

Abstract
Iridium-catalyzed borylation has been applied to various substituted thiophenes to synthesize poly-functionalized thiophenes in good to excellent yields. Apart from common functionalities compatible with iridium-catalyzed borylations, additional functional group tolerance to acyl (COMe), and trimethylsilyl (TMS) groups was also observed. High regioselectivities were observed in borylation of 3-and 2,5-di-substituted thiophenes. Electrophilic aromatic C–H/C-Si bromination on thiophene boronate esters is shown to take place without breaking the C–B bond, and one-pot C–H borylation/Suzuki-Miyaura cross-coupling has been accomplished on 2- and 3-borylated thiophenes.
Keywords: C–H activation, Iridium, Catalysis, Borylation, Thiophenes, One-pot reactions, Suzuki-Miyaura cross-coupling, Bromination
1. Introduction
Thiophenes comprise an important heterocyclic class with diverse applications ranging from the design of advanced materials1–3 to the treatment of various diseases4–7 (Figure 1). Consequently, their synthesis has garnered keen interest.
Figure 1.
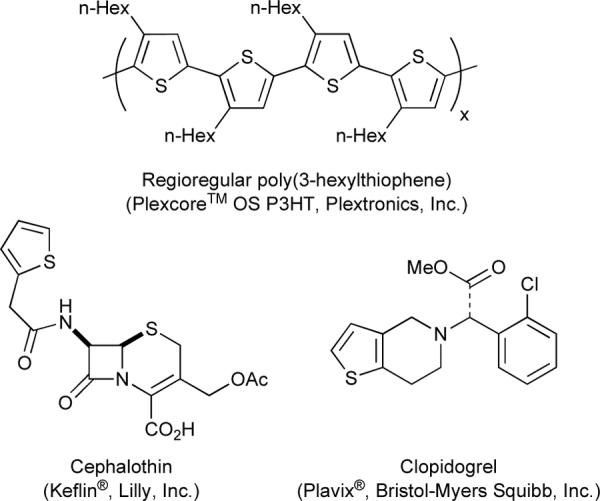
Selected thiophene-derived articles of commerce.
There are two fundamentally different approaches for synthesizing substituted thiophenes. The first entails construction of the thiophene ring from appropriate precursors with the most common examples stemming from early syntheses of thiophene from C4 carbonyl compounds and P2S5.8,9 The second general approach to substituted thiophenes involves derivatizing an existing thiophene core. Examples of the latter case include halogenations, alkylations, and metalations. The first metalation examples were mercurations reported by Volhard in 1892,10 and Thomas described the generation and reactivity of 2-thiophenylmagnesium iodide by magnesium reduction of 2-iodothiophene in 1908.11 Organolithiation reactions of thiophenes, pioneered by Gilman,12 have proved to be particularly versatile because, like mercuration, the thiophene C–H bond can be functionalized directly. The thienyl lithium intermediates that result react readily with various electrophiles.13 While biological systems can assemble, as well as functionalize, thiophenes,14,15 their synthetic utility is limited when compared to non-biochemical methods.
Lithiation reactions are some of the oldest and most prevalent means for functionalizing C–H bonds in heterocycles. More recently, attention has turned to other methods for derivatizing C–H16–18 and C–X18–22 bonds in heterocycles with an emphasis on transition metal catalyzed processes that obviate the requirement for stoichiometric metal. While the emerging methodologies can sometimes bypass intermediates for certain syntheses, they can also offer selectivities that complement metalation reactions. In this contribution we examine the scope and limitations of Ir-catalyzed C–H borylation applied to the synthesis of thiophene boronate esters.
2. Results and Discussion
Aryl boronate esters are versatile reagents that are widely used in the construction of carbon–carbon and carbon heteroatom bonds. Prior to 1995, aryl boronate esters were typically prepared by reacting an organometallic intermediate, generated from an arene or aryl halide and stoichiometric quantities of a metalating agent, with a boron electrophile (Figure 2). In 1995, Miyaura and co-workers reported the synthesis of arylboronates by the Pd catalyzed cross-coupling of tetraalkoxydiboron reagents with haloarenes, including 3-iodobenzothiophene.23 Subsequently, Masuda and co-workers devised related conversions using dialkoxyboranes,24 including the synthesis of 2-thienyl boronate esters.25 These transformations were important advances because (i) substoichiometric quantities of Pd catalysts served as the metalating agents, and (ii) the mild reaction conditions can accommodate functional groups that are incompatible with organomagnesium or organolithium reagents.
Figure 2.
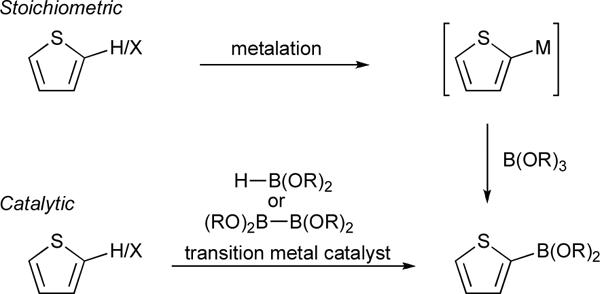
Routes to 2-thiophenyl boronate esters via stoichiometric or catalytic metalation protocols.
In 1999, we reported an Ir-catalyzed reaction that coupled benzene and pinacolborane (HBPin) to yield PhBPin generating hydrogen gas as the sole byproduct.26 Recognizing that this transformation's simplicity could offer advantages over traditional routes to arylboron compounds, we explored the generality of this reaction with arenes, including the first extensions to heterocyclic substrates.27–29 Despite improvements in catalyst generation,28,30 application of this methodology to substituted thiophenes is limited to five substrates: 2-methylthiophene,31–33 2-cyanothiophene,32 2-bromothiophene,32 2-methoxythiophene,33 and 2-trifluoromethylthiophene.33 These reactions yield 5-borylated products exclusively in accord with the preference for borylation of C–H bonds adjacent to formally sp3-hybridized heteroatoms in 5-membered heterocycles. Clearly, the restriction of previous studies to 2-substituted substrates raises questions regarding the feasibility of this reaction when the substitution pattern is varied and the range of substituents is expanded.
2.1. Borylation of 2-substituted thiophenes
As noted above, the reported borylations of 2-substituted thiophenes display excellent regioselectivity for 5-borylated products. Nevertheless, the range of substituents that have been surveyed is limited compared to aromatic substrates. Before detailing our findings, comments regarding the catalyst system are warranted. Most of the chemistry in this paper utilizes a dipyridyl-ligated catalyst that is generated in situ. However, this catalyst system was ineffective for electron-rich substrates. For these cases, phosphine supported catalysts gave better results. Comparisons between the ligand systems were not made when the dipyridyl system was effective.
For phosphine based catalysis, the phosphine (typically a bidentate ligand) and the Ir precatalyst, (η5-Ind)Ir(cod)) (Ind = indenyl, cod = 1,4-cyclooctadiene), are simply combined with the borane, substrate, and solvent (if used) in a reaction vessel. The resulting mixture is then heated to effect borylation.28 While the dtbpy system operates at room temperature, generation of the catalysts must be done as follows.34,35 First, the HBPin and the Ir precatalyst, [Ir(μ2-OMe)(η4-cod)]2 are combined. Then the dipyridyl ligand, in most cases 4,4′-di-tert-butyl-2,2′-dipyridyl (dtbpy), is dissolved in a suitable solvent and the resulting solution is added to the HBPin/[Ir(μ2-OMe)(η4-cod)]2 mixture, generating a deep orange solution. The order of operations is critical as addition of dtbpy to [Ir(μ2-OMe)(η4-cod)]2 produces a pale green solution that exhibits diminished activity upon addition of HBPin followed by substrate. Although HBPin was used exclusively in this study, it should be noted that B2Pin2, while a more active borylating agent, is less effective for catalyst generation. Should circumstances warrant use of B2Pin2, it is critical that the catalyst generation be carried out with HBPin. Lastly, we note that despite being touted for its air-stability, solid samples of [Ir(μ2-OMe)(η4-cod)]2 gradually darken when stored on the bench top and catalytic activity of “aged” precatalyst is diminished relative to pristine samples.
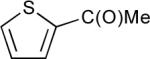
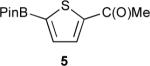
Table 1 displays borylation results for an expanded slate of 2-substituted thiophenes. Entries 1–3 show that the tolerance for heavier halogens and esters exhibited for arenes extends to thiophenes. Entry 4 is noteworthy in that the TMS group can be transformed while leaving the BPin intact (vide infra). It seemed likely that substituents that compromise arene borylations, might be compatible for thiophenes since heterocyclic substrates are usually more susceptible to borylation than arenes. This indeed proved to be the case for entry 5 where the acyl product 5 was obtained. This appears to be a limit for compatibility as the analogous product 6, though generated in small quantities, was not isolated from the borylation of 2-formylthiophene owing to reduction of the formyl group by HBPin. In the absence of Ir catalyst, HBPin does not reduce 2-formylthiophene at room temperature. It is noteworthy that Christophersen and co-workers have successfully performed a Pd-catalyzed Masuda coupling of HBPin with 2-bromo-3-formylthiophene without complications arising from reduction.36
Table 1.
Ir-catalyzed borylations of 2-substituted thiophenes.a
| Entry | Substrate | Reaction Time | Product | Yield %b |
|---|---|---|---|---|
| 1 |

|
1 h |

|
92 |
| 2 |

|
10 min |

|
78 |
| 3 |

|
30 min |

|
94 |
| 4 |

|
30 min |

|
93 |
| 5 |
|
30 min |
|
85 |
| 6 |

|
-- |

|
--c |
Reactions were carried out with 3 mol% Ir catalyst in n-hexane at room temperature with 1.5–2.0 equiv HBPin. For details see the Experimental Section.
Yields are for isolated products
GC/MS data show that 6 (or an isomer) accounts for ~10% of the reaction mixture. GC/MS and NMR data indicate that significant reduction of the formyl group occurs.
2.2. Borylation of 3-substituted thiophenes
In contrast to 2-substituted thiophenes, both C–H bonds flanking S in 3-substituted thiophenes are potentially accessible for borylation. In the absence of electronic effects, borylation at the 5-position should be generally favoured. However, selectivities will likely be lower than those for arenes since the distance between neighbouring substituents increases as the number of ring atoms decreases.37
Indeed, some of these expectations are born by the data in Table 2. In cases where isomer mixtures resulted (Table 2 entries 1–4 and 8) 2.0 equiv of thiophene was used to minimize losses arising from diborylation. Regiochemical assignment is straightforward from the magnitudes of |4JHH| (~ 2 Hz) and |3JHH| (~ 5 Hz) for the respective a and b isomers. 3-Cyanothiophene gave the poorest regioselectivity with 2-borylated product 7b being the major isomer. While CN is one of the smallest substituents, previous work shows that borylation ortho to H is preferred relative to CN for arenes,37 and the results for entry 1 are the first where borylation ortho to CN appears to be favoured. Contrary to a literature report noting its instability,367b was sufficiently stable to be persistent in the isolated isomer mixture.
Table 2.
Borylations of 3-substituted thiophenes.a
| Entry | Substrate | Conditions | 5-Borylated Product | 3-Borylated Product | a:b b | Yield %c |
|---|---|---|---|---|---|---|
| 1 |

|
0.5 equiv HBPin, 1 h |

|

|
1:1.13 | 54d |
| 2 |

|
0.5 equiv HBPin, 1 h |

|

|
3.5:1 | 66d |
| 3 |

|
0.5 equiv HBPin, 1 h |

|

|
8.9:1 | 72d |
| 4 |
|
0.5 equiv HBPin, 1 h |

|

|
8.9:1 | 67d |
| 5 |

|
1.2 equiv HBPin, 15 min |

|
-- | >99:1 | 82 |
| 6 |

|
1.2 equiv HBPin, 1 h |

|
-- | >99:1 | 95 |
| 7 |

|
1.2 equiv HBPin, 30 min |

|
-- | >99:1 | 79 |
| 8 |  |
0.9 equiv HBPin, 1 h |

|

|
>32:1 | 74d |
Reactions were carried out with 3 mol% or pregenerated Ir catalyst in n-hexane at room temperature with 1.5–2.0 equiv HBPin. For details see the Experimental Section.
Isomer ratios were determined by GC analysis of the crude reaction mixture.
Yields are reported for isolated products and are based on starting thiophene unless otherwise noted. Isomers were not separated.
Yield based on HBPin.
Isomer mixtures were also observed for Cl, Br, Me, and ptolyl (p-Tol) substituents (entries 2–4, and 8). For these substrates, the 5-borylated isomers are the major products and the relative isomer ratios (a:b) for Cl, Br, and Me follow the trend seen in arenes.37 For the p-Tol substituted substrate (entry 8) the selectivity is sufficiently high for 14a to be synthetically useful. As compared to Rh and Pd catalysts that favour borylation of benzylic C–H bonds,38–40 Ir catalysts are highly selective for aromatic over benzylic C–H bond functionalization,27,28 even for substrates with hindered arene C–H bonds like p-xylene.41 Thus, it is noteworthy that borylation of the thiophene C–H bonds is favoured. In particular, formation of 14b indicates that functionalization of relatively hindered thiophene C–H bonds is possible when arene C–H bonds are present. This might not prove to be general, particularly for compounds where electron-deficient o- or m-substituted aryl groups are present.
The selectivity for acyl, ester, and trimethylsilyl substituents was excellent and 2-borylated products were not detected. For the methyl ester substrate (entry 6), the selectivity is consistent with that observed in borylations of 4-benzonitriles, and the steric profiles of acyl and TMS groups are likely similar or greater.
Certainly, the closest comparison to our work is the related Ir-catalyzed silylations described by Ishiyama and Miyaura.42 Even though these reactions require much higher temperatures, which we frankly consider to be a very minor drawback, their selectivities for silylation at the 5- relative to the 2-position of 3-methyl- and 3-chlorothiophene (99:1 and 49:1, respectively) are better than those for borylation, while regioselectivity for silylation of methyl 3-thiophenecarboxylate (49:1) was marginally worse than that for borylation (Table 2, entry 6) It must be emphasized that 2-tert-butyl-1,10-phenanthroline was the ligand that engendered these selectivities. The silylation selectivity for dtbpy-ligated catalysts is more appropriate for directly comparing silylation and borylation. Though limited to a single example, the silylation regioselectivity for 3-methylthiophene (5-isomer:2-isomer = 2.5:1) is considerably worse than the 8.9:1 selectivity for borylation using the same precatalyst and ligand (Table 2, entry 4). Borylations using 2-tert-butyl-1,10-phenanthroline were not attempted because the ligand is not commercially available. Nevertheless, regioselectivities for thiophene borylations can be improved by altering the catalyst's coordination sphere.
In spite of the regioselectivity that silylation offers, two factors limit its synthetic utility. First, the silylating agent (tert-Bu2F2Si2) is not commercially available. Second, the synthetic elaborations of aryl and heteroaryl silanes are less well developed compared to the analogous boron chemistry. Certainly, future developments in arylsilane chemistry could change this situation.43,44
There are other existing methods for selectively functionalizing 3-substituted thiophenes at the 5-position. The two most common approaches are (i) electrophilic substitutions that are selective for 5-substitution when the 3-substituent is electron withdrawing and/or the electrophile is sterically hindered,45,46 and (ii) directed ortho metalations (DoMs) where the 3-substituent is a poor directed metalation group (DMG).47–49
When compared to DoMs, the selectivities for entries 2, 3, 5, and 6, are atypical. Even with relatively poor DMGs like Cl or Br at C-3, DoM at C-2 for thiophenes is often favoured. Consequently, protection/deprotection at C-2 can be required for selective synthesis of 3,5-substituted compounds via DoM.50 Since Ir-catalyzed borylation favours functionalization at the 5-position, it complements DoM nicely.
2.3. Borylation of 2,5-disubstituted thiophenes
Borylation of 2,5-disubstituted thiophenes are more challenging for two reasons. First, the 3- and 4-C–H bonds are less reactive towards borylation even in the absence of steric constraints, as evidenced by the results in Table 1. Second, the 2- and 5-substituents will further impede borylation.
The results for borylations of 2,5-disubstituted thiophenes are shown in Table 3. For the symmetrically substituted substrates in entries 1–3, regioselectivity is not an issue, making them the logical starting points for discussion. The first obvious difference from the data in Tables 1 and 2 is that borylation requires prolonged reactions times with Cl > Br >> Me. Ortho substituents impede borylations of C–H bonds of substituted arenes, with steric effects almost certainly being responsible.
Table 3.
Borylations of 2,5-disubstituted thiophenes.
| Entry | Substrate | Conditions | 3-Borylated Product | 4-Borylated Product | a:b a | Yield %b |
|---|---|---|---|---|---|---|
| 1 |

|
3 mol% Ir/dtbpy, 1.5 equiv HBPin, rt, 20 h |

|
-- | -- | 86 |
| 2 |

|
9 mol% Ir/dtbpy, 2.5 equiv HBPin, rt, 48 hc |

|
-- | -- | 56 |
| 3 |

|
3 mol% Ir/dtbpy, 1.5 equiv HBPin, rt, 20 h |

|
-- | -- | -- |
| 4 |

|
2 mol% Ir/dmpe, 1.5 equiv HBPin, 150 °C, 16 h |

|
-- | -- | 97 |
| 5 |

|
6 mol% Ir/dtbpy, 2.0 equiv HBPin, rt, 28 hd |

|

|
2.0:1e,f | 87 |
| 6 |

|
3 mol% Ir/dtbpy, 1.5 equiv HBPin, rt, 18 h |

|

|
2.3:1g | 86 |
| 7 |

|
3 mol% Ir/dtbpy, 1.5 equiv HBPin, rt, 20 h |

|

|
5.7:1h | 89 |
| 8 |

|
3 mol% Ir/dtbpy, 1.5 equiv HBPin, rt, 6 h |

|
-- | >99:1 | 93 |
| 9 |
|
-- | -- | -- | -- | -- |
| 10 |
|
-- | -- | -- | -- | -- |
Isomer ratios were determined by GC analysis of the crude reaction mixtures.
Yields are reported for isolated products and are based on starting thiophene unless otherwise noted. Isomers were not separated
initially, 6 mol% Ir/dtbpy catalyst loading and 1.5 equiv HBPin was used. After 36 h, conversion had ceased and the reaction flask was charged with an additional 3 mol% Ir/dtbpy and 1.0 equiv HBPin.
Initially, 3 mol% Ir/dtbpy catalyst loading and 1.5 equiv HBPin was used. After 8 h, the reaction flask was charged with an additional 3 mol% Ir/dtbpy and 0.5 equiv HBPin.
Assignment of isomers based on the 1H chemical shifts of the methine protons: 18a, 7.10 ppm, 18b, 6.94 ppm.
18a was prepared independently. See Scheme 2.
19a was identified by |4JHH| = 1.2 Hz for the coupling between the methine and methyl protons. |5JHH| was not resolved for 19b.
Assignment of isomers based on the 1H chemical shifts of the methine protons: 20a, 7.31 ppm, 18b, 6.87 ppm.
The relative ordering of the rates for thiophenes may not be a simple matter of steric effects. For example, borylation of 2,5-di-chlorothiophene slowed markedly after an initial conversion surge. This rate diminution was accompanied by precipitation of brown particles, suggesting that catalyst may be decomposing. Nevertheless the conversion was complete in 20 h and product 15 was isolated in good yield (Table 3, entry 1). The borylation of 2,5-dibromothiophene was more problematic, and only 89% conversion of the substrate was observed after 48 h at room temperature with 9 mol% Ir catalyst loadings. Consequently, compound 16 was isolated in modest yield (Table 3, entry 2). Given the highly reactive nature of C–X bonds in α-halogenated thiophenes, it would not be surprising if C–X scission led to catalyst deactivation for these substrates.
The potential for C–X activation also raises questions regarding the regiochemistry of the monoborylated products. Even though halogen tolerance is a hallmark of Ir-catalyzed C–H borylations, the observation of a single regioisomer from the borylation does not prove that the halogen regiochemistry is maintained. Assumptions of this type have lead to mischaracterization of products arising from directed metalations of 2,5-dihalothiophenes,51 where rearrangement of the metalated intermediates via “halogen dance” mechanisms can lead to 2,4-dihalogenated products.52
13C NMR data offer the first line of evidence against a similar rearrangement occurring in C–H borylations. By comparing the 13C chemical shifts of monosubstituted thiophene I to thiophene and 17 (vide infra) to 2,5-dimethylthiophene (Figure 3), increments for the 13C chemical shifts (IC-2B and IC-3B) for the BPin group can be estimated (Table 4). While we are not aware of previous reports of IC-3B values, the magnitudes and trends for the BPin IC-2B values are in line with those in the literature.53
Figure 3.

Compounds and 13C NMR chemical shift data used to estimate (a) IC–2B and (b) IC–3B values.
Table 4.
Carbon-13 chemical shifts δ(13C) (ppm) and BPin increments Ic-2 for I and Ic-3B for 17.
| I |
17 |
|||
|---|---|---|---|---|
| Position | δ | I c-2 B | δ | I c-3 B |
| C-2 | --a | -- | 150.8 | 13.5 |
| C-3 | 137.1 | 10.4 | --a | -- |
| C-4 | 128.2 | 1.5 | 130.7 | 6.0 |
| C-5 | 132.3 | 7.4 | 136.1 | −1.2 |
Cipso resonance was not observed.
Using IC-2B and IC-3B for the BPin group and the 13C increment values for Br substitution on a thiophene nucleus,52 the δ(13C) values for 16 and isomers A-D, which are generated by permuting H, Br, and BPin positions, have been calculated and the data are listed in Table 5. The Cipso resonances attached to BPin are not observed and assignment of the quaternary carbons is not certain. However, the methine carbon can be unambiguously assigned and the calculated methine shifts are indicated by boldface type. Isomer 16 gives the best fit to the data with the largest deviation observed for the C-2. This error is considerably smaller than the magnitude of the corresponding IC-2B value. The two remaining calculated shifts for 16, which include the methine resonance, fit the data very well. The fit to calculated shifts for isomer A, the analogue to the regioisomer that arises when lithiated 2,5-dibromothiophene rearranges, is poor with a large error in the shift for C-3, which is adjacent to BPin substituted carbon. Of the remaining isomers B-D, isomer C is the only candidate whose calculated values approach the fit found for 16. We discount this possibility because (i) there is no precedent for “halogen dance” rearrangement to this regioisomer, and (ii) the error in the methine shift, which is assigned unambiguously, is large. The final piece of confirming evidence comes from a chemical reaction of 16. We have observed that borylated products in crude reaction mixtures are susceptible to protodeborylation when heated with a source of acidic protons.54 Protodeborylation of 16 regenerates 2,5-dibromothiophene, which is consistent with the assigned regiochemistry.
Table 5.
Calculated Carbon-13 chemical shifts δ(13C) (ppm) for 16 and isomers A-D (methine resonances indicated in bold).
| Position | 16 | A | B | C | D |
|---|---|---|---|---|---|
| C-2 | 125.0 (3.1) | --a | 118.5 (−3.4) | 109.9 (−1.0) | --a |
| C-3 | --a | 119.9 (9.0) | 115.4 (4.5) | 119.9 (−2.0) | 124.4 (2.5) |
| C-4 | 136.2 (0.4) | 133.3 (−2.5) | 140.4 (4.6) | --a | 115.5 (4.6) |
| C-5 | 110.3 (−0.6) | 120.3 (−1.6) | --a | 140.3 (4.5) | 131.2 (−4.6) |
No data was calculated for Cipso resonances.
Deviations from fit to experimental data are shown in parentheses.
For 2,5-dimethylthiophene, electronic effects likely impact the borylation rates since the steric energies of methyl and bromine substituents are similar. The overall rate reduction in this case is consistent with results from arene borylations, where electron rich substrates are significantly less reactive than electron poor ones. Thus, borylation at room temperature with the dtbpy-ligated catalyst is impractical (Table 3, entry 3). Although this catalyst system has been reported to operate effectively at elevated temperatures, only 12% conversion was achieved after 16 h when the borylation was carried out with the Ir/dtbpy catalyst at 80 °C. We find that phosphine ligated catalysts are well-suited for substrates of this type, even though elevated temperatures are required for borylation. Indeed, the combination of the Ir precatalyst (Ind)Ir(cod) and 1,2-bis(dimethylphosphino)ethane (dmpe) promoted smooth borylation at 150 °C, and compound 17 was isolated in excellent yield (Table 3, entry 4).
For unsymmetrical chlorothiophene substrates, borylations typically gave isomer mixtures (Table 3, entries 5–7). The borylation regiochemistries were assigned either from the relative chemical shifts of the methine protons (18a and b and 20a and b) or by |JHH| values (19a and b). Compound 18a was also prepared independently (vide infra). The variations in isomer ratios reflect relative differences in steric energies for the substituents. When the relative steric are sufficiently great, single isomers can be attained as indicated by compound 21 (Table 3, entry 8). The acyl compatibility seen in Tables 1 and 3 did not extend to the 2-acyl-5-halothiophenes in entries 9 and 10.
The synthetic utility for the unsymmetrically 2,5-disubstituted thiophenes is more limited than for the other substrates that have been discussed to this point; however, it should be noted that certain substrates for which we would expect good selectivities (e.g. 2-fluoro compounds) were not surveyed because of their limited commercial availability.
2.4. Attempted borylation of 2,3,5-trisubstituted thiophenes
For 2,3,5-trisubstituted thiophenes, the 4-position is flanked by two ortho substituents. Since the H–C–C bond angles in 5-membered heterocycles are larger than those in 6-membered rings, the 4-position in 2,3,5-trisubstituted thiophenes should be more accessible for borylation. However, only about 2% borylation was observed for 3-bromo-2,5-di-methylthiophene (22) for attempted room temperature borylation with the [Ir(μ2-OMe)(COD)]2/dtbpy catalyst (Figure 5). The outcome was similar for the (Ind)Ir(COD)/dmpe system at 150 °C. Apart from steric hindrance for borylation, the electron-rich nature of 3-bromo-2,5-di-methylthiophene could also be responsible for this low reactivity. Thus, borylation of an electron deficient substrate, 3-bromo-2,5-di-chlorothiophene (23), was attempted using the [Ir(μ2-OMe)(COD)]2/dtbpy system at room temperature, the borylation reaction stalled after ~5% conversion. The borylation of this substrate was also tested using (Ind)Ir(COD) and dmpe at 150 °C. This reaction gave a 73% isolated yield of a single product, which surprisingly proved to be compound 2.
Figure 5.

Attempted borylations of 2,3,5-trisubstituted thiophenes.
The conversion of 23 to 2 is noteworthy in that the least hindered chloride is selectively cleaved. Although there is no direct supporting evidence, a plausible mechanism for the formation of 2 involves selective reduction of the 5-chloro substituent in 23 by HBPin to afford 3-bromo-2-chlorothiophene, which is then borylated to give 2. Alternatively, the transformation could proceed via C–Cl oxidative addition of 23 to Ir, C–B reductive elimination, and Ir–Cl reduction by HBPin to regenerate the active catalyst. Either scenario requires 2 equiv of HBPin for each equiv of compound 2 that is produced. Hence, the 73% yield for 1.5 equiv of HBPin indicates that the transformation is nearly quantitative.
2.5. Synthetic elaborations of borylated thiophenes
The synthetic utility of the thiophene boronate esters in Tables 1–3 hinges on their ability to participate in subsequent transformations. One attractive feature of Ir-catalyzed borylations is their amenability to one-pot reactions where subsequent transformations of the crude boronate esters can be accomplished without having to remove the spent Ir catalysts.
Preliminary studies show that one-pot C–H borylation/Suzuki-Miyaura cross-couplings can be accomplished on 2- and 3-borylated intermediates 21 and 24 (Scheme 1). The Suzuki-Miyaura couplings utilized aryl bromides to avoid the potential homocoupling of intermediates 21 and 24, and Pd(PPh3)4 was used as the catalyst. The yields for the two-step sequences in Scheme 1 are respectable and may improve with use of more efficient Pd catalysts.
Scheme 1.

One-pot C–H borylation/Suzuki-Miyaura cross-couplings of substituted thiophenes.
Although aromatic C–H bromination of aryl boronic esters (to synthesize brominated aryl boronic esters) is unknown, there are examples where aryl/heteroaryl boronic acids have been brominated.55–57 Thus, we reasoned that the products of the inefficient borylations of 2,3,5-trisubstituted thiophenes could conceivably be obtained by brominating borylation products 15 and 17.
Attempted bromination of 15 with Br2 in CHCl3 was ineffective even after 24 h at 100 °C, and the reaction between 15 and NBS in acetonitrile58 yielded a mixture of products. In addition to the desired C–H brominated product, GC/MS analysis indicated products where resulting from C–BPin scission. In contrast, compound 17 reacted rapidly with Br2 in CHCl3 to give tetrasubstituted 27 in 82% yield (Scheme 2). Excess bromine led to bromination of the thiophene methyl groups and should hence be avoided. The enhanced reactivity of 17 relative 15 arises from replacing chlorides with more electron donating methyl groups.
Scheme 2.

Bromination reactions of thiophenyl boronate esters.
Although we have evaluated the scope of C–H brominations of other substrates, related brominations of trimethylsilyl groups offer synthetic utility as indicated in Scheme 2. The TMS group in 21 is selectively cleaved by N-bromosuccinimide (NBS) yielding boronate ester 18a as a single isomer. This route is clearly preferable to borylation of 5-bromo-2-chlorothiophene, which yielded appreciable quantities of isomer 18b (Table 3, entry 5). We also find that compound 26 reacts with NBS in similar fashion to afford thiophene 28 in excellent yield, indicating that α-TMS cleavage is selective over benzylic and aromatic bromination. This is significant because the C(sp2)–Si bonds in TMS substituted arenes and heterocycles do not readily undergo cross-coupling reactions. Thus, the bromination of 26 confers synthetic utility for the selective coupling of BPin over TMS in compound 21. Compound 28 can be further derivatized as indicated by the Suzuki-Miyaura cross-coupling that yields 29 in Scheme 3. 2-halo-3,5-diarylthiophenes are quite rare.59–61 Nevertheless, related compounds substituted at the 2-position exhibit interesting physical62 and biological63,64 properties.
Scheme 3.

A Suzuki-Miyaura cross-coupling reaction of thiophene 29.
3. Conclusions
From this study, Ir-catalyzed borylations offer significant versatility for derivatizing thiophene scaffolds. In general, regioselectivities complement those established for DoM, making the combination of these two methodologies particularly attractive. In addition, the results concerning the elaboration of these boronate esters are encouraging, even though subsequent transformations are not extensively surveyed in this contribution. It should be emphasized that even though the procedures reported herein were carried out using a glovebox, this chemistry is amenable to more standard laboratory settings. We plan on publishing these details separately. We are actively exploring the chemistry of these compounds, as well as applying these synthetic approaches to other heterocyclic systems.
4. Experimental
4.1. General considerations
Materials
[Ir(μ2-OMe)(COD)]2 and (Ind)Ir(COD) were prepared per the literature procedures.65,66 Pinacolborane (HBPin) was generously supplied by BASF and was distilled before use. 2-Trimethylsilylthiophene,67 3-trimethylsilylthiophene,68 and 3-p-tolylthiophene69 were prepared per the literature procedures. 2-Chloro-5-trimethylsilylthiophene was prepared by following the literature procedure for the synthesis of 2-bromo-5-trimethylsilylthiophene.70 All other commercially available chemicals were purified before use. Solid substrates were sublimed under vacuum. Liquid substrates were distilled before use. n-Hexane was refluxed over sodium, distilled, and degassed. Dimethoxy ethane (DME), ether, and tetrahydrofuran were obtained from dry stills packed with activated alumina and degassed before use. Silica gel (230–400 Mesh) was purchased from EMD™.
General methods
All reactions were monitored by GC-FID (Varian CP-3800; column type: WCOT Fused silica 30m × 0.25mm ID coating CP-SIL 8 CB), GC-FID method: 70 °C, 2 min.; 20 °C/min, 9 min.; 250 °C, 20 min. All reported yields are for isolated materials. 1H and 13C-NMR spectra were recorded on a Varian Inova-300 (300.11 and 75.47 MHz respectively), Varian VXR-500 or Varian Unity-500-Plus spectrometer (499.74 and 125.67 MHz respectively) and referenced to residual solvent signals (7.24 ppm and 77.0 ppm for CDCl3, respectively). 11B-NMR spectra were recorded on a Varian VXR-300 operating at 96.29 MHz and were referenced to neat BF3·Et2O as the external standard. All coupling constants are apparent J values measured at the indicated field strengths. Elemental analyses were performed at Michigan State University using a Perkin Elmer Series II 2400 CHNS/O Analyzer. GC-MS data were obtained using a Varian Saturn 2200 GC/MS (column type: WCOT Fused silica 30m × 0.25mm ID coating CP-SIL 8 CB). High-resolution mass spectra were obtained at the Mass Spectrometry Core of the Research Technology Support Facility (RTSF) at Michigan State University. Melting points were measured on a MEL-TEMP® capillary melting apparatus and are uncorrected.
Regioisomer assignment of borylation products of 3-substituted thiophenes by 1H-NMR spectroscopy
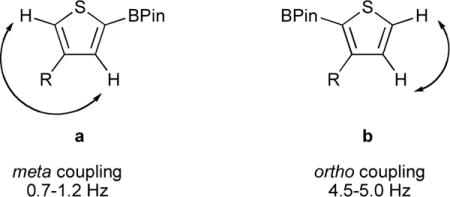
From the 1H-NMR coupling constants J, the two regioisomers obtained by the borylation of 3-substituted thiophenes can be distinguished unambiguously. In case of the 2,4-borylated product, the value of the four-bond (meta) coupling constant 4JH–H is usually around 0.7–1.2 Hz. While in case of the 2,3-borylated product, the value of the three-bond (ortho) coupling constant 3JH–H is usually around 4.5–5.0 Hz. Since these two ranges of coupling constants are quite far apart, the two regioisomers can easily be distinguished by the value of 1H-NMR coupling constant.
Catalytic borylation of substituted thiophenes
General procedure A (borylation with heteroaromatic substrate as the limiting reactant)
The [Ir] catalyst was generated by a modified literature protocol,34 where in a glove box, two separate test tubes were charged with [Ir(μ2-OMe)(COD)]2 (10 mg, 0.015 mmol, 3 mol% Ir) and dtbpy (8 mg, 0.03 mmol, 3 mol%). Excess HBPin (1.5 to 2 equiv) was added to the [Ir(μ2-OMe)(COD)]2 test tube. n-Hexane (1 mL) was added to the dtbpy containing test tube in order to dissolve the dtbpy. The dtbpy solution was then mixed with the [Ir(μ2-OMe)(COD)]2 and HBPin mixture. After mixing for one minute, the resulting solution was transferred to the 20 mL scintillation vial equipped with a magnetic stirring bar. Additional n-hexane (2 × 1 mL) was used to wash the test tubes and the washings were transferred to the scintillation vial. Substituted thiophene (1 mmol, 1 equiv) was added to the scintillation vial. The reaction was stirred at room temperature and was monitored by GC-FID/MS. After completion of the reaction, the volatile materials were removed on a rotary evaporator. The crude material was dissolved in CH2Cl2 and passed through a short plug of silica to afford the corresponding borylated product.
General procedure B (borylation with HBPin as the limiting reactant)
In a glove box, two separate test tubes were charged with [Ir(μ2-OMe)(COD)]2 (10 mg, 0.015 mmol, 3 mol% Ir) and dtbpy (8 mg, 0.03 mmol, 3 mol%). HBPin (1 mmol, 1 equiv) was added to the [Ir(μ2-OMe)(COD)]2 test tube. n-Hexane (1 mL) was added to the dtbpy containing test tube in order to dissolve the dtbpy. The dtbpy solution was then mixed with the [Ir(μ2-OMe)(COD)]2 and HBPin mixture. After mixing for one minute, the resulting solution was transferred to the 20 mL scintillation vial equipped with a magnetic stirring bar. Additional n-hexane (2 × 1 mL) was used to wash the test tubes and the washings were transferred to the scintillation vial. Excess 3-substituted thiophene (2–4 equiv) was added to the scintillation vial. The reaction was stirred at room temperature and was monitored by GC-FID/MS. After completion of the reaction, the volatile materials were removed on a rotary evaporator. The crude material was dissolved in CH2Cl2 and passed through a short plug of silica to afford the corresponding borylated product/products.
General procedure C
In a glove box, (Ind)Ir(COD) (8.3 mg, 0.02 mmol, 2.00 mol% Ir) and dmpe (3 mg, 0.02 mmol, 2.00 mol%) were weighed in two separate test tubes. HBPin (218 μL, 190 mg, 1.50 mmol, 1.50 equiv) was added to the dmpe test tube and the resulting solution was than mixed with (Ind)Ir(COD). This catalyst solution was added to a Schlenk flask equipped with a magnetic stirring bar. Substituted thiophene (1 mmol, 1 equiv) was added to the Schlenk flask. The Schlenk flask was closed, brought out of the glove box, and was heated at 150 °C in an oil bath. The reaction was monitored by GC-FID/MS. After completion of the reaction, the volatile materials were removed on a rotary evaporator. The crude material was dissolved in CH2Cl2 and passed through a short plug of silica to afford the corresponding borylated product.
2-(5-iodothiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (1)
The general borylation procedure A was applied to 2-iodothiophene (111 μL, 210 mg, 1 mmol, 1 equiv) and HBPin (218 μL, 192 mg, 1.50 mmol, 1.50 equiv) for 1 h. The product was isolated as a white solid (310 mg, 92% yield, mp 48–49 °C). 1H-NMR (CDCl3, 500 MHz): δ 7.27 (d, J = 3.5 Hz, 1 H), 7.25 (d, J = 3.5 Hz, 1 H), 1.31 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 138.5 (CH), 138.3 (CH), 84.3 (2 C), 81.5 (C), 24.7 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 28.7; FT-IR (neat) : 2978, 2932, 1522, 1418, 1314, 1267, 1142, 1064, 1018, 853, 663 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 336 (100), 321 (13), 250 (6), 236 (14), 209 (12), 167 (43); Anal. Calcd for C10H14BIO2S: C, 35.75; H, 4.20. Found: C, 36.04; H, 4.24.
2-(4-bromo-5-chlorothiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2)
The general borylation procedure A was applied to 2-chloro-3-bromothiophene (110 μL, 197 mg, 1 mmol, 1 equiv) and HBPin (192 μL, 218 mg, 1.50 mmol, 1.50 equiv) for 10 minutes. The product was isolated as a white solid (253 mg, 78% yield, mp 60–61°C). 1H-NMR (CDCl3, 500 MHz): δ 7.38 (s, 1 H), 1.30 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 138.9 (CH), 133.2 (C), 112.0 (C), 84.6 (2 C), 24.7 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 28.5; FT-IR (neat) : 2980, 2932, 1523, 1425, 1340, 1267, 1142, 1041, 852, 661 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 324 (100), 322 (73), 309 (45), 281 (26), 264 (29), 243 (38); Anal. Calcd for C10H13BBrClO2S: C, 37.13; H, 4.05. Found: C, 37.20; H, 4.16.
Note: Attempted borylation of 2,5-dichloro-3-bromothiophene with borylation procedure C also gave the same product where C-Cl bond was borylated and the single monoborylated product was isolated in 73% yield (see attempted borylation of tri-substituted thiophene). Only one of the two C-Cl bonds is activated with chemoselectivity greater than 99%. The NMR data matched with the borylated product of 2-chloro-3-bromothiophene as described above.
Methyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl) thiophene-2-carboxylate (3)
The general borylation procedure A was applied to methyl-2-thiophenecarboxylate (116 μL, 142 mg, 1 mmol, 1 equiv) and HBPin (192 μL, 218 mg, 1.50 mmol, 1.50 equiv) for 0.5 h. The product was isolated as a white solid (252 mg, 94% yield, mp 114–117 °C). 1H-NMR (CDCl3, 500 MHz): δ 7.79 (d, J = 3.7 Hz, 1 H), 7.53 (d, J = 3.7 Hz, 1 H), 3.87 (s, 3 H, CO2CH3), 1.33 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 162.6 (C=O), 139.4 (C), 136.9 (CH), 133.9 (CH), 84.6 (2 C), 52.2 (CO22CH3), 24.7 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 29.1; FT-IR (neat) : 2970, 1719, 1527, 1354, 1248, 1145, 1097, 852, 832, 752, 665 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 268 (71), 253 (91), 237 (56), 182 (100); Anal. Calcd for C12H17BO4S: C, 53.75; H, 6.39. Found: C, 53.44; H, 6.44.
Trimethyl(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl)silane (4)
The general borylation procedure A was applied to 2-trimethylsilylthiophene (312 mg, 2 mmol, 1 equiv) and HBPin (435 μL, 384 mg, 3.00 mmol, 1.50 equiv) for 30 minutes. The product was isolated as a white solid (523 mg, 93% yield, mp 61–62 °C). 1H-NMR (CDCl3, 500 MHz): δ 7.67 (d, J = 3.3 Hz, 1 H), 7.31 (d, J = 3.3 Hz, 1 H), 1.32 (br s, 12 H, 4 CH3 of BPin), 0.30 (s, 9 H, 3 CH3 of TMS); 13C-NMR {1H} (CDCl3, 75 MHz): δ 148.4 (C), 137.8 (CH), 135.0 (CH), 84.0 (2 C), 24.8 (4 CH3 of BPin), −0.1 (3 CH3 of TMS); 11B-NMR (CDCl3, 96 MHz): δ 29.6; FT-IR (neat) : 3054, 2980, 2957, 1514, 1435, 1346, 1331, 1259, 1250, 1142, 1072, 981, 841, 821, 758, 699 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 282 (14), 267 (100), 239 (31), 167 (8); Anal. Calcd for C13H23BO2SSi: C, 55.31; H, 8.21. Found: C, 54.85; H, 8.74; HRMS (EI): m/z 282.1285 [(M+); Calcd for C13H23BO2SSi: 282.1281].
1-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl)ethanone (5)
The general borylation procedure A was applied to 2-acetylthiophene (108 μL, 126 mg, 1 mmol, 1 equiv) and HBPin (175 μL, 154 mg, 1.20 mmol, 1.20 equiv) for 0.5 h. The product was isolated as a white solid (213 mg, 85% yield, mp 64–66 °C). 1H-NMR (CDCl3, 500 MHz): δ 7.69 (d, J = 3.8 Hz, 1 H), 7.54 (d, J = 3.8 Hz, 1 H), 2.53 (s, 3 H, COCH3), 1.31 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 190.6 (C=O), 149.4 (C), 137.2 (CH), 132.6 (CH), 84.6 (2 C), 27.4 (COCH3), 24.7 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 29.2; FT-IR (neat) : 2980, 2934, 1669, 1520, 1348, 1288, 1267, 1142, 1020, 852, 667 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 252 (77), 237 (100), 209 (15), 195 (8), 179 (5), 166 (33), 153 (14) 137 (12) 109 (6); Anal. Calcd for C12H17BO3S: C, 57.16; H, 6.80. Found: C, 56.88; H, 7.06.
Borylation of 3-cyanothiophene (7a and 7b)
The general borylation procedure B was applied to 3-cyanothiophene (182 μL, 218 mg, 2.00 mmol, 2.00 equiv) and HBPin (145 μL, 128 mg, 1 mmol, 1 equiv) for 1 h. The ratio of two borylated products at the end of reaction was 1:1.13 by GC-FID. The borylated product mixture was isolated as a white solid (126 mg, 54% yield). 1H-NMR (CDCl3, 300 MHz): δ (7a) 8.13 (d, J = 1.2 Hz, 1 H), 7.75 (d, J = 1.2 Hz, 1 H), 1.33 (br s, 12 H, 4 CH3 of BPin), (7b) 7.62 (d, J = 4.9 Hz, 1 H), 7.38 (d, J = 4.9 Hz, 1 H), 1.36 (br s, 12 H, CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ (7a) 140.8 (CH), 138.1 (CH), 114.7 (C), 111.9 (C), 85.1 (2 C), 24.7 (4 CH3 of BPin), (7b) 132.7 (CH), 131.3 (CH), 118.2 (C), 115.1 (C), 84.8 (2 C), 24.7 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 28.6; FT-IR (neat) : 2980, 2231, 1429, 1319, 1142, 1039, 850, 628 cm−1; GC-MS (EI) m/z (% relative intensity): (7a) M+ 235 (7), 220 (100), 192 (9), 149 (37), 136 (15), (7b) M+1 236 (100), 220 (78), 194 (51), 178 (33), 149 (36), 136 (31); Anal. Calcd for C11H14BNO2S: C, 56.19; H, 6.00; N, 5.96. Found: C, 55.74; H, 5.99; N, 6.00.
Borylation of 3-chlorothiophene (8a and 8b)
The general borylation procedure B was applied to 3-chlorothiophene (186 μL, 237 mg, 2.00 mmol, 2.00 equiv) and HBPin (145 μL, 128 mg, 1 mmol, 1 equiv) for 1 h. The ratio of two borylated products at the end of reaction was 3.5:1 by GC-FID. The borylated product mixture was isolated as a white solid (160 mg, 66% yield). 1H-NMR (CDCl3, 300 MHz): δ (8a) 7.43 (d, J = 1.0 Hz, 1 H), 7.35 (d, J = 1.0 Hz, 1 H), 1.32 (br s, 12 H, 4 CH3 of BPin), (8b) 7.51 (d, J = 5.0 Hz, 1 H), 7.01 (d, J = 5.0 Hz, 1 H), 1.34 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ (8a) 136.9 (CH), 131.8 (C), 126.7 (CH), 84.4 (2 C), 24.7 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 29.0; FT-IR (neat) : 3107, 2980, 2932, 1522, 1421, 1356, 1336, 1142, 1026, 854, 665cm−1; GC-MS (EI) m/z (% relative intensity): M+ 244 (100), 246 (38), 231 (15), 229 (38), 209 (24), 158 (27); Anal. Calcd for C10H14BClO2S: C, 49.11; H, 5.77. Found: C, 49.33; H, 5.81.
Borylation of 3-bromothiophene (9a and 9b)
The general borylation procedure B was applied to 3-bromothiophene (190 μL, 326 mg, 2.00 mmol, 2.00 equiv) and HBPin (145 μL, 128 mg, 1 mmol, 1 equiv) for 1 h. The ratio of two borylated products at the end of reaction was 8.9:1 by GC-FID. The borylated product mixture was isolated as a white solid (209 mg, 72% yield). 1H-NMR (CDCl3, 300 MHz): δ (9a) 7.49 (d, J = 1.2 Hz, 1 H), 7.46 (d, J = 1.2 Hz, 1 H), 1.32 (br s, 12 H, 4 CH3 of BPin), (9b) 7.48 (d, J = 5.0 Hz, 1 H), 7.08 (d, J = 5.0 Hz, 1 H), 1.34 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ (9a) 139.3 (CH), 129.5 (CH), 111.2 (C), 84.4 (2 C), 24.7 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 29.0; FT-IR (neat) : 2980, 1518, 1415, 1350, 1143, 1026, 852, 665 cm−1; GC-MS (EI) m/z (% relative intensity): (9a) M+ 289 (51), 290 (98), 288 (100), 275 (61), 273 (55), 247 (18), 245 (21), 230 (19) 204 (41), (9b) M+ 289 (13), 290 (25), 288 (27), 275 (10), 273 (9), 209 (100), 189 (11), 167 (67); Anal. Calcd for C10H14BBrO2S: C, 41.56; H, 4.88. Found: C, 41.74; H, 4.88.
Borylation of 3-methylthiophene (10a and 10b)
The general borylation procedure B was applied to 3-methylthiophene (194 μL, 196 mg, 2.00 mmol, 2.00 equiv) and HBPin (145 μL, 128 mg, 1 mmol, 1 equiv) for 1 h. The ratio of two borylated products at the end of reaction was 8.9:1 by GC-FID. The borylated product mixture was isolated as colorless oil (150 mg, 67% yield). 1H-NMR (CDCl3, 300 MHz): δ (10a) 7.42 (d, J = 0.7 Hz, 1 H), 7.17 (t, J = 1.1 Hz, 1 H), 2.27 (d, J = 0.5 Hz, 1 H), 1.32 (br s, 12 H, 4 CH3 of BPin), (10b) 7.46 (d, J = 4.6 Hz, 1 H), 6.95 (d, J = 4.6 Hz, 1 H), 2.47 (s, 1 H), 1.30 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ (10a) 139.4 (CH), 138.9 (C), 128.0 (CH), 83.9 (2 C), 24.7 (4 CH3 of BPin), 14.9 (CH3); 11B-NMR (CDCl3, 96 MHz): δ 29.5; FT-IR (neat) : 2978, 2930, 1550, 1441, 1371, 1327, 1302, 1271, 1143, 1028, 962, 854 665 cm−1; GC-MS (EI) m/z (% relative intensity): (10a) M+ 224 (100), 209 (27), 181 (18), 138 (44), (10b) M+ 224 (100), 209 (68), 167 (64), 138 (54), 124 (61); Anal. Calcd for C11H17BO2S: C, 58.95; H, 7.65. Found: C, 58.65; H, 8.09.
1-(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-3-yl)ethanone (11a)
The general borylation procedure A was applied to 3-acetylthiophene (126 mg, 1 mmol, 1 equiv) and HBPin (174 μL, 154 mg, 1.20 mmol, 1.20 equiv) for 15 minutes. The product was isolated as colorless oil (206 mg, 82% yield). 1H-NMR (CDCl3, 500 MHz): δ 8.26 (d, J = 1.1 Hz, 1 H), 8.00 (d, J = 1.1 Hz, 1 H), 2.50 (s, 3 H, COCH3) 1.32 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 192.0 (C=O), 143.8 (C), 138.1 (CH), 137.0 (CH), 84.5 (2 C), 27.8 (COCH3), 24.8 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 29.2; FT-IR (neat) : 3098, 2980, 2934, 1680, 1530, 1448, 1381, 1373, 1340, 1305, 1215, 1143, 1024, 850, 667 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 252 (21), 237 (55), 209 (100), 195 (9), 153 (22), 137 (19); Anal. Calcd for C12H17BO3S: C, 57.16; H, 6.80. Found: C, 56.77; H, 7.19.
Methyl 5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophene-3-carboxylate (12a)
The general borylation procedure A was applied to methyl 3-thiophenecarboxylate (121 μL, 142 mg, 1 mmol, 1 equiv) and HBPin (174 μL, 154 mg, 1.20 mmol, 1.20 equiv) for 1 h. The product was isolated as a white solid (256 mg, 95% yield, mp 84–85 °C). 1H-NMR (CDCl3, 500 MHz): δ 8.31 (d, J = 1.0 Hz, 1 H), 8.01 (d, J = 1.0 Hz, 1 H), 3.84 (s, 3 H, CO2CH3) 1.33 (br s, 12 H, CH3 of BPin); 13C-NMR {1H} (CDCl3, 75 MHz): δ 163.1 (C=O), 138.8 (CH), 137.9 (CH), 134.9 (C), 84.4 (2 C), 51.6 (CO2CH3), 24.7 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 29.4; FT-IR (neat) : 3107, 2980, 2951, 1722, 1537, 1458, 1431, 1388, 1373, 1336, 1307, 1224, 1143, 1024, 987, 852, 752, 667 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 268 (65), 253 (100), 237 (22), 225 (39), 211 (29), 193 (12), 182 (45), 169 (41), 137 (27); Anal. Calcd for C12H17BO4S: C, 53.75; H, 6.39. Found: C, 53.54; H, 6.66.
Trimethyl(5-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-3-yl)silane (13a)
The general borylation procedure A was applied to 3-trimethylsilylthiophene (156 mg, 1 mmol, 1 equiv) and HBPin (174 μL, 154 mg, 1.20 mmol, 1.20 equiv) for 30 minutes. The product was isolated as a white solid (222 mg, 79% yield, mp 87–89 °C). 1H-NMR (CDCl3, 300 MHz): δ 7.71 (d, J = 1.0 Hz, 1 H), 7.69 (d, J = 1.0 Hz, 1 H), 1.33 (br s, 12 H, 4 CH3 of BPin), 0.24 (s, 9 H, 3 CH3 of TMS); 13C-NMR {1H} (CDCl3, 75 MHz): δ 142.4 (C), 141.9 (CH), 138.4 (CH), 83.8 (2 C), 24.6 (4 CH3 of BPin), −10.6 (3 CH3 of TMS); 11B-NMR (CDCl3, 96 MHz): δ 29.5; FT-IR (neat) : 2980, 2955, 1510, 1410, 1325, 1263, 1250, 1143, 1105, 1028, 902, 852, 839, 754, 667 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 282 (7), 267 (100), 239 (2), 167 (7); Anal. Calcd for C13H23BO2SSi: C, 55.31; H, 8.21. Found: C, 54.68; H, 8.47; HRMS (EI): m/z 282.1283 [(M+); Calcd for C13H23BO2SSi: 282.1281].
Borylation of 3-p-tolylthiophene (14a and 14b)
The general borylation procedure B was applied to 3-p-tolylthiophene (192 mg, 1.1 mmol, 1.1 equiv) and HBPin (145 μL, 128 mg, 1.00 mmol, 1.00 equiv) for 1 h. The ratio of two borylated isomers at the end of reaction was 32:1 by GC-FID. The product was isolated as colorless oil (223 mg, 74 % yield). 1H-NMR (CDCl3, 300 MHz): δ (14a) 7.91 (d, J = 1.2 Hz, 1 H), 7.68 (d, J = 1.2 Hz, 1 H), 7.48–7.52 (m, 2 H), 7.17–7.20 (m, 2 H), 2.35 (s, 3 H, CH3) 1.36 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 75 MHz): δ (14a) 143.8 (C), 136.8 (C), 136.2 (CH), 132.9 (C), 129.5 (CH), 126.9 (CH), 126.4 (CH), 84.2 (2 C), 24.8 (4 CH3 of BPin), 21.1 (CH3); 11B-NMR (CDCl3, 96 MHz): δ 29.4; FT-IR (neat) : 3090, 2978, 2928, 1547, 1441, 1379, 1371, 1329, 1311, 1269, 1143, 1026, 850, 819, 771, 667 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 300 (100), 285 (12), 214 (12); Anal. Calcd for C17H21BO2S: C, 68.01; H, 7.05. Found: C, 68.54; H, 6.97; HRMS (EI): m/z 300.1360 [(M+); Calcd for C17H21BO2S: 300.1355].
2-(2,5-dichlorothiophen-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (15)
The general borylation procedure A was applied to 2-5-di-chlorothiophene (107 μL, 153 mg, 1 mmol, 1 equiv) and HBPin (218 μL, 192 mg, 1.50 mmol, 1.50 equiv) for 20 h. The product was isolated as a white solid (240 mg, 86% yield, mp 35–36 °C). 1H-NMR (CDCl3, 500 MHz): δ 6.94 (s, 1 H), 1.30 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 137.1 (C), 131.1 (CH), 126.2 (C), 84.0 (2 C), 24.8 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 28.5; FT-IR (neat) : 2980, 1535, 1437, 1371, 1313, 1263, 1142, 1032, 966, 889, 848, 692 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 278 (100), 280 (68), 263 (32), 265 (22), 243 M-35 (79), 245 (30), 201 (51); Anal. Calcd for C10H13BCl2O2S: C, 43.05; H, 4.70. Found: C, 43.26; H, 4.74.
2-(2,5-dibromothiophen-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (16)
The general borylation procedure A was applied to 2-5-di-bromothiophene (113 μL, 142 mg, 1 mmol, 1 equiv) and HBPin (218 μL, 192 mg, 1.50 mmol, 1.50 equiv) with 6 mol% [Ir] catalyst loading for 36 h. Additional 3 mol% [Ir] catalyst and 1 equiv of HBPin was added at this stage and the reaction was run for 12 more h at room temperature. The ratio of the starting material to product after 48 h was 11:89. The product was isolated as a white solid (206 mg, 56% yield, mp 72–73 °C). 1H-NMR (CDCl3, 500 MHz): δ 7.09 (s, 1 H), 1.31 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 135.8 (CH), 121.9 (C), 110.9 (C), 84.0 (2 C), 24.8 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 28.5; FT-IR (neat) : 2978, 1525, 1365, 1307, 1248, 1143, 991, 962, 883, 848, 690 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 368 (100), 370 (51), 366 (52), 353 (18), 287 (56), 289 (59), 268 (28), 208 (77), 166 (69); Calcd for C10H13BBr2O2S: C, 32.65; H, 3.56. Found: C, 32.92; H, 3.57.
2-(2,5-dimethylthiophen-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17)
The general borylation procedure C was applied to 2-5-di-methylthiophene (228 μL, 224 mg, 2 mmol, 1 equiv) and neat HBPin (435 μL, 384 mg, 3.00 mmol, 1.50 equiv) for 16 h at 150 °C. The product was isolated as a colorless semi solid (460 mg, 97% yield). 1H-NMR (CDCl3, 300 MHz): δ 6.81 (d, J = 1.2 Hz, 1 H), 2.59 (s, 3 H, CH3), 2.38 (d, J = 0.4 Hz, 3 H, CH3), 1.30 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 150.8 (C), 136.1 (C), 130.7 (CH), 83.0 (2 C), 24.8 (4 CH3 of BPin), 15.6 (CH3), 14.7 (CH3); 11B-NMR (CDCl3, 96 MHz): δ 29.3; FT-IR (neat) : 2978, 2924, 1493, 1394, 1304, 1265, 1145, 868, 700 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 238 (100), 223 (8), 181 (37); Anal. Calcd for C12H19BO2S: C, 60.52; H, 8.04. Found: C, 60.62; H, 8.18.
Borylation of 2-chloro-5-bromothiophene (18a and 18b)
The general borylation procedure A was applied to 2-chloro-5-bromothiophene (110 μL, 197 mg, 1 mmol, 1 equiv) and HBPin (290 μL, 256 mg, 2.00 mmol, 2.00 equiv) with 3 mol% [Ir] catalyst loading for 8 h. Additional 3 mol% [Ir] and 0.5 equiv of HBPin was added and the reaction was run for 20 more h at room temperature. The ratio of the two borylated products at the end of reaction was 2:1 by GC-FID. The borylated product mixture was isolated as a white solid (281 mg, 87% yield). 1H-NMR (CDCl3, 500 MHz): δ (18a) 7.10 (s, 1 H), 1.30 (br s, 12 H, 4 CH3 of BPin), (18b) 6.94 (s, 1 H), 1.30 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ (18a) 139.6 (C), 134.9 (CH), 108.3 (C), 84.0 (2C), 24.8 (4 CH3 of BPin), (18b) 132.0 (CH), 128.9 (C), 119.5 (C), 84.1 (2 C), 24.8 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 28.5; FT-IR (neat) : 2980, 1527, 1427, 1371, 1253, 1140, 1028, 962, 848, 693 cm−1; GC-MS (EI) m/z (% relative intensity): (18a) M+ 324 (100), 322 (78), 289 (67), 287 (64), 208 (40), 166 (34), (18b) M+ 324 (89), 322 (69), 309 (23), 245 (41), 243 (99), 203 (43), 201 (100), 166 (50); Anal. Calcd for C10H13BBrClO2S: C, 37.13; H, 4.05. Found: C, 37.25; H, 4.05.
Note: The data for the pure 18a is described in the bromination section of this experimental.
Borylation of 2-chloro-5-methylthiophene (19a and 19b)
The general borylation procedure A was applied to 2-chloro-5-methylthiophene (133 mg, 1 mmol, 1 equiv) and HBPin (218 μL, 192 mg, 1.50 mmol, 1.50 equiv) for 18 h. The ratio of two borylated products at the end of reaction was 2.3:1 by GC-FID. The borylated product mixture was isolated as a colorless semi solid (221 mg, 86% yield). 1H-NMR (CDCl3, 300 MHz): δ (19a) 6.77 (q, J = 1.2 Hz, 1 H), 2.35 (d, J = 1.2 Hz, 3 H, CH3), 1.31 (br s, 12 H, 4 CH3 of BPin), (19b) 6.95 (s, 1 H), 2.60 (s, 3 H, CH3), 1.28 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ (19a) 137.4 (C), 137.0 (C), 130.1 (CH), 83.6 (2 C), 24.8 (4 CH3 of BPin), 14.9 (CH3), (19b) 151.1 (C), 131.6 (CH), 125.4 (C), 83.4 (2 C), 24.8 (4 CH3 of BPin), 15.7 (CH3); 11B-NMR (CDCl3, 96 MHz): δ 29.1; FT-IR (neat) : 2980, 2926, 1556, 1475, 1390, 1371, 1309, 1257, 1143, 1026, 966, 898, 850, 696 cm−1; GC-MS (EI) m/z (% relative intensity): (19a) 258 M+ (100), 243 (17), 223 (51), 181 (36), 153 (37) (19b) 258 M+ (100), 243 (18), 223 (7), 201 (93), 172 (23); Anal. Calcd for C11H16BClO2S: C, 51.10; H, 6.24. Found: C, 51.66; H, 6.58; HRMS (EI): m/z 258.0653 [(M+); Calcd for C11H16BClO2S: 258.06526].
Borylation of 2-chloro-5-iodothiophene (20a and 20b)
The general borylation procedure A was applied to 2-chloro-5-iodothiophene (122 mg, 0.5 mmol, 1 equiv) and HBPin (109 μL, 96 mg, 0.75 mmol, 1.50 equiv) for 20 h. The ratio of two borylated products at the end of reaction was 5.7:1 by GC-FID. The borylated product mixture was isolated as a white solid (165 mg, 89% yield). 1H-NMR (CDCl3, 300 MHz): δ (20a) 7.31 (s, 1 H), 1.30 (br s, 12 H, 4 CH3 of BPin), (20b) 6.87 (s, 1 H), 1.31 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ (20a) 143.4 (C), 142.3 (CH), 84.0 (2 C), 69.3 (C), 24.8 (4 CH3 of BPin), (20b) 132.8 (CH), 84.2 (2 C), 81.1 (C), 24.8 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 28.3; FT-IR (neat) : 2978, 1523, 1414, 1371, 1248, 1140, 1024, 966, 881, 848 690 cm−1; GC-MS (EI) m/z (% relative intensity): (20a) M+ 370 (100), 355 (13), 335 (29), 270 (25), 208 (15), 166 (11), (20b) M+ 370 (100), 355 (10), 270 (24), 243 (13), 201 (32), 166 (21); Anal. Calcd for C10H13BIClO2S: C, 32.42; H, 3.54. Found: C, 32.58; H, 3.38.
(5-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl)trimethylsilane (21)
The general borylation procedure A was applied to 2-chloro-5-trimethylsilylthiophene (382 mg, 2 mmol, 1 equiv) and HBPin (435 μL, 384 mg, 3.00 mmol, 1.50 equiv) for 6 h. The single borylated product was isolated as a solid (589 mg, 93% yield, mp 68–69 °C). 1H-NMR (CDCl3, 500 MHz): δ 7.26 (s, 1 H), 1.32 (br s, 12 H, 4 CH3 of BPin), 0.26 (s, 9 H, 3 CH3 of TMS); 13C-NMR {1H} (CDCl3, 125 MHz): δ 144.7 (C), 139.42 (CH), 139.37 (C), 83.7 (2 C), 24.8 (4 CH3 of BPin), −0.24 (3 CH3 of TMS); 11B-NMR (CDCl3, 96 MHz): δ 29.1; FT-IR (neat) : 2980, 1525, 1415, 1363, 1307, 1253, 1238, 1143, 993, 841, 758, 696 cm−1; GC-MS (EI) m/z (% relative intensity) 316 (33), 301 (100), 281 (6), 201 (15): M+; Anal. Calcd for C13H22BClO2SSi: C, 49.30; H, 7.00; Found: C, 49.16; H, 7.16.
Attempted borylation of tri-substituted thiophene
2-(4-bromo-5-chlorothiophen-2-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2)
The general borylation procedure C was applied to 2–5-di-chloro-3-bromo-thiophene (232 mg, 1 mmol, 1 equiv) and neat HBPin (218 μL, 192 mg, 1.50 mmol, 1.50 equiv) for 2 h at 150 °C. The product was isolated as a colorless solid (233 mg, 73% yield). The spectroscopic data of this product matched with the data of borylated product obtained from 2-chloro-3-bromothiophene as described earlier.
One-pot borylation/Suzuki coupling of substituted thiophenes
2-Methyl-5-(3-(trifluoromethyl)phenyl)thiophene (25)
The general borylation procedure A was applied to 2-methylthiophene (484 μL, 491 mg, 5 mmol, 1 equiv) and HBPin (870 μL, 768 mg, 6.00 mmol, 1.20 equiv) in a Schlenk flask for 0.5 h. The reaction mixture was pumped down under high vacuum for 0.5 h to remove the volatile materials. Pd(PPh3)4 (116 mg, 0.10 mmol, 2 mol%), 3-bromo-benzotrifluoride (837 μL, 1350 mg, 6.00 mmol, 1.2 equiv), and DME (6 mL) were added to the Schlenk flask inside the glove box. The Schlenk flask was then brought out of the glove box and attached to a Schlenk line. K3PO4·nH2O (1592 mg, 1.50 equiv) was added under N2 counter flow to the Schlenk flask. The flask was stoppered and the mixture was heated at 80 °C for 8 h. The flask was cooled down to room temperature and 20 mL of water were added to the reaction mixture. The reaction mixture was extracted with ether (3 × 20 mL). The combined ether extractions were washed with brine (20 mL), followed by water (10 mL), dried over MgSO4 before being concentrated under reduced pressure on a rotary evaporator. Column chromatography (hexanes, Rf 0.5) furnished the product as white semi solid (1026 mg, 85% yield). 1HNMR (CDCl3, 500 MHz): δ 7.77 (t, J = 0.8 Hz, 1 H), 7.68 (d, J = 7.6 Hz, 1 H), 7.42–7.48 (m, 2 H), 7.15 (d, J = 3.5 Hz, 1 H), 6.73–6.75 (m, 1 H), 2.51 (s, 3 H, CH3); 13C-NMR {1H} (CDCl3, 125 MHz): δ 140.7 (C), 140.1 (C), 135.5 (C), 131.2 (q, 2JC-F = 32.6 Hz, C), 129.3 (CH), 128.5 (CH), 126.4 (CH), 124.1 (q, 1JC-F = 273 Hz, CF3), 124.0 (CH), 123.4 (q, 3JC-F = 3.6 Hz, CH), 122.0 (q, 3JC-F = 3.6 Hz, CH), 15.4 (CH3); FT-IR (neat) 3073, 2922, 2865, 1497, 1340, 1325, 1165, 1126, 1074, 790, 694 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 242 (100), 223 (4), 173 (6); Anal. Calcd for C12H9F3S: C, 59.49; H, 3.74; Found: C, 59.38; H, 3.56.
(5-Chloro-4-m-tolylthiophen-2-yl)trimethylsilane (26)
The general borylation procedure A was applied to 2-chloro-5-trimethylsilylthiophene (382 mg, 2 mmol, 1 equiv) and HBPin (435 μL, 384 mg, 3.00 mmol, 1.50 equiv) in a Schlenk flask for 10 h. The reaction mixture was pumped down under high vacuum for 1 h to remove the volatile materials. Pd(PPh3)4 (46 mg, 2 mol%), 3-bromo-toluene (291 μL, 410 mg, 2.40 mmol, 1.2 equiv), and DME (3 mL) were added to the Schlenk flask inside the glove box. The Schlenk flask was then brought out of the glove box and attached to a Schlenk line. K3PO4·nH2O (637 mg, 1.50 equiv) was added under N2 counter flow to the Schlenk flask. The flask was stoppered and the mixture was heated at 80 °C for 6 h. The flask was cooled down to room temperature and 10 mL of water were added to the reaction mixture. The reaction mixture was extracted with ether (3 × 10 mL). The combined ether extractions were washed with brine (10 mL), followed by water (10 mL), dried over MgSO4 before being concentrated under reduced pressure on a rotary evaporator. Column chromatography (hexanes, Rf 0.5) furnished the product as a colorless liquid (369 mg, 66% yield). 1H-NMR (CDCl3, 300 MHz): δ 7.27–7.37 (m, 3 H), 7.13–7.16 (m, 1 H), 7.12 (s, 1 H), 2.39 (s, 3 H, CH3), 0.31 (s, 9 H, 3 CH3 of TMS); 13C-NMR {1H} (CDCl3, 75 MHz): δ 139.6 (C), 138.2 (C), 138.0 (C), 135.3 (CH), 134.3 (C), 129.3 (C), 129.1 (CH), 128.29 (CH), 128.27 (CH), 125.6 (CH), 21.5 (CH3), −0.3 (3 CH3 of TMS); FT-IR (neat) 3040, 2957, 2922, 1606, 1408, 1252, 993, 839, 781, 756, 700, 630 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 280 (49), 282 (19), 266 (100), 267 (48); Anal. Calcd for C14H17ClSSi: C, 59.86; H, 6.10; Found: C, 59.56; H, 6.21.
Bromination
2-(4-bromo-2,5-dimethylthiophen-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (27)
2-(2,5-dimethylthiophen-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (17) (238 mg, 1 mmol, 1 equiv) was dissolved in 2 mL of CHCl3 in a 20 mL scintillation vial equipped with a magnetic stirring bar. Bromine (160 mg, 1 mmol, 1 equiv, dissolved in 2 mL of CHCl3) was added drop-wise during two minutes. The reaction was then quenched with water. The product was extracted with CH2Cl2 (3 × 20 mL) and dried over MgSO4. Column chromatography (hexane/CH2Cl2 1:1, Rf 0.7) furnished the desired product as a white solid (260 mg, 82%, mp 55–56 °C). 1H-NMR (CDCl3, 300 MHz): δ 2.54 (s, 3 H, CH3), 2.29 (s, 3 H, CH3), 1.32 (br s, 12 H, 4 CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 147.9 (C), 131.2 (C), 113.1 (C), 83.5 (2 C), 24.8 (4 CH3 of BPin), 16.2 (CH3), 14.5 (CH3); 11B-NMR (CDCl3, 96 MHz): δ 29.5; FT-IR (neat) : 2978, 2922, 1537, 1377, 1315, 1234, 1143, 852 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 317 (46), 318 (84), 316 (81), 303 (11) 301 (10), 261 (100), 259 (99), 237 927), 195 (38), 180 (41); Anal. Calcd for C12H18BBrO2S: C, 45.46; H, 5.72. Found: C, 45.54; H, 5.91.
General procedure D (substitution of TMS with Br)
TMS group were replaced with bromine by employing the literature conditions used for aromatic C-H bromination.58 Substrate (1 mmol, 1 equiv) was added to a 20 mL scintillation vial equipped with a magnetic stirring bar. N-Bromosuccinamide (1 mmol, 1 equiv) was added in to the vial. Acetonitrile (3–5 mL) was also added to the vial. The reaction mixture was stirred at room temperature and was monitored by GC-FID/MS. After the completion of the reaction, the volatile materials were removed on a rotary evaporator and the crude product was passed through a short silica plug to afford the brominated product.
2-(5-Bromo-2-chlorothiophen-3-yl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (18a)
The general bromination procedure D was applied to (5-chloro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)thiophen-2-yl)trimethylsilane (21) (317 mg, 1 mmol) for 12 h. The product was isolated as a white solid (295 mg, 91%, mp 51–53 °C). 1H-NMR (CDCl3, 300 MHz): δ 7.10 (s, 1 H), 1.30 (br s, 12 H, CH3 of BPin); 13C-NMR {1H} (CDCl3, 125 MHz): δ 139.6 (C), 134.9 (CH), 108.3 (C), 84.1 (2 C), 24.8 (4 CH3 of BPin); 11B-NMR (CDCl3, 96 MHz): δ 28.5; FT-IR (neat) 2978, 1530, 1427, 1373, 1311, 1253, 1142, 1028, 962, 848, 883, 848, 692 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 323 (48), 324 (100), 322 (81), 309 (21), 307 (14), 289 (38), 287 (36) 208 (23), 166 (22); Anal. Calcd for C10H13BBrClO2S: C, 37.13; H, 4.05; Found: C, 37.25; H, 4.19.
5-Bromo-2-chloro-3-m-tolylthiophene (28)
The general bromination procedure D was applied to (5-chloro-4-m-tolylthiophen-2-yl)trimethylsilane (26) (280 mg, 1 mmol) for 12 h. The product was isolated as a colorless liquid (261 mg, 91%). 1H-NMR (CDCl3, 300 MHz): δ 7.29–7.31 (m, 3 H), 7.15–7.18 (m, 1 H), 7.02 (s, 1 H), 2.38 (s, 3 H, CH3); 13C-NMR {1H} (CDCl3, 75 MHz): δ 139.3 (C), 138.2 (C), 133.1 (C), 131.2 (CH), 129.1 (CH), 128.8 (CH), 128.4 (CH), 125.5 (CH), 124.0 (C), 108.3 (C), 21.4 (CH3); FT-IR (neat) 3042, 2920, 2858, 1604, 1487, 1028, 972, 831, 789, 779, 700 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 287 (63), 288 (100), 290 (29), 287 (63), 251 (5), 171 (19); Anal. Calcd for C11H9BrClS: C, 45.94; H, 2.80; Found: C, 45.96; H, 2.79.
5-(3,5-bis(trifluoromethyl)phenyl)-2-chloro-3-m-tolylthiophene (29)
In a glove box, a Schlenk flask, equipped with a magnetic stirring bar, was charged with 2-(3,5-bis(trifluoromethyl)phenyl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (82 mg, 0.24 mmol, 1.0 equiv). Two separate test tubes were charged with Pd(PPh3)4 (5.5 mg, 0.0048 mmol, 2 mol %) and 5-bromo-2-chloro-3-m-tolylthiophene (28) (69 mg, 0.24 mmol, 1.0 equiv). DME (2 mL) was used to transfer the contents of the test tubes into the Schlenk flask. The Schlenk flask was then brought out of the glove box and attached to a Schlenk line. K3PO4·nH2O (319 mg, 1.50 equiv) was added under N2 counter flow to the Schlenk flask. The flask was stoppered and the mixture was heated at 80 °C for 7 h. At this point the reaction mixture was allowed to cool to room temperature. The reaction solution was then filtered through a thin pad of silica gel (eluting with CH2Cl2) and the eluent was concentrated under reduced pressure. The crude material so obtained was purified via flash chromatography on silica gel (hexanes, Rf 0.5) to provide the Suzuki product as a white solid (85 mg, 84% yield, mp 77–79 °C). 1H-NMR (CDCl3, 500 MHz): δ 7.94 (s, 2 H), 7.80 (s, 1 H), 7.41–7.40 (m, 2 H), 7.38 (s, 1 H), 7.37–7.33 (t, J = 7.8 Hz, 1 H), 7.22–7.20 (d, J = 7.3 Hz, 1 H), 2.43 (s, 3 H, CH3); 13C-NMR {1H} (CDCl3, 125 MHz): δ 140.1 (C), 138.3 (C), 137.1 (C), 135.6 (C), 133.4 (C), 132.6 (q, 2JC–F = 33.6 Hz, 2 C), 129.1(CH), 128.9 (CH), 128.5 (CH), 126.4 (CH), 126.2 (C), 125.5 (CH), 125.2 (q, 3JC–F = 3.8 Hz, 2 CH), 123.1 (q, 1JC–F = 272.8 Hz, CF3), 121.1 (septet, 3JC–F = 3.9 Hz, CH), 21.4 (CH3); FT-IR (neat) : 3048, 2926, 1618, 1474, 1433, 1369, 1330, 1279, 1227, 1181, 1136, 1109, 1011, 891, 845, 789, 698, 684 cm−1; HRMS (FAB+): m/z 420.0174 [M+; Calcd for C19H11ClF6S: 420.0177].
Figure 4.
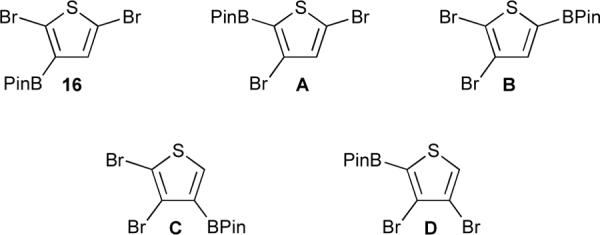
Compound 16 and its regioisomers A–D.
Acknowledgments
We thank BASF, Inc. for a generous gift of HBPin, and the Michigan Economic Development Corp., NIH (GM63188), and the Astellas USA Foundation for their generous financial support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Huynh WU, Dittmer JJ, Alivisatos AP. Science. 2002;295:2425–2427. doi: 10.1126/science.1069156. [DOI] [PubMed] [Google Scholar]
- 2.Li G, Shrotriya V, Huang J, Yao Y, Moriarty T, Emery K, Yang Y. Nature Materials. 2005;4:864–868. [Google Scholar]
- 3.Street RA. Nature Materials. 2006;5:171–172. [Google Scholar]
- 4.Fox KAA, Chelliah R. Expert Opin. Drug Metab. Toxicol. 2007;3:621–631. doi: 10.1517/17425225.3.4.621. [DOI] [PubMed] [Google Scholar]
- 5.Sperry JB, Wright DL. Curr. Opin. Drug Discov. Dev. 2005;8:723–740. [PubMed] [Google Scholar]
- 6.De Francesco R, Migliaccio G. Nature. 2005;436:953–960. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- 7.Chauvette RR, Flynn EH, Jackson BG, Lavagnino ER, Morin RB, Mueller RA, Pioch RP, Roeske RW, Ryan CW, Spencer JL, Van Heyningen E. J. Am. Chem. Soc. 1962;84:3401–2. [Google Scholar]
- 8.Volhard J, Erdmann H. Ber. Dtsch. Chem. Ges. 1885;454 [Google Scholar]
- 9.Meyer V. Ber. Dtsch. Chem. Ges. 1885;217 [Google Scholar]
- 10.Volhard J. J. Liebigs Ann. Chem. 1892;267:180. [Google Scholar]
- 11.Thomas V. C. R. Hebd. Seances Acad. Sci. 1908;146:642–645. [Google Scholar]
- 12.Gilman H, Shirley DA. J. Am. Chem. Soc. 1949;71:1870–1871. [Google Scholar]
- 13.Snieckus V. Chem. Rev. 1990;90:879–933. [Google Scholar]
- 14.Arroo RRJ, Jacobs J, VanGestel JAM, Kenkel H, Jannink W, Croes AF, Wullems GJ. New Phytologist. 1997;135:175–181. [Google Scholar]
- 15.Mosurkal R, Kumar R, Bruno FF, Nagarajan R, Samuelson L, Kumar J. Pure Appl. Chem. 2005;77:263–272. [Google Scholar]
- 16.Campos KR. Chem. Soc. Rev. 2007;36:1069–1084. doi: 10.1039/b607547a. [DOI] [PubMed] [Google Scholar]
- 17.Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007;36:1173–1193. doi: 10.1039/b606984n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wolfe JP, Thomas JS. Curr. Org. Chem. 2005;9:625–655. [Google Scholar]
- 19.Anderson KW, Tundel RE, Ikawa T, Altman RA, Buchwald SL. Angew. Chem.-Int. Edit. 2006;45:6523–6527. doi: 10.1002/anie.200601612. [DOI] [PubMed] [Google Scholar]
- 20.Fairlamb IJS. Chem. Soc. Rev. 2007;36:1036–1045. doi: 10.1039/b611177g. [DOI] [PubMed] [Google Scholar]
- 21.Alessi M, Larkin AL, Ogilvie KA, Green LA, Lai S, Lopez S, Snieckus V. J. Org. Chem. 2007;72:1588–1594. doi: 10.1021/jo0620359. [DOI] [PubMed] [Google Scholar]
- 22.Kalinin VN. Synthesis-Stuttgart. 1992:413–432. [Google Scholar]
- 23.Ishiyama T, Murata M, Miyaura N. J. Org. Chem. 1995;60:7508–7510. [Google Scholar]
- 24.Murata M, Watanabe S, Masuda Y. J. Org. Chem. 1997;62:6458–6459. doi: 10.1021/jo971143f. [DOI] [PubMed] [Google Scholar]
- 25.Murata M, Oyama T, Watanabe S, Masuda Y. J. Org. Chem. 2000;65:164–168. doi: 10.1021/jo991337q. [DOI] [PubMed] [Google Scholar]
- 26.Iverson CN, Smith MR., III J. Am. Chem. Soc. 1999;121:7696–7697. [Google Scholar]
- 27.Cho JY, Iverson CN, Smith MR., III J. Am. Chem. Soc. 2000;122:12868–12869. [Google Scholar]
- 28.Cho JY, Tse MK, Holmes D, Maleczka RE, Jr., Smith MR., III Science. 2002;295:305–308. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]
- 29.Tse MK, Cho JY, Smith MR., III Org. Lett. 2001;3:2831–2833. doi: 10.1021/ol0162668. [DOI] [PubMed] [Google Scholar]
- 30.Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew. Chem.-Int. Edit. 2002;41:3056–3058. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 31.Takagi J, Sato K, Hartwig JF, Ishiyama T, Miyaura N. Tetrahedron Lett. 2002;43:5649–5651. [Google Scholar]
- 32.Ishiyama T, Takagi J, Yonekawa Y, Hartwig JF, Miyaura N. Adv. Synth. Catal. 2003;345:1103–1106. [Google Scholar]
- 33.Miyaura N, Ishiyama T. PCT Int. Appl.: WO 2003074533. 2003. p. 42. [Google Scholar]
- 34.Boller TM, Murphy JM, Hapke M, Ishiyama T, Miyaura N, Hartwig JF. J. Am. Chem. Soc. 2005;127:14263–14278. doi: 10.1021/ja053433g. [DOI] [PubMed] [Google Scholar]
- 35.Paul S, Chotana GA, Holmes D, Reichle RC, Maleczka RE, Jr., Smith MR., III J. Am. Chem. Soc. 2006;128:15552–15553. doi: 10.1021/ja0631652. [DOI] [PubMed] [Google Scholar]
- 36.Christophersen C, Begtrup M, Ebdrup S, Petersen H, Vedso P. J. Org. Chem. 2003;68:9513–9516. doi: 10.1021/jo034919n. [DOI] [PubMed] [Google Scholar]
- 37.Chotana GA, Rak MA, Smith MR., III J. Am. Chem. Soc. 2005;127:10539–10544. doi: 10.1021/ja0428309. [DOI] [PubMed] [Google Scholar]
- 38.Mertins K, Zapf A, Beller M. J. Mol. Catal. A: Chem. 2004;207:21–25. [Google Scholar]
- 39.Ishiyama T, Ishida K, Takagi J, Miyaura N. Chem. Lett. 2001:1082–1083. [Google Scholar]
- 40.Shimada S, Batsanov AS, Howard JAK, Marder TB. Angew. Chem. Int. Ed. 2001;40:2168–2171. doi: 10.1002/1521-3773(20010601)40:11<2168::AID-ANIE2168>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 41.Ishiyama T, Takagi J, Ishida K, Miyaura N, Anastasi NR, Hartwig JF. J. Am. Chem. Soc. 2002;124:390–391. doi: 10.1021/ja0173019. [DOI] [PubMed] [Google Scholar]
- 42.Ishiyama T, Sato K, Nishio Y, Saiki T, Miyaura N. Chem. Commun. 2005:5065–5067. doi: 10.1039/b511171d. [DOI] [PubMed] [Google Scholar]
- 43.Strotman NA, Sommer S, Fu GC. Angew. Chem.-Int. Edit. 2007;46:3556–3558. doi: 10.1002/anie.200700440. [DOI] [PubMed] [Google Scholar]
- 44.Denmark SE, Baird JD. Chem.-Eur. J. 2006;12:4954–4963. doi: 10.1002/chem.200600034. [DOI] [PubMed] [Google Scholar]
- 45.Karlsson O. Synth. Commun. 1981;11:29–34. [Google Scholar]
- 46.Meth-Cohn O, Ashton M. Tetrahedron Lett. 2000;41:2749–2752. [Google Scholar]
- 47.Smith K, Barratt ML. J. Org. Chem. 2007;72:1031–1034. doi: 10.1021/jo062024f. [DOI] [PubMed] [Google Scholar]; Highly selective functionalization of 3-methylthiophene by DoM typically requires a hindered electrophile trap. Recently it has been shown that LiTMP metalates the 5-position of 3-methyl thiophene with high selectivity:
- 48.Lomas JS, Lacroix JC, Vaissermann J. J. Chem. Soc., Perkin Trans. 1999;2:2001–2010. [Google Scholar]
- 49.Espaze F, Hamon J, Hirbec H, Vignon J, Kamenka J-M. Eur. J. Med. Chem. 2000;35:323–331. doi: 10.1016/s0223-5234(00)00135-5. [DOI] [PubMed] [Google Scholar]
- 50.Miyasaka M, Rajca A. Synlett. 2004:177–181. [Google Scholar]
- 51.Pham Chiem V, Macomber RS, Mark HB, Jr., Zimmer H. J. Org. Chem. 1984;49:5250–3. [Google Scholar]
- 52.Frohlich H, Kalt W. J. Org. Chem. 1990;55:2993–2995. [Google Scholar]
- 53.Odom JD, Moore TF, Goetze R, Noeth H, Wrackmeyer B. J. Organomet. Chem. 1979;173:15–32. [Google Scholar]
- 54.Feng S, Maleczka RE, Jr., Smith MR., III manuscript in preparation. [Google Scholar]
- 55.Lawesson S-O. Arkiv foer Kemi. 1957;11:387–395. [Google Scholar]
- 56.Kuivila HG, Benjamin LE, Price AD, Polevy JH, Murphy CJ. J. Org. Chem. 1962;27:825–829. [Google Scholar]
- 57.Szumigala RH, Devine PN, Gauthier DR, Volante RP. J. Org. Chem. 2004;69:566–569. doi: 10.1021/jo035184p. [DOI] [PubMed] [Google Scholar]
- 58.Carreno MC, Ruano JLG, Sanz G, Toledo MA, Urbano A. J. Org. Chem. 1995;60:5328–5331. [Google Scholar]
- 59.Gallant M, Lachance N, Labelle M, Zamboni R, Juteau H, Gareau Y, Lacombe P. PCT Int. Appl.: WO 2001081312. 2001. p. 77. [Google Scholar]
- 60.Talley JJ, Bertenshaw SR, Carter JS, Collins PW, Docter S, Graneto MJ, Isakson PC, Lee LF, Malecha JW, Miyashiro JM, Penning TD, Rogers RS, Rogier DJ, Yu SS, Anderson GD, Burton EG, Cogburn JN, Gregory SA, Koboldt CM, Perkins WE, Seibert K, Veehuizen AW, Zhang YY. Actualites de Chimie Therapeutique. 1999;25:123–134. [Google Scholar]
- 61.Parham WE, Nicholson I, Traynelis VJ. J. Am. Chem. Soc. 1956;78:850–4. [Google Scholar]
- 62.Almutairi A, Yoon K, Tham F, Marsella MJ. Pure Appl. Chem. 2006;78:777–781. [Google Scholar]
- 63.Sudarsanam V, Giordano A, Vasu KK, Thakar HM, Giri RS, Yerande SG, Inamdar GS. PCT Int. Appl: WO 2007118149. 2007. p. 172. [Google Scholar]
- 64.Pillai AD, Rathod PD, Xavier FP, Vasu KK, Padh H, Sudarsanam V. Biorg. Med. Chem. 2004;12:4667–4671. doi: 10.1016/j.bmc.2004.06.028. [DOI] [PubMed] [Google Scholar]
- 65.Merola JS, Kacmarcik RT. Organometallics. 1989;8:778–784. [Google Scholar]
- 66.Uson R, Oro LA, Cabeza JA. Inorg. Synth. 1985;23:126–30. [Google Scholar]
- 67.Barton TJ, Hussmann GP. J. Org. Chem. 1985;50:5881–5882. [Google Scholar]
- 68.Liska R. Heterocycles. 2001;55:1475–1486. [Google Scholar]
- 69.Wu XM, Rieke RD. J. Org. Chem. 1995;60:6658–6659. [Google Scholar]
- 70.Wu RL, Schumm JS, Pearson DL, Tour JM. J. Org. Chem. 1996;61:6906–6921. doi: 10.1021/jo960897b. [DOI] [PubMed] [Google Scholar]