAT5-dependent fashion.125 Our group recently reported that GATA3 expression and IL-5 production was not impaired in STAT6−/− ILC2.51 However, after airway challenges with the fungal allergen Alternaria, STAT6 was shown to control ILC2 proliferation as well as airway eotaxin levels critical to eosinophil chemotaxis to the lung. The precise role of STAT6 in ILC2 has not been evaluated in vitro; therefore indirect effects from STAT6 in other cell types in vivo may be contributing to ILC2 proliferation.
Lung epithelial cell mucus and chemokine production
Widespread mucous plugging of bronchi and bronchioles is a key feature in the pathogenesis of respiratory diseases such as asthma and COPD.126,127 As a direct consequence of inflammation or respiratory viral infection, lung epithelial metaplasia into mucous (goblet) cells leads to mucus hypersecretion, which can in turn lead to fatal airway lumen obstruction.128 IL-13 has been shown to directly drive mucin gene expression in epithelial cells and IL-13 is often overexpressed in the setting of mucous cell metaplasia in asthma.129-131
Mucin 5A (MUC5AC) is a major airway mucin that is highly expressed in goblet cells and is induced by IL-13 in a STAT6-dependent manner.132 Expression of MUC5A as well as calcium-dependent chloride channel 1 (hCLCA1) or mouse homolog, Gob 5 (mCLCA3), is increased in airway mucus producing cells in patients with asthma.133 The induction of MUC5A and Gob 5 in airway epithelial cells was shown to be completely abrogated in STAT6-deficient mice.26 Mucin 4 (MUC4) has also been shown to modulate epithelial cell proliferation in asthmatic airways, and its upregulation in vivo by IL-4 is mediated by JAK3-STAT6 signaling.134
In addition to mucus production, instillation of interleukin-13 has direct effects on airway hyperresponsiveness.135 In an elegant study by Kuperman et al., STAT6-deficient mice were protected from all pulmonary effects of IL-13. However, reconstitution of STAT6 only in epithelial cells was sufficient for IL-13-induced AHR and mucus production in the absence of other lung pathology.136
STAT6-dependent eotaxins are expressed in airway epithelial cells after cytokine stimulation and contribute to eosinophilic infiltration and Th2 inflammatory responses.137 Eotaxin has been shown to be expressed in the airway epithelium of asthmatics.138 Further, TNF-α and IL-4 have been shown to act synergistically to stimulate eotaxin gene expression in airway epithelial cells through activation of NFκB and STAT6.28 Expression of eotaxin is upregulated in a STAT6-dependent manner in the airways of mice that overexpress IL-4 and as well as in wild-type mice following intranasal administration of IL-4.139 These reports have added an important link between STAT6, the airway epithelium, and eosinophilic infiltration.
A number of other proteins and chemoattractants regulated by STAT6 likely play a role in the molecular link between Th2 activation and recruitment of effector cells in lung inflammation. A study by Shum et al. showed that adipocyte fatty acid-binding protein, aP2, is expressed in human airway epithelial cells and is upregulated following stimulation with Th2 cytokines IL-4 and IL-13.140 Further, the regulation of aP2 mRNA expression was highly dependent on STAT6, and aP2-deficient mice lacked eosinophilic infiltration in a model of allergic airway inflammation.
Our group has demonstrated that STAT6 is required for epithelial expression of a resistin-like molecule, found in inflammatory zone 1 (FIZZ1/Retnla), in mice after a single challenge with the allergen Alternaria.52 Exogenous administration of FIZZ1 was shown to bind to inflammatory cells in the lungs, consisting of macrophages and dendritic cells, as well as structural fibroblasts, leading to initiation of eosinophil influx, peribronchial fibrosis, and increased epithelial thickness. These findings suggest a role for STAT6 in FIZZ1-mediated airway remodeling, collagen deposition, and fibrosis in Alternaria-associated asthma. Taken together, STAT6 regulates many epithelial functions in lung inflammatory response including mucus production, chemokine generation, and pro-remodeling factors.
Smooth muscle cell contractility and chemokine production
IL-13-mediated bronchial smooth muscle contractility is thought to be one mechanism by which airway constriction occurs in human asthma. In vitro, IL-13 has been shown to induce bronchial smooth muscle (BSM) contraction and upregulation of RhoA/Rho-kinase signaling pathway.30 RhoA, a monomeric GTP binding protein, and its downstream target Rho-kinase have been shown to promote calcium sensitization via inactivation of myosin phosphatase, resulting in smooth muscle contraction.141 Mutational studies revealed that STAT6 and NFκB are required for the upregulation of RhoA induced by IL-13 and TNFα in human BSM.29 Another report showed that inhibition of STAT6 with a small molecule prevented IL-13-induced Rho induction.142 Finally, a very interesting finding in IL-13 stimulated airway smooth cells is induction of eotaxins. Both IL-4 and IL-13 have been shown to induce eotaxin release from smooth muscle cells in vitro, dependent on IL-4Rα and STAT6.31,143
Summary
STAT6 is activated by IL-4 and IL-13 and plays a critical role in Th2 lung inflammatory responses including clearance of parasitic infections and in the pathogenesis of asthma. Studies from humans and animal models have demonstrated that STAT6 has diverse and complex functions in mediating distinct gene expression profiles in a variety of cell types involved in lung inflammation. While STAT6 is required for normal immune function, it has been implicated as a crucial factor in the development of pathologic features of asthma including airway eosinophilia, mucus production, Th2 cell accumulation, smooth muscle contraction, airway remodeling, and hyperresponsiveness. More recently, STAT6 has been shown to regulate lung anti-viral responses and likely has a role in the development of pulmonary fibrosis. Currently, experimental therapeutics that target the IL-4/IL-13/STAT6 pathway are being tested in clinical trials for the treatment of asthma. Ongoing investigations into the role of pathogenic STAT6-mediated responses in the lung may lead to future treatments for a variety of human lung diseases.
Acknowledgments
Support was provided by NIH grant 1K08AI080938-01A1 and ALA/AAAI Allergic Respiratory Diseases Award to TA Doherty.
Glossary
Abbreviations:
- AECs
alveolar type II epithelial cells
- AHR
airway hyperresponsiveness
- aP2
adipocyte fatty acid-binding protein
- Arg1
arginase 1
- Bcl-6
B-cell lymphoma 6
- BSM
bronchial smooth muscle
- γc
common gamma chain
- CCL17
chemokine ligand 17
- CCR
chemokine receptor
- CD4
cluster of differentiation 4
- Cd206
cluster of differentiation 206
- C/EBP-β
CCAAT-enhancer-binding proteins, beta
- CFA
cryptogenic fibrosing alveolitis
- chi3l3
chitinase 3-like 3
- cMAF
c-musculoaponeurotic fibrosarcoma
- CRTH2
chemoattractant receptor-homologous molecule expressed on Th2 cells
- DNA
deoxyribonucleic acid
- FIZZ1
found in inflammatory zone 1
- GATA3
transacting T cell specific transcription factor 3
- hCLCA1
human calcium dependent chloride channel 1
- HIMF
hypoxia induced mitogenic factor
- IL-4
interleukin-4
- IL-5
interleukin-5
- IL-9
interleukin-9
- IL-13
interleukin-13
- ILC2s
innate lymphoid cells type 2
- IL-4Rα
interleukin-4 receptor alpha
- IL-13Rα1
interleukin-13 receptor alpha 1
- IL-13Rα2
interleukin-13 receptor alpha 2
- IIP
idiopathic interstitial pneumonia
- IgE
immunoglobulin E
- IgD
immunoglobulin D
- IFN-γ
interferon gamma
- iNOS
inducible nitric oxide synthase
- IPF
interstitial pulmonary fibrosis
- IRF
interferon-regulatory factor
- JAK
Janus kinase
- MCP-1
monocyte chemotactic protein 1
- MITA
mediator of IRF3 activation
- MPYS
membrane tetraspanner associated with MHC class II
- mTARC
murine thymus and activation-regulated chemokine
- Mrc1
macrophage mannose receptor 1
- MUC
mucin
- mCLCA3
murine calcium dependent chloride channel 1
- NFκB
nuclear factor kappa light chain enhancer of activated B cells
- ORMDL-3
orosomucoid-like 3
- OVA
ovalbumin
- PI3K
phosphoinositide 3-kinase
- PPAR-γ
peroxisome proliferator-activated receptor-γ
- RhoA
RAS homolog gene family member A
- RELMα
resistin-like molecule α
- Retnla
resistin-like-α
- RNA
ribonucleic acid
- SH2
Src homology 2
- α-SMA
alpha-smooth muscle actin
- SNPs
single nucleotide polymorphisms
- SOCS
suppressor of cytokine signaling
- STAT
signal transducer and activator of transcription
- STING
stimulator of interferon genes
- TARC
thymus and activation regulated chemokine
- TGF-β1
transforming growth factor beta 1
- Th2
T helper type 2
- Th9
T helper type 9
- TNF-α
tumor necrosis factor alpha
- UIP
usual interstitial pneumonia
- VCAM-1
vascular cell adhesion molecule-1
- VEGF
vascular endothelial growth factor
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/jak-stat/article/25301
References
- 1.Hershey GK. IL-13 receptors and signaling pathways: an evolving web. J Allergy Clin Immunol. 2003;111:677–90, quiz 691. doi: 10.1067/mai.2003.1333. [DOI] [PubMed] [Google Scholar]
- 2.Zimmermann N, Hershey GK, Foster PS, Rothenberg ME. Chemokines in asthma: cooperative interaction between chemokines and IL-13. J Allergy Clin Immunol. 2003;111:227–42, quiz 243. doi: 10.1067/mai.2003.139. [DOI] [PubMed] [Google Scholar]
- 3.Wurster AL, Tanaka T, Grusby MJ. The biology of Stat4 and Stat6. Oncogene. 2000;19:2577–84. doi: 10.1038/sj.onc.1203485. [DOI] [PubMed] [Google Scholar]
- 4.Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunol Res. 2011;50:87–96. doi: 10.1007/s12026-011-8205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ingram JL, Kraft M. IL-13 in asthma and allergic disease: asthma phenotypes and targeted therapies. J Allergy Clin Immunol. 2012;130:829–42, quiz 843-4. doi: 10.1016/j.jaci.2012.06.034. [DOI] [PubMed] [Google Scholar]
- 6.Doherty T, Broide D. Cytokines and growth factors in airway remodeling in asthma. Curr Opin Immunol. 2007;19:676–80. doi: 10.1016/j.coi.2007.07.017. [DOI] [PubMed] [Google Scholar]
- 7.Mathew A, MacLean JA, DeHaan E, Tager AM, Green FH, Luster AD. Signal transducer and activator of transcription 6 controls chemokine production and T helper cell type 2 cell trafficking in allergic pulmonary inflammation. J Exp Med. 2001;193:1087–96. doi: 10.1084/jem.193.9.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuperman D, Schofield B, Wills-Karp M, Grusby MJ. Signal transducer and activator of transcription factor 6 (Stat6)-deficient mice are protected from antigen-induced airway hyperresponsiveness and mucus production. J Exp Med. 1998;187:939–48. doi: 10.1084/jem.187.6.939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Akimoto T, Numata F, Tamura M, Takata Y, Higashida N, Takashi T, et al. Abrogation of bronchial eosinophilic inflammation and airway hyperreactivity in signal transducers and activators of transcription (STAT)6-deficient mice. J Exp Med. 1998;187:1537–42. doi: 10.1084/jem.187.9.1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Finkelman FD, Shea-Donohue T, Morris SC, Gildea L, Strait R, Madden KB, et al. Interleukin-4- and interleukin-13-mediated host protection against intestinal nematode parasites. Immunol Rev. 2004;201:139–55. doi: 10.1111/j.0105-2896.2004.00192.x. [DOI] [PubMed] [Google Scholar]
- 11.Nelms K, Keegan AD, Zamorano J, Ryan JJ, Paul WE. The IL-4 receptor: signaling mechanisms and biologic functions. Annu Rev Immunol. 1999;17:701–38. doi: 10.1146/annurev.immunol.17.1.701. [DOI] [PubMed] [Google Scholar]
- 12.Mueller TD, Zhang JL, Sebald W, Duschl A. Structure, binding, and antagonists in the IL-4/IL-13 receptor system. Biochim Biophys Acta. 2002;1592:237–50. doi: 10.1016/S0167-4889(02)00318-X. [DOI] [PubMed] [Google Scholar]
- 13.Galizzi JP, Zuber CE, Harada N, Gorman DM, Djossou O, Kastelein R, et al. Molecular cloning of a cDNA encoding the human interleukin 4 receptor. Int Immunol. 1990;2:669–75. doi: 10.1093/intimm/2.7.669. [DOI] [PubMed] [Google Scholar]
- 14.Oh CK, Geba GP, Molfino N. Investigational therapeutics targeting the IL-4/IL-13/STAT-6 pathway for the treatment of asthma. Eur Respir Rev. 2010;19:46–54. doi: 10.1183/09059180.00007609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cho WK, Lee CM, Kang MJ, Huang Y, Giordano FJ, Lee PJ, et al. IL-13 receptor α2-arginase 2 pathway mediates IL-13-induced pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol. 2013;304:L112–24. doi: 10.1152/ajplung.00101.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fichtner-Feigl S, Strober W, Kawakami K, Puri RK, Kitani A. IL-13 signaling through the IL-13alpha2 receptor is involved in induction of TGF-beta1 production and fibrosis. Nat Med. 2006;12:99–106. doi: 10.1038/nm1332. [DOI] [PubMed] [Google Scholar]
- 17.Wang HY, Paul WE, Keegan AD. IL-4 function can be transferred to the IL-2 receptor by tyrosine containing sequences found in the IL-4 receptor alpha chain. Immunity. 1996;4:113–21. doi: 10.1016/S1074-7613(00)80676-7. [DOI] [PubMed] [Google Scholar]
- 18.Pernis A, Witthuhn B, Keegan AD, Nelms K, Garfein E, Ihle JN, et al. Interleukin 4 signals through two related pathways. Proc Natl Acad Sci U S A. 1995;92:7971–5. doi: 10.1073/pnas.92.17.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryan JJ, McReynolds LJ, Keegan A, Wang LH, Garfein E, Rothman P, et al. Growth and gene expression are predominantly controlled by distinct regions of the human IL-4 receptor. Immunity. 1996;4:123–32. doi: 10.1016/S1074-7613(00)80677-9. [DOI] [PubMed] [Google Scholar]
- 20.Hebenstreit D, Wirnsberger G, Horejs-Hoeck J, Duschl A. Signaling mechanisms, interaction partners, and target genes of STAT6. Cytokine Growth Factor Rev. 2006;17:173–88. doi: 10.1016/j.cytogfr.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Lee JH, Kaminski N, Dolganov G, Grunig G, Koth L, Solomon C, et al. Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am J Respir Cell Mol Biol. 2001;25:474–85. doi: 10.1165/ajrcmb.25.4.4522. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan MH, Schindler U, Smiley ST, Grusby MJ. Stat6 is required for mediating responses to IL-4 and for development of Th2 cells. Immunity. 1996;4:313–9. doi: 10.1016/S1074-7613(00)80439-2. [DOI] [PubMed] [Google Scholar]
- 23.Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, et al. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–30. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- 24.Shimoda K, van Deursen J, Sangster MY, Sarawar SR, Carson RT, Tripp RA, et al. Lack of IL-4-induced Th2 response and IgE class switching in mice with disrupted Stat6 gene. Nature. 1996;380:630–3. doi: 10.1038/380630a0. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence T, Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat Rev Immunol. 2011;11:750–61. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- 26.Thai P, Chen Y, Dolganov G, Wu R. Differential regulation of MUC5AC/Muc5ac and hCLCA-1/mGob-5 expression in airway epithelium. Am J Respir Cell Mol Biol. 2005;33:523–30. doi: 10.1165/rcmb.2004-0220RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dabbagh K, Takeyama K, Lee HM, Ueki IF, Lausier JA, Nadel JA. IL-4 induces mucin gene expression and goblet cell metaplasia in vitro and in vivo. J Immunol. 1999;162:6233–7. [PubMed] [Google Scholar]
- 28.Matsukura S, Stellato C, Plitt JR, Bickel C, Miura K, Georas SN, et al. Activation of eotaxin gene transcription by NF-kappa B and STAT6 in human airway epithelial cells. J Immunol. 1999;163:6876–83. [PubMed] [Google Scholar]
- 29.Goto K, Chiba Y, Matsusue K, Hattori Y, Maitani Y, Sakai H, et al. The proximal STAT6 and NF-kappaB sites are responsible for IL-13- and TNF-alpha-induced RhoA transcriptions in human bronchial smooth muscle cells. Pharmacol Res. 2010;61:466–72. doi: 10.1016/j.phrs.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiba Y, Nakazawa S, Todoroki M, Shinozaki K, Sakai H, Misawa M. Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am J Respir Cell Mol Biol. 2009;40:159–67. doi: 10.1165/rcmb.2008-0162OC. [DOI] [PubMed] [Google Scholar]
- 31.Hirst SJ, Hallsworth MP, Peng Q, Lee TH. Selective induction of eotaxin release by interleukin-13 or interleukin-4 in human airway smooth muscle cells is synergistic with interleukin-1beta and is mediated by the interleukin-4 receptor alpha-chain. Am J Respir Crit Care Med. 2002;165:1161–71. doi: 10.1164/ajrccm.165.8.2107158. [DOI] [PubMed] [Google Scholar]
- 32.Maier E, Duschl A, Horejs-Hoeck J. STAT6-dependent and -independent mechanisms in Th2 polarization. Eur J Immunol. 2012;42:2827–33. doi: 10.1002/eji.201242433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nguyen VT, Benveniste EN. IL-4-activated STAT-6 inhibits IFN-gamma-induced CD40 gene expression in macrophages/microglia. J Immunol. 2000;165:6235–43. doi: 10.4049/jimmunol.165.11.6235. [DOI] [PubMed] [Google Scholar]
- 34.Bennett BL, Cruz R, Lacson RG, Manning AM. Interleukin-4 suppression of tumor necrosis factor alpha-stimulated E-selectin gene transcription is mediated by STAT6 antagonism of NF-kappaB. J Biol Chem. 1997;272:10212–9. doi: 10.1074/jbc.272.15.10212. [DOI] [PubMed] [Google Scholar]
- 35.Elo LL, Järvenpää H, Tuomela S, Raghav S, Ahlfors H, Laurila K, et al. Genome-wide profiling of interleukin-4 and STAT6 transcription factor regulation of human Th2 cell programming. Immunity. 2010;32:852–62. doi: 10.1016/j.immuni.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 36.Losman JA, Chen XP, Hilton D, Rothman P. Cutting edge: SOCS-1 is a potent inhibitor of IL-4 signal transduction. J Immunol. 1999;162:3770–4. [PMC free article] [PubMed] [Google Scholar]
- 37.Hebenstreit D, Luft P, Schmiedlechner A, Duschl A, Horejs-Hoeck J. SOCS-1 and SOCS-3 inhibit IL-4 and IL-13 induced activation of Eotaxin-3/CCL26 gene expression in HEK293 cells. Mol Immunol. 2005;42:295–303. doi: 10.1016/j.molimm.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 38.Fujimoto M, Tsutsui H, Yumikura-Futatsugi S, Ueda H, Xingshou O, Abe T, et al. A regulatory role for suppressor of cytokine signaling-1 in T(h) polarization in vivo. Int Immunol. 2002;14:1343–50. doi: 10.1093/intimm/dxf094. [DOI] [PubMed] [Google Scholar]
- 39.Hebenstreit D, Luft P, Schmiedlechner A, Regl G, Frischauf AM, Aberger F, et al. IL-4 and IL-13 induce SOCS-1 gene expression in A549 cells by three functional STAT6-binding motifs located upstream of the transcription initiation site. J Immunol. 2003;171:5901–7. doi: 10.4049/jimmunol.171.11.5901. [DOI] [PubMed] [Google Scholar]
- 40.Knosp CA, Carroll HP, Elliott J, Saunders SP, Nel HJ, Amu S, et al. SOCS2 regulates T helper type 2 differentiation and the generation of type 2 allergic responses. J Exp Med. 2011;208:1523–31. doi: 10.1084/jem.20101167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, et al. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol. 2011;12:295–303. doi: 10.1038/ni.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venkataraman C, Leung S, Salvekar A, Mano H, Schindler U. Repression of IL-4-induced gene expression by IFN-gamma requires Stat1 activation. J Immunol. 1999;162:4053–61. [PubMed] [Google Scholar]
- 43.Dent AL, Shaffer AL, Yu X, Allman D, Staudt LM. Control of inflammation, cytokine expression, and germinal center formation by BCL-6. Science. 1997;276:589–92. doi: 10.1126/science.276.5312.589. [DOI] [PubMed] [Google Scholar]
- 44.Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136:2348–57. [PubMed] [Google Scholar]
- 45.Dent AL, Hu-Li J, Paul WE, Staudt LM. T helper type 2 inflammatory disease in the absence of interleukin 4 and transcription factor STAT6. Proc Natl Acad Sci U S A. 1998;95:13823–8. doi: 10.1073/pnas.95.23.13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chapoval SP, Dasgupta P, Smith EP, DeTolla LJ, Lipsky MM, Kelly-Welch AE, et al. STAT6 expression in multiple cell types mediates the cooperative development of allergic airway disease. J Immunol. 2011;186:2571–83. doi: 10.4049/jimmunol.1002567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rankin JA, Picarella DE, Geba GP, Temann UA, Prasad B, DiCosmo B, et al. Phenotypic and physiologic characterization of transgenic mice expressing interleukin 4 in the lung: lymphocytic and eosinophilic inflammation without airway hyperreactivity. Proc Natl Acad Sci U S A. 1996;93:7821–5. doi: 10.1073/pnas.93.15.7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–88. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kuperman DA, Schleimer RP. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr Mol Med. 2008;8:384–92. doi: 10.2174/156652408785161032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bush RK, Prochnau JJ. Alternaria-induced asthma. J Allergy Clin Immunol. 2004;113:227–34. doi: 10.1016/j.jaci.2003.11.023. [DOI] [PubMed] [Google Scholar]
- 51.Doherty TA, Khorram N, Chang JE, Kim HK, Rosenthal P, Croft M, et al. STAT6 regulates natural helper cell proliferation during lung inflammation initiated by Alternaria. Am J Physiol Lung Cell Mol Physiol. 2012;303:L577–88. doi: 10.1152/ajplung.00174.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Doherty TA, Khorram N, Sugimoto K, Sheppard D, Rosenthal P, Cho JY, et al. Alternaria induces STAT6-dependent acute airway eosinophilia and epithelial FIZZ1 expression that promotes airway fibrosis and epithelial thickness. J Immunol. 2012;188:2622–9. doi: 10.4049/jimmunol.1101632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Blease K, Schuh JM, Jakubzick C, Lukacs NW, Kunkel SL, Joshi BH, et al. Stat6-deficient mice develop airway hyperresponsiveness and peribronchial fibrosis during chronic fungal asthma. Am J Pathol. 2002;160:481–90. doi: 10.1016/S0002-9440(10)64867-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goenka S, Kaplan MH. Transcriptional regulation by STAT6. Immunol Res. 2011;50:87–96. doi: 10.1007/s12026-011-8205-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Finkelman FD, Wynn TA, Donaldson DD, Urban JF. The role of IL-13 in helminth-induced inflammation and protective immunity against nematode infections. Curr Opin Immunol. 1999;11:420–6. doi: 10.1016/S0952-7915(99)80070-3. [DOI] [PubMed] [Google Scholar]
- 56.Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erle DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A. 2010;107:11489–94. doi: 10.1073/pnas.1003988107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature. 2010;464:1367–70. doi: 10.1038/nature08900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liang HE, Reinhardt RL, Bando JK, Sullivan BM, Ho IC, Locksley RM. Divergent expression patterns of IL-4 and IL-13 define unique functions in allergic immunity. Nat Immunol. 2012;13:58–66. doi: 10.1038/ni.2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kopf M, Le Gros G, Bachmann M, Lamers MC, Bluethmann H, Köhler G. Disruption of the murine IL-4 gene blocks Th2 cytokine responses. Nature. 1993;362:245–8. doi: 10.1038/362245a0. [DOI] [PubMed] [Google Scholar]
- 60.McKenzie GJ, Fallon PG, Emson CL, Grencis RK, McKenzie AN. Simultaneous disruption of interleukin (IL)-4 and IL-13 defines individual roles in T helper cell type 2-mediated responses. J Exp Med. 1999;189:1565–72. doi: 10.1084/jem.189.10.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Urban JF, Jr., Noben-Trauth N, Donaldson DD, Madden KB, Morris SC, Collins M, et al. IL-13, IL-4Ralpha, and Stat6 are required for the expulsion of the gastrointestinal nematode parasite Nippostrongylus brasiliensis. Immunity. 1998;8:255–64. doi: 10.1016/S1074-7613(00)80477-X. [DOI] [PubMed] [Google Scholar]
- 62.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–92. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen H, Jiang Z. The essential adaptors of innate immune signaling. Protein Cell. 2013;4:27–39. doi: 10.1007/s13238-012-2063-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen H, Sun H, You F, Sun W, Zhou X, Chen L, et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell. 2011;147:436–46. doi: 10.1016/j.cell.2011.09.022. [DOI] [PubMed] [Google Scholar]
- 65.Holtzman MJ. Asthma as a chronic disease of the innate and adaptive immune systems responding to viruses and allergens. J Clin Invest. 2012;122:2741–8. doi: 10.1172/JCI60325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Fulkerson PC, Fischetti CA, Rothenberg ME. Eosinophils and CCR3 regulate interleukin-13 transgene-induced pulmonary remodeling. Am J Pathol. 2006;169:2117–26. doi: 10.2353/ajpath.2006.060617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fulkerson PC, Fischetti CA, Hassman LM, Nikolaidis NM, Rothenberg ME. Persistent effects induced by IL-13 in the lung. Am J Respir Cell Mol Biol. 2006;35:337–46. doi: 10.1165/rcmb.2005-0474OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Doucet C, Brouty-Boyé D, Pottin-Clémenceau C, Canonica GW, Jasmin C, Azzarone B. Interleukin (IL) 4 and IL-13 act on human lung fibroblasts. Implication in asthma. J Clin Invest. 1998;101:2129–39. doi: 10.1172/JCI741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jakubzick C, Choi ES, Joshi BH, Keane MP, Kunkel SL, Puri RK, et al. Therapeutic attenuation of pulmonary fibrosis via targeting of IL-4- and IL-13-responsive cells. J Immunol. 2003;171:2684–93. doi: 10.4049/jimmunol.171.5.2684. [DOI] [PubMed] [Google Scholar]
- 70.Katsuma S, Nishi K, Tanigawara K, Ikawa H, Shiojima S, Takagaki K, et al. Molecular monitoring of bleomycin-induced pulmonary fibrosis by cDNA microarray-based gene expression profiling. Biochem Biophys Res Commun. 2001;288:747–51. doi: 10.1006/bbrc.2001.5853. [DOI] [PubMed] [Google Scholar]
- 71.Liu T, Dhanasekaran SM, Jin H, Hu B, Tomlins SA, Chinnaiyan AM, et al. FIZZ1 stimulation of myofibroblast differentiation. Am J Pathol. 2004;164:1315–26. doi: 10.1016/S0002-9440(10)63218-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yamaji-Kegan K, Su Q, Angelini DJ, Myers AC, Cheadle C, Johns RA. Hypoxia-induced mitogenic factor (HIMF/FIZZ1/RELMalpha) increases lung inflammation and activates pulmonary microvascular endothelial cells via an IL-4-dependent mechanism. J Immunol. 2010;185:5539–48. doi: 10.4049/jimmunol.0904021. [DOI] [PubMed] [Google Scholar]
- 73.Liu T, Jin H, Ullenbruch M, Hu B, Hashimoto N, Moore B, et al. Regulation of found in inflammatory zone 1 expression in bleomycin-induced lung fibrosis: role of IL-4/IL-13 and mediation via STAT-6. J Immunol. 2004;173:3425–31. doi: 10.4049/jimmunol.173.5.3425. [DOI] [PubMed] [Google Scholar]
- 74.Saha SK, Berry MA, Parker D, Siddiqui S, Morgan A, May R, et al. Increased sputum and bronchial biopsy IL-13 expression in severe asthma. J Allergy Clin Immunol. 2008;121:685–91. doi: 10.1016/j.jaci.2008.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Liu CT, Wang G, Luo FM, Wang ZL, Liu R, Wang CL. [Imbalanced T cell-specific transcription factors T-bet and GATA-3 contributes to type 2 T helper cell polarization in asthmatic patients] Zhonghua Jie He He Hu Xi Za Zhi. 2004;27:398–402. [PubMed] [Google Scholar]
- 76.Brightling CE, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID, Bradding P. Interleukin-4 and -13 expression is co-localized to mast cells within the airway smooth muscle in asthma. Clin Exp Allergy. 2003;33:1711–6. doi: 10.1111/j.1365-2222.2003.01827.x. [DOI] [PubMed] [Google Scholar]
- 77.Kotsimbos TC, Ghaffar O, Minshall EM, Humbert M, Durham SR, Pfister R, et al. Expression of the IL-4 receptor alpha-subunit is increased in bronchial biopsy specimens from atopic and nonatopic asthmatic subjects. J Allergy Clin Immunol. 1998;102:859–66. doi: 10.1016/S0091-6749(98)70029-6. [DOI] [PubMed] [Google Scholar]
- 78.Nakamura Y, Ghaffar O, Olivenstein R, Taha RA, Soussi-Gounni A, Zhang DH, et al. Gene expression of the GATA-3 transcription factor is increased in atopic asthma. J Allergy Clin Immunol. 1999;103:215–22. doi: 10.1016/S0091-6749(99)70493-8. [DOI] [PubMed] [Google Scholar]
- 79.Christodoulopoulos P, Cameron L, Nakamura Y, Lemière C, Muro S, Dugas M, et al. TH2 cytokine-associated transcription factors in atopic and nonatopic asthma: evidence for differential signal transducer and activator of transcription 6 expression. J Allergy Clin Immunol. 2001;107:586–91. doi: 10.1067/mai.2001.114883. [DOI] [PubMed] [Google Scholar]
- 80.Tomita K, Caramori G, Ito K, Sano H, Lim S, Oates T, et al. STAT6 expression in T cells, alveolar macrophages and bronchial biopsies of normal and asthmatic subjects. J Inflamm (Lond) 2012;9:5. doi: 10.1186/1476-9255-9-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tamura K, Suzuki M, Arakawa H, Tokuyama K, Morikawa A. Linkage and association studies of STAT6 gene polymorphisms and allergic diseases. Int Arch Allergy Immunol. 2003;131:33–8. doi: 10.1159/000070432. [DOI] [PubMed] [Google Scholar]
- 82.Shirakawa I, Deichmann KA, Izuhara I, Mao I, Adra CN, Hopkin JM. Atopy and asthma: genetic variants of IL-4 and IL-13 signalling. Immunol Today. 2000;21:60–4. doi: 10.1016/S0167-5699(99)01492-9. [DOI] [PubMed] [Google Scholar]
- 83.Tamura K, Arakawa H, Suzuki M, Kobayashi Y, Mochizuki H, Kato M, et al. Novel dinucleotide repeat polymorphism in the first exon of the STAT-6 gene is associated with allergic diseases. Clin Exp Allergy. 2001;31:1509–14. doi: 10.1046/j.1365-2222.2001.01191.x. [DOI] [PubMed] [Google Scholar]
- 84.Gao PS, Heller NM, Walker W, Chen CH, Moller M, Plunkett B, et al. Variation in dinucleotide (GT) repeat sequence in the first exon of the STAT6 gene is associated with atopic asthma and differentially regulates the promoter activity in vitro. J Med Genet. 2004;41:535–9. doi: 10.1136/jmg.2003.015842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242:10–30. doi: 10.1111/j.1600-065X.2011.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Miller M, Tam AB, Cho JY, Doherty TA, Pham A, Khorram N, et al. ORMDL3 is an inducible lung epithelial gene regulating metalloproteases, chemokines, OAS, and ATF6. Proc Natl Acad Sci U S A. 2012;109:16648–53. doi: 10.1073/pnas.1204151109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qiu R, Yang Y, Zhao H, Li J, Xin Q, Shan S, et al. Signal transducer and activator of transcription 6 directly regulates human ORMDL3 expression. FEBS J. 2013;280:2014–26. doi: 10.1111/febs.12225. [DOI] [PubMed] [Google Scholar]
- 88.Wenzel S, Wilbraham D, Fuller R, Getz EB, Longphre M. Effect of an interleukin-4 variant on late phase asthmatic response to allergen challenge in asthmatic patients: results of two phase 2a studies. Lancet. 2007;370:1422–31. doi: 10.1016/S0140-6736(07)61600-6. [DOI] [PubMed] [Google Scholar]
- 89.Corren J, Lemanske RF, Hanania NA, Korenblat PE, Parsey MV, Arron JR, et al. Lebrikizumab treatment in adults with asthma. N Engl J Med. 2011;365:1088–98. doi: 10.1056/NEJMoa1106469. [DOI] [PubMed] [Google Scholar]
- 90.Hancock A, Armstrong L, Gama R, Millar A. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am J Respir Cell Mol Biol. 1998;18:60–5. doi: 10.1165/ajrcmb.18.1.2627. [DOI] [PubMed] [Google Scholar]
- 91.Wallace WA, Ramage EA, Lamb D, Howie SE. A type 2 (Th2-like) pattern of immune response predominates in the pulmonary interstitium of patients with cryptogenic fibrosing alveolitis (CFA) Clin Exp Immunol. 1995;101:436–41. doi: 10.1111/j.1365-2249.1995.tb03131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jakubzick C, Choi ES, Carpenter KJ, Kunkel SL, Evanoff H, Martinez FJ, et al. Human pulmonary fibroblasts exhibit altered interleukin-4 and interleukin-13 receptor subunit expression in idiopathic interstitial pneumonia. Am J Pathol. 2004;164:1989–2001. doi: 10.1016/S0002-9440(10)63759-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jakubzick C, Choi ES, Kunkel SL, Evanoff H, Martinez FJ, Puri RK, et al. Augmented pulmonary IL-4 and IL-13 receptor subunit expression in idiopathic interstitial pneumonia. J Clin Pathol. 2004;57:477–86. doi: 10.1136/jcp.2003.012799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–89. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–35. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mishima H, Hojo M, Watanabe A, Hamid QA, Martin JG. CD4+ T cells can induce airway hyperresponsiveness to allergen challenge in the brown norway rat. Am J Respir Crit Care Med. 1998;158:1863–70. doi: 10.1164/ajrccm.158.6.9709123. [DOI] [PubMed] [Google Scholar]
- 97.Watanabe A, Mishima H, Renzi PM, Xu LJ, Hamid Q, Martin JG. Transfer of allergic airway responses with antigen-primed CD4+ but not CD8+ T cells in brown Norway rats. J Clin Invest. 1995;96:1303–10. doi: 10.1172/JCI118165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhu J, Yamane H, Cote-Sierra J, Guo L, Paul WE. GATA-3 promotes Th2 responses through three different mechanisms: induction of Th2 cytokine production, selective growth of Th2 cells and inhibition of Th1 cell-specific factors. Cell Res. 2006;16:3–10. doi: 10.1038/sj.cr.7310002. [DOI] [PubMed] [Google Scholar]
- 99.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–96. doi: 10.1016/S0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 100.Ouyang W, Ranganath SH, Weindel K, Bhattacharya D, Murphy TL, Sha WC, et al. Inhibition of Th1 development mediated by GATA-3 through an IL-4-independent mechanism. Immunity. 1998;9:745–55. doi: 10.1016/S1074-7613(00)80671-8. [DOI] [PubMed] [Google Scholar]
- 101.Ouyang W, Löhning M, Gao Z, Assenmacher M, Ranganath S, Radbruch A, et al. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 2000;12:27–37. doi: 10.1016/S1074-7613(00)80156-9. [DOI] [PubMed] [Google Scholar]
- 102.Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, et al. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol. 2004;5:1157–65. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 103.Wirnsberger G, Hebenstreit D, Posselt G, Horejs-Hoeck J, Duschl A. IL-4 induces expression of TARC/CCL17 via two STAT6 binding sites. Eur J Immunol. 2006;36:1882–91. doi: 10.1002/eji.200635972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liddiard K, Welch JS, Lozach J, Heinz S, Glass CK, Greaves DR. Interleukin-4 induction of the CC chemokine TARC (CCL17) in murine macrophages is mediated by multiple STAT6 sites in the TARC gene promoter. BMC Mol Biol. 2006;7:45. doi: 10.1186/1471-2199-7-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cote-Sierra J, Foucras G, Guo L, Chiodetti L, Young HA, Hu-Li J, et al. Interleukin 2 plays a central role in Th2 differentiation. Proc Natl Acad Sci U S A. 2004;101:3880–5. doi: 10.1073/pnas.0400339101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhu J, Cote-Sierra J, Guo L, Paul WE. Stat5 activation plays a critical role in Th2 differentiation. Immunity. 2003;19:739–48. doi: 10.1016/S1074-7613(03)00292-9. [DOI] [PubMed] [Google Scholar]
- 107.Takatori H, Nakajima H, Hirose K, Kagami S, Tamachi T, Suto A, et al. Indispensable role of Stat5a in Stat6-independent Th2 cell differentiation and allergic airway inflammation. J Immunol. 2005;174:3734–40. doi: 10.4049/jimmunol.174.6.3734. [DOI] [PubMed] [Google Scholar]
- 108.Kurowska-Stolarska M, Kewin P, Murphy G, Russo RC, Stolarski B, Garcia CC, et al. IL-33 induces antigen-specific IL-5+ T cells and promotes allergic-induced airway inflammation independent of IL-4. J Immunol. 2008;181:4780–90. doi: 10.4049/jimmunol.181.7.4780. [DOI] [PubMed] [Google Scholar]
- 109.Soroosh P, Doherty TA. Th9 and allergic disease. Immunology. 2009;127:450–8. doi: 10.1111/j.1365-2567.2009.03114.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chang HC, Sehra S, Goswami R, Yao W, Yu Q, Stritesky GL, et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat Immunol. 2010;11:527–34. doi: 10.1038/ni.1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Goswami R, Jabeen R, Yagi R, Pham D, Zhu J, Goenka S, et al. STAT6-dependent regulation of Th9 development. J Immunol. 2012;188:968–75. doi: 10.4049/jimmunol.1102840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Lebman DA, Coffman RL. Interleukin 4 causes isotype switching to IgE in T cell-stimulated clonal B cell cultures. J Exp Med. 1988;168:853–62. doi: 10.1084/jem.168.3.853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Helming L, Gordon S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009;19:514–22. doi: 10.1016/j.tcb.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 114.Brombacher F, Arendse B, Peterson R, Hölscher A, Hölscher C. Analyzing classical and alternative macrophage activation in macrophage/neutrophil-specific IL-4 receptor-alpha-deficient mice. Methods Mol Biol. 2009;531:225–52. doi: 10.1007/978-1-59745-396-7_15. [DOI] [PubMed] [Google Scholar]
- 115.Gray MJ, Poljakovic M, Kepka-Lenhart D, Morris SM., Jr. Induction of arginase I transcription by IL-4 requires a composite DNA response element for STAT6 and C/EBPbeta. Gene. 2005;353:98–106. doi: 10.1016/j.gene.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 116.Takeda K, Kamanaka M, Tanaka T, Kishimoto T, Akira S. Impaired IL-13-mediated functions of macrophages in STAT6-deficient mice. J Immunol. 1996;157:3220–2. [PubMed] [Google Scholar]
- 117.Szanto A, Balint BL, Nagy ZS, Barta E, Dezso B, Pap A, et al. STAT6 transcription factor is a facilitator of the nuclear receptor PPARγ-regulated gene expression in macrophages and dendritic cells. Immunity. 2010;33:699–712. doi: 10.1016/j.immuni.2010.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, et al. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–82. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- 119.Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6:137–43. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 120.Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell-type cytokines in protease allergen-induced airway inflammation. Immunity. 2012;36:451–63. doi: 10.1016/j.immuni.2011.12.020. [DOI] [PubMed] [Google Scholar]
- 121.Chang YJ, Kim HY, Albacker LA, Baumgarth N, McKenzie AN, Smith DE, et al. Innate lymphoid cells mediate influenza-induced airway hyper-reactivity independently of adaptive immunity. Nat Immunol. 2011;12:631–8. doi: 10.1038/ni.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188:1503–13. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129:191–8, e1-4. doi: 10.1016/j.jaci.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 124.Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, et al. Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur J Immunol. 2012;42:1106–16. doi: 10.1002/eji.201142018. [DOI] [PubMed] [Google Scholar]
- 125.Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity. 2012;37:649–59. doi: 10.1016/j.immuni.2012.08.015. [DOI] [PubMed] [Google Scholar]
- 126.Fanta CH. Clinical aspects of mucus and mucous plugging in asthma. J Asthma. 1985;22:295–301. doi: 10.3109/02770908509087113. [DOI] [PubMed] [Google Scholar]
- 127.Turner J, Jones CE. Regulation of mucin expression in respiratory diseases. Biochem Soc Trans. 2009;37:877–81. doi: 10.1042/BST0370877. [DOI] [PubMed] [Google Scholar]
- 128.Tyner JW, Kim EY, Ide K, Pelletier MR, Roswit WT, Morton JD, et al. Blocking airway mucous cell metaplasia by inhibiting EGFR antiapoptosis and IL-13 transdifferentiation signals. J Clin Invest. 2006;116:309–21. doi: 10.1172/JCI25167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Atherton HC, Jones G, Danahay H. IL-13-induced changes in the goblet cell density of human bronchial epithelial cell cultures: MAP kinase and phosphatidylinositol 3-kinase regulation. Am J Physiol Lung Cell Mol Physiol. 2003;285:L730–9. doi: 10.1152/ajplung.00089.2003. [DOI] [PubMed] [Google Scholar]
- 130.Laoukili J, Perret E, Willems T, Minty A, Parthoens E, Houcine O, et al. IL-13 alters mucociliary differentiation and ciliary beating of human respiratory epithelial cells. J Clin Invest. 2001;108:1817–24. doi: 10.1172/JCI13557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kondo M, Tamaoki J, Takeyama K, Nakata J, Nagai A. Interleukin-13 induces goblet cell differentiation in primary cell culture from Guinea pig tracheal epithelium. Am J Respir Cell Mol Biol. 2002;27:536–41. doi: 10.1165/rcmb.4682. [DOI] [PubMed] [Google Scholar]
- 132.Yu H, Li Q, Kolosov VP, Perelman JM, Zhou X. Interleukin-13 induces mucin 5AC production involving STAT6/SPDEF in human airway epithelial cells. Cell Commun Adhes. 2010;17:83–92. doi: 10.3109/15419061.2010.551682. [DOI] [PubMed] [Google Scholar]
- 133.Hoshino M, Morita S, Iwashita H, Sagiya Y, Nagi T, Nakanishi A, et al. Increased expression of the human Ca2+-activated Cl- channel 1 (CaCC1) gene in the asthmatic airway. Am J Respir Crit Care Med. 2002;165:1132–6. doi: 10.1164/ajrccm.165.8.2107068. [DOI] [PubMed] [Google Scholar]
- 134.Damera G, Xia B, Sachdev GP. IL-4 induced MUC4 enhancement in respiratory epithelial cells in vitro is mediated through JAK-3 selective signaling. Respir Res. 2006;7:39. doi: 10.1186/1465-9921-7-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, et al. Interleukin-13: central mediator of allergic asthma. Science. 1998;282:2258–61. doi: 10.1126/science.282.5397.2258. [DOI] [PubMed] [Google Scholar]
- 136.Kuperman DA, Huang X, Koth LL, Chang GH, Dolganov GM, Zhu Z, et al. Direct effects of interleukin-13 on epithelial cells cause airway hyperreactivity and mucus overproduction in asthma. Nat Med. 2002;8:885–9. doi: 10.1038/nm734. [DOI] [PubMed] [Google Scholar]
- 137.Chen W, Khurana Hershey GK. Signal transducer and activator of transcription signals in allergic disease. J Allergy Clin Immunol. 2007;119:529–41, quiz 542-3. doi: 10.1016/j.jaci.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 138.Lamkhioued B, Renzi PM, Abi-Younes S, Garcia-Zepada EA, Allakhverdi Z, Ghaffar O, et al. Increased expression of eotaxin in bronchoalveolar lavage and airways of asthmatics contributes to the chemotaxis of eosinophils to the site of inflammation. J Immunol. 1997;159:4593–601. [PubMed] [Google Scholar]
- 139.Zimmermann N, Hogan SP, Mishra A, Brandt EB, Bodette TR, Pope SM, et al. Murine eotaxin-2: a constitutive eosinophil chemokine induced by allergen challenge and IL-4 overexpression. J Immunol. 2000;165:5839–46. doi: 10.4049/jimmunol.165.10.5839. [DOI] [PubMed] [Google Scholar]
- 140.Shum BO, Mackay CR, Gorgun CZ, Frost MJ, Kumar RK, Hotamisligil GS, et al. The adipocyte fatty acid-binding protein aP2 is required in allergic airway inflammation. J Clin Invest. 2006;116:2183–92. doi: 10.1172/JCI24767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Chiba Y, Matsusue K, Misawa M. RhoA, a possible target for treatment of airway hyperresponsiveness in bronchial asthma. J Pharmacol Sci. 2010;114:239–47. doi: 10.1254/jphs.10R03CR. [DOI] [PubMed] [Google Scholar]
- 142.Chiba Y, Todoroki M, Nishida Y, Tanabe M, Misawa M. A novel STAT6 inhibitor AS1517499 ameliorates antigen-induced bronchial hypercontractility in mice. Am J Respir Cell Mol Biol. 2009;41:516–24. doi: 10.1165/rcmb.2008-0163OC. [DOI] [PubMed] [Google Scholar]
- 143.Peng Q, Matsuda T, Hirst SJ. Signaling pathways regulating interleukin-13-stimulated chemokine release from airway smooth muscle. Am J Respir Crit Care Med. 2004;169:596–603. doi: 10.1164/rccm.200307-888OC. [DOI] [PubMed] [Google Scholar]