

Abstract

Ir-catalyzed C–H borylation is found to be compatible with Boc protecting groups. Thus, pyrroles, indoles, and azaindoles can be selectively functionalized at C–H positions β to N. The Boc group can be removed on thermolysis or left intact during subsequent transformations.
Ir-catalyzed borylation of C–H bonds is emerging as a new methodology for functionalizing aromatic and heteroaromatic hydrocarbons.1 For aromatic substrates, steric effects dictate the regioselectivity, giving access to regiochemistry that is difficult to obtain using traditional synthetic methods. While for heterocyclic substrates, the origins of regioselectivity are less apparent, it has been shown borylation of pyrroles and indoles occurs adjacent to the heteroatom (Chart 1).1
Chart 1.
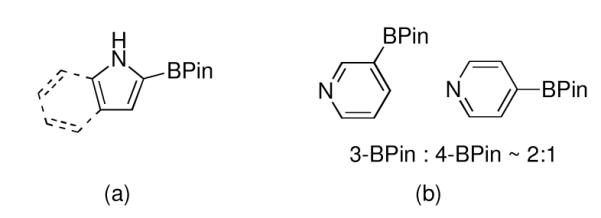
Borylation regioselectivities for (a) unprotected pyrrole, indole, and (b) pyridine.
We had previously shown that the borylation regioselectivity for pyrrole can be shifted to the 3-position if the nitrogen is protected with a triisopropysilyl (TIPS) group.2 Removal of the TIPS group provided a regioisomer that is the complement to the product obtained from borylation of unprotected pyrrole. Unfortunately, trimethylsilyl protection, the more economical alternative, was impractical as the N–SiMe3 bond is prone to hydrolysis. For general synthetic utility, we sought an economical, robust, yet readily removable protecting group to impart regioselectivity that TIPS protection provided. The compatibility of amides in aromatic borylations suggested that tert-butoxycarbonyl (Boc) protecting groups might be inert to the borylation conditions. Indeed, Gaunt and coworkers borylated N-Boc-pyrrole under microwave conditions3a and Shibasaki described the borylation of an N-Boc protected aniline.3b We sought to establish Boc compatibility beyond these limited examples, including in the Ir-catalyzed borylation of a Boc protected amino acid.4 Furthermore, we wanted to establish Boc removal protocols that would leave the boryl intact.
N–Boc-pyrrole was a logical starting substrate for comparing Boc and TIPS protecting groups. We were pleased to find that borylation proceeded smoothly with effectively complete regioselectivity for the 3-position (Table 1, entry 1) The yields are reproducible and scale reasonably well. For example, 100 g of the pyrrole and 1.25 equiv of pinacolborane (HBPin) afford product in 85% yield using an Ir catalyst loading of 0.5 mol%.
Table 1.
Borylation of N-Boc protected heterocycles and L-tryptophan.a
| entry | product | conditions, yield |
entry | product | conditions, yield |
|---|---|---|---|---|---|
| 1 |

|
THF, 55 °C, 13 h, 90% |
7c |

|
THF, 55 °C, 20 h, 14% |
| 2 |

|
THF 60 °C, 6 h, 82% |
8 |
|
--d |
| 3 |

|
n-hexane, rt, 5 h, 75% |
9e |
|
Et2O, rt, 65 h, 82% |
| 4 |
|
n-hexane, 60 °C, 8 h, 65% |
10 |

|
n-pentane, rt, 1.5 h, 76% |
| 5 |
|
n-hexane, rt, 5 h, 56% |
11e,f |
|
MTBE, rt, 45 min, 43% |
| 6b,c |

|
n-hexane, rt, 96 h, 54% |
See Supporting Information for details.
3.5 equiv HBPin used.
3.0 mol% [Ir(OMe)(COD)]2, 6.0 mol% dtbpy used.
Approximately 90% conversion achieved, but the product decomposed on attempted isolation.
1.0 equiv B2Pin2 used as the borylating agent.
31% of starting material recovered.
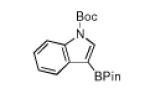
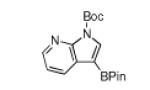
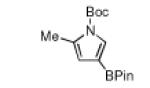
N-Boc compatibility is reasonably general as indicated by the other entries in Table 1. In addition to substituted pyrrole (entries 2 and 3), N-Boc-indole (entry 4) and N-Boc-7-azaindole (entry 5) afford acceptable yields of 3-borylated products. The outcome for N-Boc-7-azaindole reflects a preference for the 3-position of a 5-membered nitrogen heterocycle over sterically accessible sites in the 6-membered N-heterocyclic moiety. A second borylation of N-Boc-7-azaindole proceeds selectively at the 5-position (entry 6), presumably because C5 is less hindered than C4.4
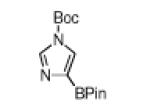
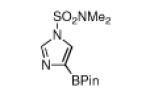
The yield for N-Boc-6-azaindole was low and the N-Boc-imidazole reacted slowly (entry 8). In the latter case, rate diminution from N3 coordination to Ir is compounded by that fact that borylations adjacent to sp2-hybridized N are difficult. For N-Boc-imidazole, extensive decomposition occurred on workup. A stable imidazole analog can be isolated in good yield if the more robust dimethylsulfonamide protecting group is used (entry 9). Entry 10 shows that N-Boc pyrazole affords the 4-borylated product, whereas borylation of N-methyl pyrazole gives the 5-borylated isomer as the major species.5
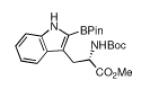
N-Boc amino acids are a very important class of Boc-protected compounds for consideration. As shown in Table 1, the indole nucleus of protected tryptophan can be monoborylated. While preparation of the monoborylated compound (entry 11) was complicated by competing diborylation, the pure monoborylated compound could be obtained with no loss of stereochemistry in 43% yield with 31 % of the starting material recovered. Stereospecificity was assayed by borylating d and l isomers of N-Boc tryptophan methyl ester in separate experiments. In both cases the opposite enantiomer was not detected by chiral HPLC analysis. Of the compounds in Table 1, those in entries 1 and 6 have been described in the primary literature, entries 4, 5, 7, 9 and 10 appear in patents, and entries 2, 3, 8, and 11 are new.6 To the best of our knowledge this entry represents the first example of protected α-amino acid C-H Borylation.
We, and others, have developed one-pot processes where Ir-catalyzed borylations are followed by one or more chemical transformations.1a,c-f To assess the potential for using the N-Boc protected substrates in one-pot processes, the borylation of N-Boc pyrrole was followed by the removal of volatiles. Without further purification, the cross-coupling of 1 with 3-chlorothiophene, afforded biheterocycle 2 in 76% isolated yield after 48 h (Scheme 1). Several aspects of this sequence deserve further comment. Compound 2 was previously prepared by Buchwald and Billingsley, whose coupling of purified 1 with 3-chlorothiophene afforded 2 in 51% yield after 12 hours.7 While our one-pot yield of 21% after 12 h was considerably lower than 51%, the real advantage of the C–H borylation/cross-coupling approach is that 1 is prepared in a single step from N-Boc pyrrole. In contrast, synthesis of 1 from pyrrole using standard chemistry requires multiple steps and protective group swapping.7 As such, the total synthesis of 2 from pyrrole via conventional methods proceeds in only 15% overall yield. On the other hand, the one-pot C–H borylation/Suzuki based approach allows for the transformation of pyrrole to 2 in far fewer steps and 72% overall yield.8
Scheme 1.
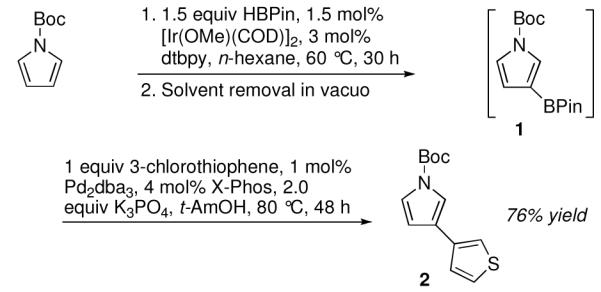
One-pot borylation/C–C crosscoupling of N-Boc pyrrole with 3-chlorothiophene.
Of course a protective group must be removable. In this regard, Boc group deprotections that leave the C–B bond of borylated heterocycles intact are unprecedented. Thus, deprotection of 1 was investigated. Attempts to remove the Boc-group with HCl or TFA resulted in unidentifiable decomposition products, whereas TBAF was ineffective in deprotection.9 Treatment with NaOMe yielded 42% of the desired product on 0.5 mmol scale, but at 4.0 mmol yields varied significantly. In contrast, the Boc groups of 1 and all other heterocycles (except the azaindole products) could be cleaved thermally (Table 2) at scales up to 10 mmol. This reagent free deprotection is not only economical but is also in keeping with principles of green chemistry 1 and 8, namely preventing waste and avoiding solvent use.10 Significantly, the products in Table 2 are regioisomers of the compounds obtained by borylating the unprotected heterocycles. Of the compounds in Table 2, those in entries 1, 4 and 5 have been described in the primary literature, and entries 2 and 3 are new.6
Table 2.
| entry | substrate | Conditions | product | % yield |
|---|---|---|---|---|
| 1 |

|
180 °C, 35 min |
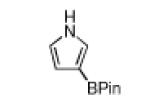
|
80 |
| 2 |
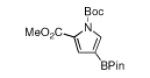
|
180 °C, 18 min |
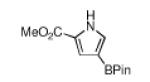
|
76 |
| 3 |
|
140 °C, 16 h |

|
72 |
| 4 |
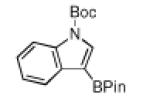
|
180 °C, 45 min |
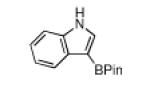
|
64 |
| 5 |
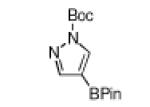
|
180 °C, 5 min |
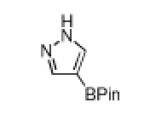
|
72 |
N-Boc protected substrates were placed in a flask and heated in air.
In summary, compatibility with Boc protecting groups allows for manipulating the regioselectivities for Ir-catalyzed borylations of nitrogen heterocycles. In addition, Ir-catalyzed borylations of protected tryptophan is shown to be feasible for the first time, which augurs favorably for similar functionalizations of peptides. Importantly, this work also establishes heat as a clean agent for Boc deprotection of BPin substituted heteroarenes.
Experimental Section
General Procedure for C-H Borylation
The reaction was set up in a glove box, where a Schlenk flask, equipped with a magnetic stirring bar, was charged with the corresponding substrate (1 mmol, 1 equiv). Two separate test tubes were charged with [Ir(OMe)(COD)]2 (10 mg, 0.015 mmol, 3 mol % Ir) and dtbpy (8 mg, 0.03 mmol, 3 mol %). Excess HBPin (1.1 to 2 equiv) was added to the [Ir(OMe)(COD)]2 containing test tube. Solvent (1 mL) was added to the dtbpy containing test tube in order to dissolve the dtbpy. The dtbpy solution was then mixed with the [Ir(OMe)(COD)]2 and HBPin mixture. After mixing for one minute, the resulting solution was transferred to the Schlenk flask. Additional solvent (2 × 1 mL) was used to wash the test tubes and the washings were transferred to the Schlenk flask. The flask was stoppered, brought out of the glove box, and attached to a Schlenk line in a fume hood. The Schlenk flask was placed under N2 and the reaction was carried out at the specified temperature. The reaction was monitored by GC FID/MS. After completion of the reaction, the volatile materials were removed on a rotary evaporator. The crude material was purified by column chromatography or dissolved in CH2Cl2 and passed through a plug of silica. Evaporation of solvent afforded the product.
N-Boc-3-(4,4,5,5-tetramethyl-1,3,2-dioxaboryl)pyrrole
White solid in 90% yield. 1H NMR (CDCl3, 500 MHz): δ 7.61 (t, J = 1.7 Hz, 1 H), 7.23 (dd, J = 3.2, 2.1 Hz, 1H), 6.44 (dd, J = 3.2, 1.5 Hz, 1 H), 1.56 (br s, 9 H), 1.30 (br s, 12 H); 13C NMR {1H} (CDCl3, 125 MHz): δ 148.6 (C=O), 128.8 (CH), 120.7 (CH), 116.2 (CH), 83.8 (C), 83.3 (C), 28.0 (3 CH3 of tBu), 24.8 (4 CH3 of BPin); 11B NMR (CDCl3, 96 MHz): δ 30.2; FT-IR (neat) Ṽmax: 3150, 2980, 2934, 1748, 1563, 1491, 1372, 1329, 1292, 1217, 1183, 1144, 1067, 976, 936, 857, 775, 691 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 293 (13), 237 (55), 194 (39), 193 (35), 178 (76), 107 (100), 57 (14); Anal. Calc’d for C15H24BNO4: C, 61.45; H, 8.25; N, 4.78. Found: C, 61.68; H, 8.53; N, 4.70.
General Procedure for Boc Deprotection
A Schlenk flask, equipped with a magnetic stirring bar, was charged with the substrate and heated in air at specified temperature until bubbling ceased. The crude material was dissolved in CH2Cl2 and passed through a plug of silica. Evaporation of solvent afforded the product.
3-(4,4,5,5-tetramethyl-1,3,2-dioxaboryl)-pyrrole
White solid in 80% yield. 1H NMR (CDCl3, 500 MHz): δ 8.61 (br s, 1 H), 7.23 (ddd, J =1.5, 1.7, 2.7 Hz, 1 H), 6.82 (dd, J =1.7, 2.5 Hz, 1 H), 6.55 (ddd, J =1.5, 2.5, 2.6 Hz, 1 H), 1.31 (br s, 12 H); 13C NMR {1H} (CDCl3, 125 MHz): δ 127.0 (CH), 118.6 (CH), 113.8 (CH), 82.9 (C), 24.8 (4 CH3 of BPin); 11B NMR (CDCl3, 96 MHz): δ 30.6; FT-IR (neat) Ṽmax: 3372, 3121, 2980, 2930, 1549, 1495, 1429, 1418, 1383, 1371, 1318, 1291, 1165, 1140, 1107, 966, 930, 860, 737, 691, 592 cm−1; GC-MS (EI) m/z (% relative intensity): M+ 193 (100), 178 (20), 150 (9), 107 (21); Anal. Calc’d for C10H16BNO2: C, 62.22; H, 8.35; N, 7.26. Found: C, 62.46; H, 8.35; N, 7.35.
Supplementary Material
Acknowledgement
We thank BASF, Inc. for gifts of HBPin and B2Pin2, and the Michigan Economic Development Corp., the NIH (GM63188 to M.R.S.), the ACS Green Chemistry Institute’s Pharmaceutical Roundtable, and Pfizer for kind financial support.
Footnotes
Supporting Information Available: Spectral data for all new compounds, as well as general experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org
References
- (1) (a).Cho JY, Tse MK, Holmes D, Maleczka RE, Jr., Smith MR., III Science. 2002;295:305–308. doi: 10.1126/science.1067074. [DOI] [PubMed] [Google Scholar]; (b) Ishiyama T, Takagi J, Hartwig JF, Miyaura N. Angew. Chem. Int. Ed. 2002;41:3056–3058. doi: 10.1002/1521-3773(20020816)41:16<3056::AID-ANIE3056>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]; (c) Ishiyama T, Takagi J, Yonekawa Y, Hartwig JF, Miyaura N. Adv. Synth. Catal. 2003;345:1103–1106. [Google Scholar]; (d) Maleczka RE, Jr., Shi F, Holmes D, Smith MR., III J. Am. Chem. Soc. 2003;125:7792–7793. doi: 10.1021/ja0349857. [DOI] [PubMed] [Google Scholar]; (e) Mkhalid IAI, Coventry DN, Albesa-Jove D, Batsanov AS, Howard JAK, Perutz RN, Marder TB. Angew. Chem. Int. Ed. 2006;45:489–491. doi: 10.1002/anie.200503047. [DOI] [PubMed] [Google Scholar]; (f) Kikuchi T, Nobuta Y, Umeda J, Yamamoto Y, Ishiyama T, Miyaura N. Tetrahedron. 2008;64:4967–4971. [Google Scholar]
- (2).Tse MK, Cho JY, Smith MR., III Org. Lett. 2001;3:2831–2833. doi: 10.1021/ol0162668. [DOI] [PubMed] [Google Scholar]
- (3) (a).Beck EM, Hatley R, Gaunt MJ. Angew. Chem. Int. Ed. 2008;47:3004–3007. doi: 10.1002/anie.200705005. [DOI] [PubMed] [Google Scholar]; (b) Tomita D, Yamatsugu K, Kanai M, Shibasaki M. J. Am. Chem. Soc. 2009;131:6946–6948. doi: 10.1021/ja901995a. [DOI] [PubMed] [Google Scholar]
- (4).After this manuscript was prepared Marder and Steel reported microwave-accelerated iridium-catalyzed borylations of two Boc-protected heterocycles and one example of a one-pot cross-coupling reaction: Harrisson P, Morris J, Marder TB, Steel PG. Org. Lett. 2009;11:3586–3589. doi: 10.1021/ol901306m.
- (5).Smith MR, III, Maleczka RE, Jr., Kallepalli V, Onyeozili E. 2008-0091027 U.S. Patent Application. 2008 Apr 17;
- (6).As per a search of the CAS database with SciFinder Scholar™ on October 8, 2009.
- (7).Billingsley K, Buchwald SL. J. Am. Chem. Soc. 2007;129:3358–3366. doi: 10.1021/ja068577p. [DOI] [PubMed] [Google Scholar]
- (8).N-Boc pyrrole is prepared in 95% yield from pyrrole: Salman H, Abraham Y, Tal S, Meltzman S, Kapon M, Tessler N, Speiser S, Eichen Y. Eur. J. Org. Chem. 2005:2207–2212.
- (9).Greene TW, Wuts PGM. Protective Groups in Organic Synthesis. 3rd ed John Wiley & Sons, Inc.; New York: 1999. pp. 520–522.pp. 618 [Google Scholar]
- (10). [accessed Aug 2009]. http://www.epa.gov/greenchemistry/pubs/principles.html.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.