

Abstract
We describe herein the hydrogen-atom transfer (HAT)/ proton-coupled electron-transfer (PCET) reactivity for FeIV-oxo and FeIII-oxo complexes (1–4) that activate C-H, N-H, and O-H bonds in 9,10 dihydroanthracene (S1), dimethylformamide (S2), 1,2 diphenylhydrazine (S3), p-methoxyphenol (S4), and 1,4-cyclohexadiene (S5). In 1–3, the iron is pentacoordinated by tris[N'-tert-butylureaylato)-N-ethylene]aminato ([H3buea]3−) or its derivatives. These complexes are basic, in the order 3 >> 1 > 2. Oxidant 4, [FeIVN4Py(O)]2+ (N4Py: N,N-bis(2-pyridylmethyl)-bis(2-pyridyl) methylamine), is the least basic oxidant. The DFT results match experimental trends and exhibit a mechanistic spectrum ranging from concerted HAT and PCET reactions to concerted-asynchronous proton transfer (PT) / electron transfer (ET) mechanisms, all the way to PT. The singly occupied orbital along the O---H---X (X= C, N, O) moiety in the TS shows clearly that in the PCET cases, the electron is transferred separately from the proton. The Bell-Evans-Polanyi principle does not account for the observed reactivity pattern, as evidenced by the scatter in the plot of calculated barrier vs. reactions driving forces. However, a plot of the deformation energy in the TS vs. the respective barrier provides a clear signature of the HAT/PCET dichotomy. Thus, in all C-H bond activations, the barrier derives from the deformation energy required to create the TS, whereas in N-H/O-H bond activations, the deformation energy is much larger than the corresponding barrier, indicating the presence of stabilizing interaction between the TS fragments. A valence bond model is used to link the observed results with the basicity/acidity of the reactants.
1. Introduction
Hydrogen abstraction (H-abstraction) is a fundamental process in Nature that is mediated mainly by heme- and nonheme-enzymes, which employ high-valent iron(IV)-oxo complexes to activate C-H as well as O-H and N-H bonds.1–4 With nonheme iron oxygenases as inspiration, many biomimetic nonheme iron(IV)-oxo and other metal-oxo complexes have been synthesized1b–f,5 and studied for their C-H/O-H/N-H bond activation capabilities.
It is abundantly found that C-H bond activation usually follows the Bell-Evans-Polanyi (BEP) principle6 and gets faster as the C-H bonds become weaker.7,8 However, this trend is not generally observed for O-H/N-H bonds in reactions with high-valent transition metal-oxo complexes.7a,9 For some iron(IV)-oxo reagents, H-abstraction from O-H bonds is either not observed or is sluggish. Examples include the [FeIVN4Py(O)]2+ (N4Py: N,N-bis(2-pyridylmethyl)-bis(2-pyridyl) methylamine) and [FeIVBnTPEN(O)]2+(BnTPEN:N-benzyl-N,N',N'-tris(2-pyridylmethyl)-1,2-diaminoethane) complexes10 and the related tetramethylcylam (TMC) containing complexes, [FeIV(O)TMC(Lax)]z+ (z=1,2), where Lax is an axial ligand of varying donor properties.11 However, in other cases such as a recently reported diiron(IV)-μ-oxo complex, the O-H bonds exhibit much higher reactivity than the C-H bonds with the same bond dissociation energy (BDE).12 Understanding why these reactivity patterns differ is important in order to gain a complete description of the reactivity of iron-oxo species with external substrates.
The differing reactivity of O-H and N-H bonds vs. C-H bonds of the same strength could be associated with the type of mechanism, as either H-atom transfer (HAT) or proton coupled electron transfer (PCET) processes are possible.13–16 A HAT process involves a transfer of a hydrogen atom from one atomic site to the other, as shown in Scheme 1a. On the other hand, when the proton and the electron are transferred to different locations as is often the case in hydrogen abstraction reactions by transition metal (TM) complexes (Scheme 1b), the process is generically called proton-coupled electron-transfer (PCET). However, this classification by itself still does not explain the different reactivity of iron-oxo complexes with C-H vs. O-H/N-H bonds. There must be other effects that make O-H/N-H bonds more susceptible to H-abstraction than C-H bonds of equal or lesser strength.
Scheme 1.
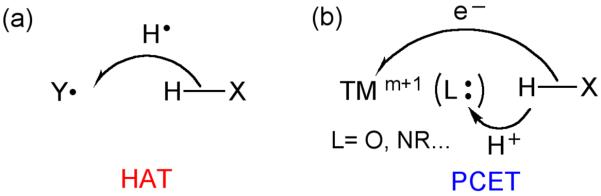
Generic representations of: (a) HAT where the H is transferred along with its electron, and (b) PCET, where the proton H+ and electron (e−) are transferred to different locations.
Because HAT and PCET are fundamental processes in chemical and biological oxidations,13–16 it is important to establish the underlying factors of this dichotomic reactivity in a manner that reveals their differences, as well as the dependence of their dichotomy on the nature of the iron-oxo reagent and the X-H bond. This is the general goal of the paper, which specifically addresses the reactivity of the iron-oxo complexes [FeIVN4Py(O)]2+ and those synthesized with the ligand tris[(N'-tert-butylureaylato)-N-ethylene]aminato ([H3buea]3−)17 towards different X-H bonds (Scheme 2).
Scheme 2.

(a) Iron-oxo species (1–4) and (b) substrates (S1–S5) studied for the hydrogen-abstraction reactions (c) involving C-H/N-H/O-H bonds. The abstracted H's are marked in red.
The targeted complexes 1–4 are shown in Scheme 2a while the substrates undergoing X-H bond activation (S1–S5) are depicted in Scheme 2b. The recently synthesized17a [FeIVH3buea(O)]− complex 1 belongs to a family of transition metal complexes in which the metal is coordinated by the tri-anionic ligand [H3buea]3−.17 As a result of the enforced trigonal symmetry, 1 has a high-spin (S = 2) ground state.17a,b This is important because non-heme iron enzymes with FeIVO intermediates have high spin (S=2) ground states, which is often invoked as the reason for their potent reactivity.18 However, the anionic [H3buea]3− ligand in 1 increases the basicity of the terminal-oxo ligand17a,f and protects it with weak intramolecular hydrogen bonds. Further protection is provided by the bulky tert-butyl substituents (But) which form a hydrophobic fence around the oxo moiety. Complex 2 utilizes a hybrid ligand5k containing one urea groups and two cyclopentylcarboxamide moiety and is included to compare the role of multiple H-bonds19 on the basicity and reactivity of 1. Complex 3 is the reduced form of 1; it has an Fe(III)-oxo moiety and is more basic than 1.17d,e Finally, 4 ([FeIVN4Py(O)]2+), is the least basic and has the least protected Fe(IV)-oxo moiety.10a,20
The double protection (But groups and H-bonds) lowers the reactivity of 1 and 3, which nevertheless exhibit some intriguing reactivity patterns (see Experimental Methods). At room temperature, 1 abstracts a hydrogen atom from 1,2-diphenyl hydrazine (DPH, S3) and experimental evidence suggests from dimethylformamide (DMF, S2); in both reactions the well-characterized [FeIIIH3buea(OH)]− species is generated.17a The reactivity of 1 towards S3 reflects the BEP principle6 because the activated N-H bond (BDE = 69 kcal/mol) is weaker than the OH bond that is formed (BDEOH ~ 87 kcal/mol).17cWith such a BDEOH value, it is surprising that 1 reacts so slowly with DHA (S1; BDECH = 77 kcal/mol) that a rate constant cannot be measured at room temperature, even with a 100-fold excess of S1 (see experimental Methods section).17c Since S1 may be too bulky to react with the protected FeO moiety of 1, we also studied S5 which is sterically less demanding and has a BDECH close to that in S1. Note that complex 3 has a much weaker FeO-H bond (BDEOH = 66(4) kcal/mol) than 1, yet it activates S1, S3, and S5, apparently by abstraction of H-atoms.17d,e However, its reactivity with S4 proceeds by proton abstraction. By analogy with the corresponding manganese complex,17g it was proposed that the reaction of 3 with S1 might be occurring by a preliminary proton abstraction step followed by electron transfer (hence stepwise PCET).17e Finally, 4 activates the strong C-H bond of cyclohexane20 but is unable to activate the O-H bond of MeOH.10
It is obvious that the compounds in Scheme 2 offer a sufficiently rich reactivity landscape, which needs to be organized into a comprehensible picture. We began our investigations by considering a recent study by Mayer-Borden and coworkers21,22 which revealed an interesting electronic effect in the self H-exchange reactions of benzyl and phenoxyl radicals. Thus, the H-abstraction by benzyl radical involves a transfer of a H-atom from one site to the other (Scheme 3a). The singly occupied orbital in the transition state is the usual σ-type nonbonding orbital (ϕσ−) with the node on the H in transit. In the reaction of phenoxyl radical with phenol, however, the electron is transferred via the π-type orbital (ϕπ−) that is perpendicular to the O---H---O axis (Scheme 3b) while the H is transferred as a proton between σ-orbitals. Moreover, for PhO•/PhOH, the `normal' HAT “transition state” in which ϕσ− is singly occupied was found to be approximately 5 kcal/mol higher in energy. In contrast, the H-atom transfer between methoxyl radicals (CH3O•/CH3OH) was shown to follow the HAT pathway with the PCET-type species being approximately 5 kcal/mol higher in energy. Thus, the singly occupied ϕπ− orbital may serve as a signature for identifying PCET processes.
Scheme 3.
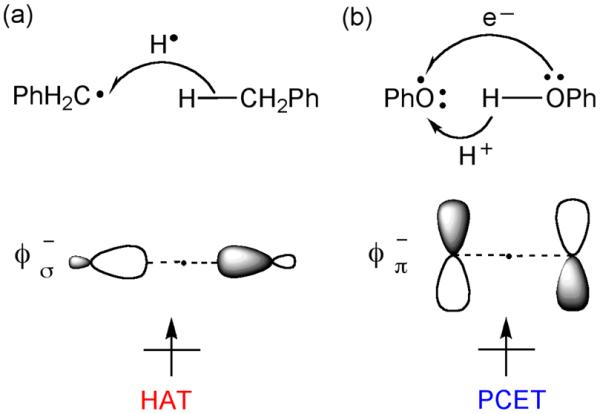
The singly occupied orbitals during H-abstraction in: (a) a normal HAT reaction, PhCH2• + PhCH3 → PhCH3 + PhCH2•, and (b) PCET reaction, PhO• + PhOH → PhOH + PhO•
We recently extended this finding of different electronic structures for PCET and HAT processes to a variety of identity reactions.14b Those that involved C-H bonds followed the HAT electronic structure (Scheme 3a) while those that involved MO-H bonds (M – transition metal) revealed a PCET electronic structure (Scheme 3b). In the tested nonidentity reactions, this orbital-based signature was not found.14b Nevertheless, we showed by usage of valence bond (VB) modeling that the VB structures of the HAT- and PCET-types mix to generate transition states with blended HAT-PCET characters, which stabilized the corresponding transition states.14 Using similar computational approaches,16,23 Gao-Wu and their coworkers arrived at a similar conclusion and showed that the mixing of the HAT and PCET type structures account also for the nonadiabatic features of PCET processes.15 Thus, the combination of the VB analysis and the orbital signature of HAT/PCET may serve as a useful approach for a broader understanding of the problem.
Another aspect of the HAT/PCET dichotomy is mechanistic. As shown in Scheme 4, the H-abstraction can occur via a concerted process in which both the electron and the proton are transferred in a single step, leading to pure HAT or a HAT/PCET blend. The process can alternatively transpire in a stepwise mechanism with distinct proton transfer (PT) and electron transfer (ET) steps. This mechanistic aspect and its underlying factors is another target in the present study.
Scheme 4.
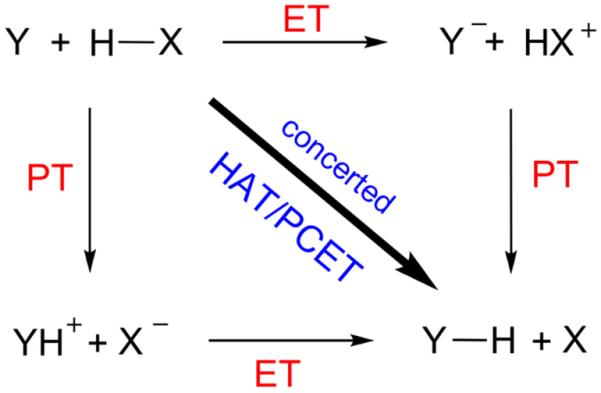
The mechanistic spectrum of HAT and PCET.
We will demonstrate that the reactivity patterns in the series of complexes investigated (Scheme 2) are linked to the HAT/PCET dichotomy, which normally typifies C-H vs. O-H/N-H bond activations.14–16 However, the PCET propensity is highly amplified by the basic nature of 1–3 such that 1 and 2 show marked propensity for PCET while 3 exhibits the stepwise PT/ET mechanism even with the C-H bond of S1. It will be further shown that the HAT/PCET reactivity patterns can be distinguished in two ways: (1) by the nature of the singly occupied orbital in the H-abstraction transition state (TS)21 and (2) by a plot of the deformation energy vs. the reaction barrier24 using the energy decomposition analysis approach to reactivity.25,26 Furthermore, VB conceptualization of the deformation energy criterion provides a global behavior that is common to many reactions involving H-atom abstraction by iron-oxo complexes14a and can serve as a physically-based classifier of the mechanistic dichotomy.
Methods
A. Experimental Properties of 1 and its Reactivity Towards S1
We have reported that the redox potential for the FeIV(O)/FeIII(O) couple, is −0.90 V vs. [FeCp2]−/0.17c Using this result in conjunction with other thermochemical data for the series of Fe-oxo and Fe-hydroxo complexes, we have constructed the thermodynamic square scheme depicted in Figure 1. Note that BDEOH is 87 kcal/mol for [FeIIIH3buea(OH)]− rather than the 102 kcal/mol value reported previously.17e We have shown previously that 1 reacts with S3 at room temperature.17a By contrast, we now report that the reactivity of 1 towards S1 is relatively slow. For instance, even with a 100-fold excess of S1, at room temperature, 1 reacts so slowly with S1 in DMSO that a rate constant could not be measured.17c
Figure 1.
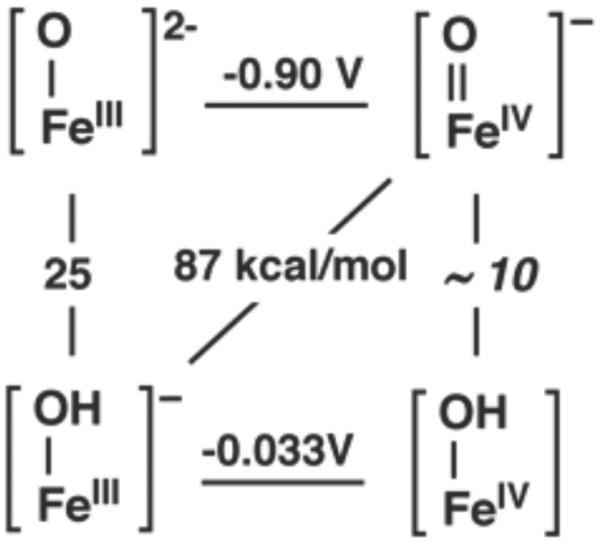
Square scheme of the thermodynamic cycle for [FeIVH3buea(O)]−. The horizontal lines represent electron transfer (V vs [FeCp2]+/0), the vertical lines represent proton transfer (pKa), and the diagonal line represents the hydroxo bond dissociation energy (BDEOH) in kcal/mol.
B. Theoretical Approach and Methods
Usage of Counter Ions During Calculations
To minimize self-interaction errors27,28 or other spurious effects caused by the anionic charges of 1–3, we neutralized the charges by adding K+ counter ions. The dipositive charge of 4 was neutralized using two perchlorate anions (ClO4)−.28 The counter ions were positioned as found in the crystal structures of 1, 317d and 4.29 During geometry optimization and throughout the reaction path, the position of the counter ions essentially remained constant.
The “Standard Computational Scheme”
We employed density functional theoretical (DFT) calculations using the B3LYP functional30 for H-abstraction reactivity of oxidant 1 with substrates S1–S5, oxidants 2 and 3 with S1 and S3–S5, and oxidant 4 with S1, S4 and S5 (Scheme 2a and 2b).
Geometries and frequencies were generally computed using the LACVP(Fe,K)/6–31G(rest) basis set (B1).31 The connection of transition states (TSs) to reactants and products was verified by intrinsic reaction coordinate (IRC) using Gaussian 03.32 The B1 energies were corrected with LACV3P+*(Fe,K)/6–311+G*(rest), hence B2,31 along with solvation corrections (ε =47.24 for 1–3 with S1 and S3–S5, ε =36.70 for 1 with S2, and ε=37.5 for 4 with S1, S4 and S5) using Poisson-Boltzmann solver33 as implemented in Jaguar 7.6.34 Thermal and entropic corrections at T = 25°C from the corresponding frequency calculations were used to augment the B2+solv energies. Empirical dispersion correction for all of the stationary points was done using DFT-D335 with zero damping.
The above standard methodology was tested in a variety of ways (see below), which revealed the robustness of the trends. The various tests and their results are summarized in the supporting information (SI) document (Tables S1–S12), while the discussion here shows only the free energies based on UB3LYP/B2 with solvation, which represents all other data sets.
Tests of Basis Sets, Functionals, Solvation models, and Dispersion models
The sensitivity of the HAT/PCET classification was tested as follows (See Table S12):
-
(a)
The effect of basis sets on the barriers: Single point calculations on B1 geometries were carried out with the large all electron basis set, Def2-TZVP36(B3), using Gaussian 09.37 The UB3LYP/B3//UB3LYP/B1 trends (Table S12a) were identical to those of UB3LYP/B2//UB3LYP/B1.
-
(b)
The effect of solvation scheme: Using the SMD solvation model,38 implemented in Gaussian 09,37 showed that the trends (see Table S12b) in the barrier heights are identical to those obtained with the “standard scheme”.
-
(c)
The effect of density functionals: Single point calculations with the B2 basis set using mPW1K,39 PBE0,40 M06-L41and M0642 showed that the trends for barrier heights were identical to those obtained with the UB3LYP/B2+Solv level (see Table S12c).
-
(d)
The steric effect of But groups on reactivity of substrates was tested by generating a model of oxidant 3 (denoted 3') in which the But groups were replaced with hydrogen atoms. 3' and 3 gave similar features and reactivity trends, and hence the data for 3' were relegated to the SI (Figure S7 and S8).
-
(e)
The effect of the geometry optimization level for the reactions of the substrates bearing the N-H and O-H bonds, S3 and S4, was probed using three different basis sets; (i) the Stuttgart ECP and basis set on Fe,43,44 ECP10MDF(Fe)/6–31G(rest), labeled B4 (ii) LACVP*(Fe)/6–31G*(rest) labeled B5, and (iii) LACVP**(Fe)/6–31G**(rest) labeled B6. For 1 with S3, we employed M06L/B4 geometry optimization, and for 2 with S4, we used B3LYP with B5 and B6 geometry optimizations. The resulting barriers and geometrical parameters (Figure S12) matched those based on B1 optimization.
Identification of HAT/PCET Characters
We found that Spin-Natural Orbitals (SNOs) are useful for characterizing the HAT/PCET features of a given TS.24,28 Spin densities and natural bond order (NBO) charges of the optimized structures at B2+Solv level (Table S14–S19) were also analyzed for the identification of the electronic states of stationary points.
Results
All data is reported in the SI document, while here we present only the free energies based on UB3LYP/B2 with solvation.
A. Oxidants, Reaction Barriers, and Transition State Features
Oxidant 1–4
Figure 2 shows key geometric and spin-density data for the iron-oxo complexes 1–4 in the lowest spin states. It is seen that 1 has an S = 2 ground state, while for 3 the ground state is S = 5/2. These results are in accord with the reported EPR and DFT studies for these species.17a,b,d,f2 is analogous to 1 and has a ground state of S = 2, as expected from Fe(IV)-oxo complexes with trigonal bipyramidal structures.18f–g;45a By comparison, 4 has the expected S = 1 ground state with a closely lying S = 2 state.20,24,28,29 The computed geometries for 1, 3, and 4 are in good accord with experiment. The computed spin density on the oxo ligand, ρO, for 1 is smaller than experiment.17b
Figure 2.

Computed and experimental (Expt)17a,b,d;29 geometric parameters (bond length in Å and angle in °) and relative free energies including solvation correction (ΔG in kcal/mol) of the lowest spin states for 1–4. The pink spheres in 1–3 represent the K+ ions. ρO is the spin density on the oxo ligand in S = 2;1 for 1, 2, 4 and in S = 5/2;3/2 for 3.
From inspection of the spin density on the oxo ligand, ρO (Figure 2), it is clear that 1 and 2 in an S = 2 state possess smaller oxo-spin densities compared to 4 in the same spin state.17b The oxo spin density of 3 is also small. Small oxo-spin density reflects the greater ionicity of the respective Fe-O bond, and is hence related to the increased basicity of this iron-oxo reagent. Interestingly, 2, which possesses fewer H-bonds to the oxo ligand than 1, also has a higher spin density, which means that the H-bonds increase the basicity of the reagents.17b Finally, all of the complexes with intramolecular H-bonds are more basic compared to 4, and the basicity increases in the order 3 >> 1 > 2 > 4.
Reactivity Patterns of 1–4 with S1–S5
Figure 3 shows generic reaction energy profiles of H-atom abstraction of 1–4. Note that 1–3 (Figure 3a) abstract hydrogen within a single spin state, which is the ground state (S = 2 for 1 and 2, and S = 5/2 for 3). On the other hand, 4 (Figure 3b) performs H-atom abstraction using two-state reactivity (TSR). TSR was demonstrated before24,28 by showing that the lowest energy TS arises from a spin crossover from an S = 1 ground state to S = 2 as the reaction begins.
Figure 3.
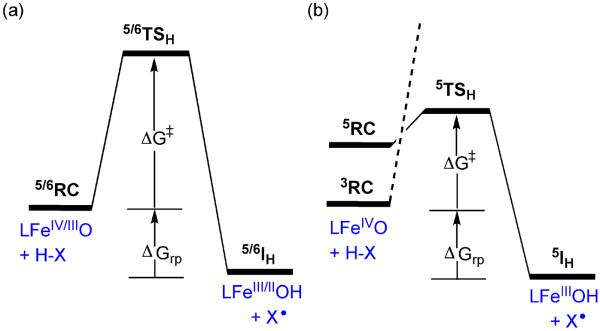
Generic energy profiles, starting from the reactant complex (RC). ΔG‡ is the free energy barrier and ΔGrp is the thermodynamic driving force of the reaction. (a) LFeIV/IIIO (of 1–3 in their ground spin states) + H-X to the intermediates LFeIII/IIOH + X•. (b) LFeIVO of 4, which involves TSR (the dashed line signifies that the S=1 process correlates to a high energy 3TSH species24,28)
Table 1 summarizes the free-energy barriers obtained for the oxidants reacting with the various substrates. For the most part, the data in Table 1 agree with the experimental results. Thus, the largest barrier found for 1 involves S1, which was also found experimentally to be non-reactive.17c In contrast, the barrier is much lower for S2 and S3 than S1, which agrees with our experimental findings that S2 and S3 react with 1.17a Between the two C-H bonds of S2, the more reactive is the C-Hformyl bond, which is also more acidic than the C-H bond of the N-CH3 moiety. Similar trends are obtained for 2 reacting with S1–S5. Compared with 1, the barriers of 2 are significantly smaller for C-H bond activation (with S1 and S5) while the barriers for N-H and O-H bond activation remain small and are less affected. Thus, the decreased basicity is beneficial primarily for C-H activation and less so for O-H/N-H activations.
Table 1.
Computed free energy barriers (ΔG‡, kcal/mol)a for H-abstraction reactions of oxidants 1–4 with substrates S1–S5.
| Oxidant |
1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Substrate | ||||
| S1 | 26.1 | 21.1 | 23.9 | 8.8 |
| S2(CH3) | 17.7 | N/A | N/A | N/A |
| S2(CHformyl) | 12.2 | N/A | N/A | N/A |
| S3 | 8.5 | 7.6 | 11.7 | N/A |
| S4 | 7.4 | 6.1 | Barrier-free | 7.7 |
| S5 | 17.1 | 14.8 | 24.7 | 9.5 |
These are free energy barriers with solvation correction relative to the reactant clusters (RCs, Figure 3).
The highly basic reagent 317d,e reacts with S1 and S3–S5 at room temperature, as observed experimentally. With S4, the reaction proceeds in a barrier-free fashion by proton abstraction, which is in accord with experimental observation of PT reactivity with phenols.17e As we discuss later, the mechanism of C-H activation by 3 is never HAT (the same applies to 3', see SI, Figures S7 and S8).
Finally, the barriers in Table 1 reveal that 4 is the most potent oxidant with all the substrates tested, which is essentially in accord with experiment.20 However, these barriers are small and are determined mostly by the triplet-quintet energy separation in Figure 3b, while on the quintet state surface, the barrier is very small.24 All in all, the barrier data fits the experimental results.
Figure 4 shows a BEP plot6 that was constructed using the barrier data in Table 1 and the corresponding free energy quantities of the H-atom abstraction reaction, ΔGrp. Figure 4 shows basically a scatter (similar scatter plots are given in the SI for all other levels, see Figure S1). The lack of BEP correlation agrees with the experimental observation that O-H bonds (and presumably also N-H bonds) are more reactive in H-abstraction than C-H bonds.
Figure 4.

A BEP plot of barriers (ΔG‡) vs. the thermodynamic driving force of the reaction (ΔGrp) for the H-abstraction reactions of S1–S5 with oxidants 1–4.
Origins of the Barriers
To understand the patterns of the barriers in Table 1, we examined origins of the barrier (Scheme 5)25e,h,i using the energy decomposition analysis,25,26 according to which the H-atom abstraction barrier (ΔE‡) can be understood as a sum of two quantities in equation 1:
| (1a) |
| (1b) |
ΔEdef is the total deformation energy, which is the energy required for distorting the oxidant and the substrate to their geometries in the TS. ΔEint is the interaction energy between the so deformed reactants as they are brought to their distance(s) in the TS. The interaction energy (eq. 1b) can be stabilizing (ΔEint < 0, scheme 5a) because of the orbital mixing term, ΔEorb, and a favorable electrostatic interaction, ΔEel. Alternatively, ΔEint can be destabilizing (ΔEint > 0, Scheme 5b) because of steric repulsions (labeled in eq. 1b as ΔEPauli(steric)).
Scheme 5.
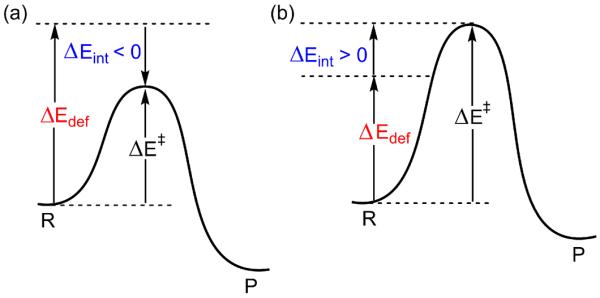
The relationship between the barrier ΔE‡, the deformation energy of reactants ΔEdef, and their interaction energy, ΔEint, at the TS: (a) ΔEint < 0, and (b) ΔEint > 0.
Figure 5a is a plot of ΔEdef vs. the corresponding gas-phase energy barriers, ΔE‡. Since the deformation energy is a gas phase quantity, we use the corresponding gas phase barriers while noting that the trends in the free energy barriers and the gas phase barriers are very similar (SI, Figure S2 and Tables S3–S11). The line in Figure 5 is drawn with a slope of unity such that points above it have ΔEdef > ΔE‡ while points below it have ΔEdef < ΔE‡.24 An informative picture emerges from this plot. The red squares show that when the oxidants are 1, 2, and/or 4 and the substrate undergoes C-H activation as in S1, S2, and/or S5, the ΔEdef values are close to the corresponding activation barriers, ΔEdef ≈ ΔE‡. In these cases, the barrier derives from the deformation energy required to establish the TS.24 The interaction energy, ΔEint, is not very significant.
Figure 5.

(a) A plot of sum of the deformation energies of the reactants in the TS (ΔEdef, kcal/mol) vs. the corresponding gas-phase barriers, ΔE‡ (kcal/mol), for the reactions of 1–4 with S1–S5. The line is drawn with a slope of unity such that the interaction energy between the reactants in the TS, ΔEint, is the vertical difference ΔEdef - ΔE‡. The red squares correspond to C-H activations while the blue squares correspond to N-H/O-H activations (except for 3 + S1 which involve C-H activation by 3). (b) An extended plot of ΔEdef vs. ΔE‡. The black circles correspond to C-H activation data from ref. 24, while the red and blue squares are the present data shown explicitly in Figure 5a. All of the points (black circles and red squares) adjacent to the line of unity slope correspond to C-H bond activations. By contrast, all of the blue squares correspond to N-H and O-H bond activations.
By contrast, for the N-H/O-H bond activations observed for S3 and S4 with 1, 2, 3, and 4, the deformation energies in Figure 5a are significantly larger than the barriers, thus revealing that in all of these cases, the corresponding TSs have very large stabilizing interactions which lower the barriers well below the corresponding deformation energies, i.e., ΔEint << 0 in equation 1a. The largest deviations are observed for the most basic iron-oxo complex, 3, while the smallest deviations are found for 4, which is the least basic. In fact, 3 exhibits the same large and negative ΔEint quantity even in the TS of S1, which reacts via C-H bond activation. As such, the comparison of the differences ΔEdef -ΔE‡ = ΔEint, for the entire set of reactions projects the dichotomy between the normal HAT reactions for S1, S2, and S5 visà-vis those for S3 and S4 and for the very basic iron-oxo 3 with S1.
Figure 5b shows an extended ΔEdef vs. ΔE‡ plot that combines the data of the present study (red and blue squares from Figure 5a) with all of the previously studied C-H activation data (black circles).24 A spectacular picture emerges from Figure 5b: all of the C-H bond activation reactions (red squares and black circles) cluster near the line for which ΔEdef = ΔE‡, whereas all of the N-H/O-H bond activations (and for 3 + S1 having TSPT) lie above the line and have ΔEdef >> ΔE‡ (blue squares). Thus, the dichotomy observed in Figure 5a is generally not limited to the chosen set of reactants. Understanding these features of the Edef/ΔE‡ maps in Figures 5a and 5b will elucidate the HAT/PCET dichotomy and the basic cause for the faster PCET processes of N-H and O-H bonds.
Transition State Features and Mechanistic Information
To complement the deformation-energy data in Figure 5a, we proceeded with structural data and other features for the respective TSs. Since we have many TS species, we show only representative ones, and the rest can be found in the supporting information (Figures S3–S11).
Figure 6 shows key geometric features and charge- and spin-distribution information on the TSs for C-H activation (Figure 6a) and for N-H/O-H activation (Figure 6b). Inspection of the TS geometries in Figure 6a shows that for C-H bond activation by 1 and 4, the TSs are quite early (small C-H cleavage and H-O bond making), which is in accord with the general findings that the respective deformation energies (Figure 5a) are relatively small and close to the corresponding barriers. In all of the cases (1+S1, 1+S5, and 4+S1), the charge on the H-abstracted moiety in the TS (QS-H) is small. This finding indicates that these TSs have dominant HAT characters, albeit with some PCET character, which is indicated by the QCT quantities that shows charge transfer from one reactant to the other.
Figure 6.

Schematic representations of bond activation TSs along with NBO charges (Q) and spin densities (ρ) on the H-abstracted moiety (S-H), the charge on the oxidant (Q1–4), and the amount of charge transferred (QCT) from the substrate to the oxidant or vice versa in the cases of (a) C-H activation (S1 and S5) and (b) N-H/O-H activation (S3 and S4).
The C-H activation TS for 3 + S1 is different than others in that it has a `late' TS species with a long C-H bond and a short H-O bond; this accords with the high deformation energy of this TS in Figure 5a. The charge on the H-abstracted moiety in the TS (QS-H) is −0.71 and the corresponding spin density is close to zero, thus making this moiety virtually a carbanion and the corresponding TS a proton transfer (PT) species, TSPT. This character was verified by following the IRC, which is displayed in Figure 7a. Thus, the reaction has a TSPT species while past the TSPT there is an electron transfer (ET) from the carbanionic moiety to the Fe(III)OH− moiety leading to the Fe(II)OH2−/X• intermediate (X• is the radical of S1-H). There is no proton abstraction intermediate and the entire process is concerted but asynchronous, having discrete PT and ET events (similar to Scheme 4 but not stepwise). The reaction 3 + S5 has some similar features but the PT character in the TS is smaller and the geometry is much earlier.
Figure 7.
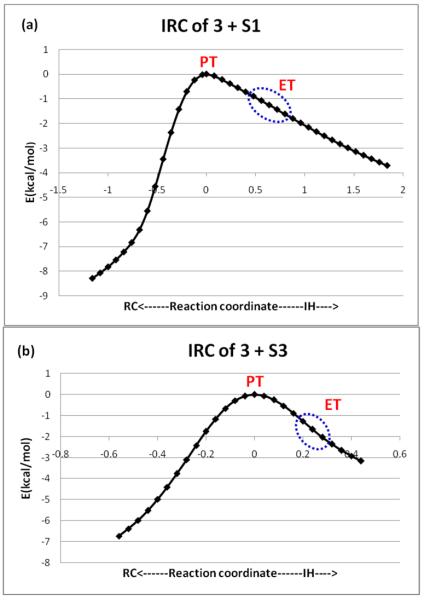
IRCs for: (a) 3 + S1, and (b) 3 + S3. The labels PT and ET indicate the nature of the species.
Inspection of the TS geometries for N-H/O-H bond activation by 1–4 (Figure 6b) reveals both similarities and differences compared to the geometries in Figure 6a. The reaction of the basic oxidant 3 with S3 has a high charge on the H-abstracted moiety in the TS (QS-H = −0.63) and has a TSPT species similar to the reaction of 3 + S1. Moreover, its IRC (Figure 7b) reveals a concerted but asynchronous H-abstraction, exhibiting discrete PT and ET events along the path. On the other hand, the reactions of 1 and 2 with S3 have low charges on the H-abstracted moiety in the TS (QS-H) but possess large degrees of charge transfer (QCT) from the substrate to the oxidant such that the oxidants acquire substantial negative charges. This finding means that the corresponding TS species have a high ET character and can be labeled as TSPCET. Furthermore, the reaction 4 + S4 has some intermediate character between PCET and a regular HAT while 1 + S4 has some PT character (QS-H = −0.37).
Characteristic Orbitals of the TSs for HAT, PCET, or PT/ET
Complementary and insightful information regarding the distinction between the HAT and PCET type TSs can be further obtained from the electronic structures of the TSs. Figures 8 and 9 show the singly occupied spin natural orbitals (SNOs) for the various transition states.
Figure 8.

SNOs in TSs: (a) ϕHAT type SNOs for 1+S1, 1+S5, 2+S1, and 4+S1, shown alongside the ϕHAT orbital in the TS (H3C---H---CH3)•. (b) ϕPCET type SNOs for 1+S3, 2+S3, and 1+S4. The hydrogen atoms not in O---H---X moiety are omitted for clarity.
Figure 9.
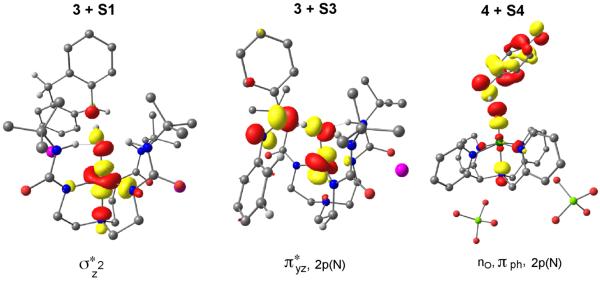
SNOs in TSs: ϕPT type SNOs for 3+S1, 3+S3, vs. the typical ϕHAT type in 4+S4. The hydrogen atoms not in O---H---X moiety are omitted for clarity.
Examination of Figure 8a shows that the C-H activation TSs for 1+S1, 1+S5, 2+S1, and 4+S1 possess a singly occupied SNO with a node on the H in transit between two σ-type lobes on the two heavy atoms O and C; this behavior is much like that in the prototypical SNO for the TS of methyl radical with methane. Thus, these TS species represent cases with a dominant HAT character. By contrast, the SNOs in Figure 8b look entirely different. We still see a σ-lobe on the oxo ligand of the FeO moiety. However, the contributing orbitals on the substrates S3 and S4 are π-type (or mix π-lone pair type) orbitals that are perpendicular to the σ-lobes of the FeO orbital. This is precisely as found by Borden and Mayer21 for the identity reaction of PhO•/PhOH (Scheme 3b) except that here, the electron is transferred between σ-and π-orbitals. This electronic structure feature shows that these are predominantly PCET TS species having some blended ET, PT, and HAT characters.
Finally, Figure 9 shows the singly occupied SNOs in the TSs of the H-abstraction from S1, S3, and S4 by the basic reagent 3. We find that the SNO in the TSPT species for 3+ S1 is largely a dz2 type orbital with hardly any contribution from the substrate. This SNO is identical to the SNO of the protonated 3/H+ reagent, which is in agreement with the PT nature of this TS species as discussed for Figure 6b. For 3+S3, the SNO in the TS has a mixed character with π-lone-pair type contributions on S3, thus having blended PT and ET character (or simply PCET). Finally, for 4+S4, the SNO looks almost like a HAT orbital with a node on the H-in transit and two σ-type lobes with opposite signs. This means that with 4 and related oxidants, O-H bond activation has a high degree of HAT character as in C-H activation.
Discussion
Since there are plenty of mechanistic details, it may be useful to usher the discussion by summarizing key results of the mechanistic spectrum, the electronic structure, and the reactivity patterns derived from the barrier-deformation energy maps.
A. Mechanistic Features and Electronic Structures
The Mechanistic Spectrum
Table 2 summarizes the mechanisms observed for all of the combinations of oxidants and substrates in Scheme 2, which are seen to exhibit a mechanistic manifold that depends largely on the acidity/basicity relationships of the oxidants and the X-H bonds of the substrates. We found concerted reactions having dominant HAT characters for the less basic oxidants 2 and 4 with the C-H bonds of S1 and S5 and for 1 reacting with the C-H bonds of S1, S2, and S5. However, the same oxidants give TSs with PCET characters in reactions with N-H/O-H bonds (of S3 and S4). With the most basic oxidant 3, we observe a TS with HAT and PT/ET characters for the C-H bond activation in S5. For the more acidic C-H bond of S1 and with the N-H bond of S3, we find that 3 reacts through a concerted asynchronous PT/ET mechanism, in which the TS involves PT followed by an ET en route to the H-abstraction product (Figure 7). Finally, a PT mechanism was characterized for the reaction of 3 with S4.
Table 2.
The Mechanistic Findings for H-abstraction Reactions of Substrates (S1 – S5) with Oxidants (1 – 4).
| Oxidant |
1 | 2 | 3 | 4 |
|---|---|---|---|---|
| Substrate | ||||
| S1 | HAT | HAT | concerted-asynchronous PT/ET | HAT |
| S2(CH3) | HAT | N/A | N/A | N/A |
| S2(CHformyl) | HAT | N/A | N/A | N/A |
| S3 | PCET (ET) | PCET (ET) | concerted-asynchronous PT/ET | N/A |
| S4 | PCET (PT) | PCET (PT) | barrier free PT | HAT with PT/ET characters |
| S5 | HAT | HAT | HAT with PT/ET character | HAT |
Electronic Structural Features During Activation of X-H Bonds by 1–4
The two key aspects that merit highlighting are 1) the electronic structure and appearance of HAT/PCET features in the d-block orbitals, and 2) the additional key orbital interactions that bring about ET and PT characters during the X-H bond activation. Scheme 6 summarizes the d-block electronic structures for the above mechanistic types.
Scheme 6.

Summaries of electronic structures for the transition states showing the d-block SNOs and the orbitals participating in the e-shift to the d shell: (a) FeIVO complexes (1, 2, 4) activating C-H bonds (left), (b) FeIVO complexes (1, 2) activating N-H/O-H bonds (right), (c) The FeIIIO complex (3) activating C-H or O-H bonds, and (d) the FeIIIO complex (3) activating N-H bond.
Schemes 6a and 6b depict the occupancy diagrams in the d-block orbitals of the TSs for the HAT and PCET mechanisms of X-H bond activation by the FeIVO complexes (with S=2 ground states) using the information obtained from the SNOs in Figure 8. For illustration, we use the orbital arrangement corresponding to the pentacoordinated complexes. Scheme 6a depicts the electronic structure for a TSHAT that is typically observed during C-H bond activation (e.g., 1, 2, 4 reacting with S1 and S5; and 4 with O-H in S4). The corresponding d-block possesses a half-filled shell that is stabilized by exchange enhancement relative to the reactant state of the FeIVO complex. In addition, there is a singly occupied orbital, denoted as ϕHAT, which corresponds to the orbital in a classical HAT mechanism that has a node on the H in transit, and this orbital is flanked by two σ-type lobes along the O---H---C axis (see Figure 8a). Underneath this orbital occupation diagram we show the electron-shift from σCH to σ*z2 that is responsible for the enrichment of the d-block by one spin-up electron in σ*z2.24,28,45b,46,47,48
During O-H and N-H activations by 1 and 2 (Scheme 6b), we obtain TSPCET species that have the same half-filled d-block as the HAT case in Scheme 6a. But now the 6th orbital that is delocalized over the O---H---X (X = N, O) axis is a ϕPCET type orbital that involves delocalization of an electron between a σ-type lobe on the oxo ligand and a π/lone-pair type orbital on the substrate (Figure 8b). Underneath the orbital occupation diagram in Scheme 6b we show the electron shift from this lone pair/π orbital (labeled as n(π)) to σ*z2 that enriches the d-block by one spin-up electron in σ*z2, and generates ϕPCET. When the substrate has high-lying lone pairs or π-orbitals like S3 and S4, these orbitals replace the low-lying σXH orbitals (X = N, O) and participate in the HAT type electron shift to σ*z2, enhancing the TS with a significant ET character.
Schemes 6c and 6d show the archetypical cases for bond activation by the highly basic reagent 3. In both cases, the TS remarkably conserves the exchange-rich d-block of the initial FeIIIO complex, having five singly occupied orbitals with S = 5/2 spin quantum number. Scheme 6c corresponds to the proton transfer-type transition state TSPT for 3 + S1, having five d-type SNOs without much contribution from the substrate. For this case, we identified a doubly occupied orbital, ϕX, that corresponds to the filled orbital of the carbanion generated at the H-abstracted substrate moiety, S1-H. The same features apply for the TS in the PT mechanism with the O-H bond of S4. The orbital diagram Scheme 6d describes the TS for the reaction 3 + S3, where one of the d-block SNOs is delocalized over the π*xz orbital and a substrate orbital, as shown in Figure 9.
The cases with the high PCET character must have other orbital interactions that are responsible for the proton abstraction from the X-H bond of the substrate. These orbital interactions contain donor orbitals on the oxidant48a and acceptor orbitals on the substrate. When the oxidant is basic, it has high-lying donor orbitals. When the substrate has O-H/N-H bonds, its antibonding σ*XH orbital is relatively low-lying. In addition, there are interactions that are responsible for charge transfer from donor orbitals of the substrate to acceptor orbitals of the oxidants (see QCT in Figure 6). Scheme 7 shows some of these interactions.
Scheme 7.

Orbital interactions responsible for: (a) proton abstraction from the X-H bond and/or a PT character in the TS, and (b) charge transfer from donor orbitals on the substrate to the oxidant and ET character in the TS.
Scheme 7a depicts the interactions that are responsible for the proton abstraction. Here, the donor orbitals are the lone pairs on the oxo ligand of the oxidant. These orbitals can be either the 2pz type lone-pair (labeled as σz2) or the 2px/y lone-pairs (πxz/yz orbitals). The orbitals interact with the σXH orbital and σ*XH orbitals of the X-H bond, resulting in bond cleavage in a PT process. Scheme 7b shows potential orbital interactions in which the substrate uses donor orbitals to transfer charge to the oxidant. These donor orbitals are either a lone pair (n), high lying π orbitals of S3 and S4, or the mixed σCH and π orbitals of S1. Thus, even for S1 and to a lesser extent for S2 and S5, we anticipate some PCET characters as manifested in the QCT quantities in Figure 6. Depending on the relative strengths of these orbital interactions, the TS can change its orientation from being approximately upright to being more sideways. Thus, when the stronger interaction depicted in Scheme 7a is with the 2px orbital, the orientation will be sideways. In contrast, when σz2 (2pz) is dominant, the orientation will be upright. The FeOH angles in Figure 6 show indeed different orientations of the respective TSs.
B. Reactivity Patterns
Having established the generality of the ΔEdef/ΔE‡ map as an organizer of the HAT/PCET dichotomy in Figure 5, we now can address the nature of these stabilizing interactions that favor PCET reactions as well as the finer details of relative reactivity of specific cases. To accomplish these studies, we shall make use of Schemes 6 and 7 and Figures 5 and 6. Inspection of Scheme 6 provides some insight into relative reactivities of the various oxidants and substrates.
Relative Reactivity Patterns in HAT between FeIVO complexes and C-H bonds
In the light of Scheme 6a, we first considered the C-H bond activation of S1, S2, and S5 by the FeIVO complexes 1, 2 and 4. In all of these cases, the d-block orbitals of the FeIVO complexes are enriched by one electron in σ*z2. These TSs should have therefore experienced exchange-enhanced reactivity (EER)24,45b,46,47, resulting in low barriers. As shown in Figure 5a, EER is observed for 4 but not for the more basic complexes 1 and 2, which have much larger barriers. It is also worthy to note that the H-abstraction barriers of these basic oxidants with S1 are higher even than that of the analogous high spin [FeIVOTMG3tren]2+ oxidant (15.6 kcal/mol)45 or of other FeIVO complexes5j,19 (Tables S20 and S21). The root cause of the higher HAT barriers for 1 and 2 is the significant basicity of these iron(IV)-oxo complexes. With an increase in basicity, the energy of the acceptor orbital, σ*z2, is raised,18a,24,48b thus offsetting the exchange enhancement for 1 and 2. Additionally, the available spin density49 on the oxo ligand varies as follows: 4 > 2 ≥ 1, and consequently, the O-H bond formation during abstraction by a basic FeIVO complex such as 1 and 2 requires higher deformation energies in order to localize a spin on the oxo ligand and make a bond in the TS. Therefore, for a given substrate, the deformation energies change in the following order: ΔEdef(4) < ΔEdef(2) ≤ ΔEdef(1) (Figure 5a). The consequence of this result is that the barriers follow the same order as the ΔEdef quantities. Of course there are different steric effects, but as we argued before,24 the steric effects are largely expressed as higher deformation energies. Because all of the C-H bond activations here fall close to the line for which ΔEdef = ΔE‡, the barriers for the sterically more encumbered cases are necessarily larger (e.g., ΔE‡(S1) > ΔE‡(S5)).
Relative Reactivity Patterns in PCET between FeIVO complexes and N-H/O-H bonds
We have employed a similar analysis using Scheme 6b for the N-H/O-H bond activation of S3 and S4 by the FeIVO complexes 1, 2, and 4. The electronic structures in Scheme 6b show that these TSs have EER. Moreover, the additional orbital interactions (Scheme 7),48a which cleave the X-H (X=N,O) bond heterolytically and transfer charge to the FeO moiety, further stabilize the TS by providing some PT and ET character. A complimentary and more comprehensive picture of the stabilizing interaction using VB theory will be presented below.
Relative Reactivity Patterns in PCET (PT/ET) between the FeIIIO complex and X-H bonds
The reactivity of 3 (and its model 3' in the SI) is dominated by a high PT character in the TS, which is in turn blended with variable degrees of HAT characters. In essence, the deformation energies and the barriers (Figure 5) follow acid-base relationships such that with the least `acidic' C-H bond (in S1, S5), both the deformation energy and the barrier are large. When the more acidic N-H bond (such as in S3) is used, the deformation energy and barriers are smaller. Finally, when the most acidic O-H bond is reacted with the most basic complex (in 3 + S4), the reaction is a barrier-free PT (Table 2). In some cases, the PT character is blended with ET and HAT (e.g., for FeIIIO reacting with S1, S3, and S5, Figure 9) as is explained in Schemes 6c and 6d. However, in all of the cases with the exception of 3 + S5, the deformation energy is large and the final low barrier reflects significant stabilization due to interaction of the reactant moieties in the TS (Figure 5a and Scheme 5). Understanding the source of the stabilization energy that lowers the highly deformed reactants to their final TS energies is warranted.
C. Origins of HAT, PCET or stepwise PT-ET mechanisms
To complete the characterization of the HAT/PCET dichotomy, we have to conceptualize the source of the large stabilization in those TSs having significant PCET character (Figures 5a and 5b). Valence bond (VB) modeling of the HAT/PCET blending in the TS14,16 provides a connection to the preceding MO-based discussions of PCET character along with a rationale for the lowering of the barrier by blending PCET character. Figure 10 shows the VB-state curves from which one can reconstruct the energy profile for the H-abstraction reaction.
Figure 10.

VB diagrams for (a) HAT between two alkyl radicals, (b) a metal-oxo (M-O) abstracting an H atom from a molecule X-H (where X is an unsaturated-alkyl moiety having π-orbitals), (c) M-O abstracting H from X-H (X = N, O), and (d) a highly basic M-O abstracting H from significantly acidic X-H bonds. The HAT/PCET dichotomy in (b)–(d) is shown by mixing of HAT states (black, unbroken lines) and proton transfer/charge transfer (PT/ET) curves (red, dotted lines), along the reaction coordinate. For simplicity, only one oxygen lone pair is shown on the M-O complex. The PCET curves are anchored in electron transfer (ET) excited states of reactants and products, indicated as ΦET,r and ΦET,p, and in corresponding PT states, ΦPT,p and ΦPT/ET,r, respectively.
Figure 10a shows a “pure” HAT case of two alkyl radicals, R• and R'•, exchanging an H• atom. The two ground states at the reactant and product sides (r and p, respectively) involve the molecule and the corresponding radical, R-H/R'• and R•/H-R'. These two states correlate to two excited states, ΦHAT,r* and ΦHAT,p*, in which the electron pairs in R-H and R'-H are decoupled and the electron on the H• is paired to the other alkyl moiety. This long-distance pairing is indicated in Figure 10a by the arched lines connecting the electrons. When these two state-curves are followed along the reaction coordinate, they cross one another and mix to form a TS species (ψ≠) on the lower energy surface. For this reaction, all other states are quite high in energy and do not significantly influence the TS, which can represent a `pure' HAT species, ψ≠HAT50.
Consider now the case in Figure 10b where a metal-oxo (M-O) complex with significant basicity abstracts an H atom from a molecule X-H in which X is an unsaturated alkyl moiety of the type present in S1 and S5 and which also possesses p orbitals (mixed with σXH). These orbitals are symbolized by an electron pair over the X moiety. To simplify the depiction, the M-O species in the TS is drawn with a radical center on the oxo ligand (as is, in fact, present in 1–4) and a single lone pair (other electrons of M-O are omitted). In such an oxidant/substrate combination, the H-abstraction process has to be described by at least four state curves.14,16 Two of these are the HAT curves shown in Figure 10b in the full black lines. The other two shown in the dotted red curves involve ET/PT states.50c,51 For example, on the reactant side (r), the ΦET,r state arises by transferring an electron from the oxo-lone pair to the X-H bond while pairing the remaining odd electrons on the O• and H• to a singlet pair (as indicated by the arched line). Along the reaction coordinate (r → p), this state goes down in energy and correlates to a PT state on the product side, ΦPT,P, involving M-OH+ and X−
Consider now the product side (p) in Figure 10b. Here, the ΦET,p state arises by transferring an electron from the π-electron pair of X to the O-H bond in MO-H. Along the reverse reaction coordinate (p → r), ΦET,p descends in energy, forming M-O:− and (HX)+. Note that the latter species has a mixed π-σ character and is therefore denoted as a mixed PT/ET state on the reactant side (ΦPT/ET,r). For X being an unsaturated-alkyl moiety, these red ET/PT curves lie higher in energy than the HAT curves. Nevertheless, they can mix slightly into the TS and generate a TS species having a blended character, that is, a TS with a dominant ψHAT character and a secondary PT/ET character ψPT/ET (λ <<1 is the relative contribution).14
For the case represented in Figure 10c, in addition to a moderately basic metal-oxo complex we considered an X-H bond that is more acidic than a C-H bond and has a high-lying lone-pair/π-pair orbital(s), as in S3. As a result of these features, the ET/PT curves go down in energy, and at the crossing geometry, these curves are lower than the HAT curves. Thus, the VB mixing yields a TS species with a blended nature but that now has dominant ΦPCET character with a significant charge transfer from the substrate to the metal-oxo unit. This low energy TS has a low barrier for H-abstraction.
Figure 10d shows the final VB diagram for a highly basic metal-oxo complex (like 3) activating a significantly acidic X-H bond (S1, S3, S4). In this case, the PT/ET and PT curves are highly stabilized relative to the HAT curve and totally dominate the VB mixing, yielding a TS species with a dominant PT character and some ET character. Furthermore, en-route to the product, the PT/ET red curve is crossed again by the HAT curve on the product side. If this crossing occurs on the down hill slope, the VB mixing will create a concerted reaction with a PT character, followed by an ET. This situation was found in the reaction of 3 + S1, and 3 + S3 (see IRCs in Fig. 7). If the ΦPT,p state is more stable than the one shown in Figure 10d, the H-abstraction will terminate as proton abstraction and the follow up ET step may not take place as in the reaction of S3 with 4.
Usage of the VB Model to Understand the Global ΔEdef/ΔE‡Map
The global organization brought about by plotting ΔEdef vs. ΔE‡ in Figure 5, can now be interpreted lucidly based on the above VB model. Most of the C-H bond activation reactions in Figure 5 obey the relationship ΔE‡ (HAT) ≈ ΔEdef. This result means that in the O---H---C TS, which is predominantly HAT in character, the VB mixing energy (the lowering of the TS relative to the crossing point in Figure 10a) balances the Pauli repulsion and any other repulsive interactions (eq. 1b)25,26 between the reacting moieties. The net effect causes the entire barrier to be close to the deformation energy of the reactants while ΔEint ≈ 0.
Whenever the PT/ET states begins to blend significantly into the pure HAT species, ψHAT, it produces a lower energy TS (ψ‡, Figure 10c), which is more stabilized than a pure HAT TS (Figure 10a). Scheme 8 shows the relation of this extra stabilization, relative to the reference pure HAT state, and the ΔEdef/ΔE‡ map. As the ψHAT species maintains ΔE‡ ≈ ΔEdef and ΔEint(HAT) ≈ 0 (Scheme 8), the extra stabilization energy due to the PT/ET mixing into the pure HAT species will corresponds to the vertical deviation from the line of slope unity in ΔEdef/ΔE‡ map in Figure 5a, i.e., as a nonzero ΔEint quantity. Therefore, the ΔEint quantity in Figures 5a and 5b gauges the excess stabilization energy of the actual TS relative to a pure HAT TS. Scheme 8a shows the resulting ΔEint for moderate mixing as in Figure 10b, while Scheme 8b shows the case where the lowest state is a ψET/PT type, as the described in Figures 10c and 10d. In the latter case (Scheme 8b), the ΔEint quantity, which is gauged relative to the HAT reference-state, becomes very large as shown in Figure 5a. In this case, the resulting ΔEint term gauges not only the interactions between the reactant moieties but also the fact that these moieties are described now by more stable fragment states than in the HAT reference.
Scheme 8.
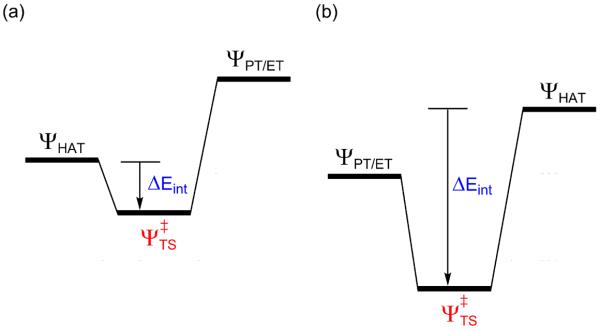
Mixing of HAT and PT/ET states and the resulting ΔEint quantities in ΔE‡ /ΔEdef maps. (a) Small stabilization as in Figure 10b. (b) Cases where the PT/ET states lie below the HAT crossing points (Figures 10c,d).
The VB model thus provides a physical basis for the origins of the stabilization interactions (ΔEint) during activation of X-H bonds. Furthermore, a simple VB language can be used to show that the oxyl character on an oxo ligand in the transition is gauged by the basicity of the Fe-O bond.52 When the oxidant is less basic, it has a higher oxyl character, it operates in typical HAT mechanisms, and acts as a powerful oxidant towards C-H bonds. In contrast, when the oxidant is more basic (nucleophilic), it cleaves bonds more heterolytically, proceeds through PCET or PT/ET mechanisms, and can activate N-H and O-H bonds as well as relatively acidic C-H bonds.
Summary and Conclusions
Our present work on the H-abstraction reactivity of four nonheme oxidants (1–4) with substrates (S1–S5) demonstrates the fundamental cause of the puzzling reactivity scenarios observed in the experimental studies.17,10–12,20 First is the higher reactivity of [FeIVH3buea(O)]1− (1) towards the stronger C-H bond in dimethylformamide (S2) compared with the weak C-H bond of 9,10 dihydroanthracene (S1). Second, despite the relatively weak FeOH bond in [FeIIIH3buea(OH)]1−, the corresponding FeIII-oxo complex (3) is nevertheless capable of activating S1 by PT. Third, the least basic complex [FeIVN4Py(O)]2+ (4) is able to activate strong C-H bonds as well as relatively weak N-H and O-H bonds in S3 and S4.
These intriguing reactivity patterns are mainly linked to the HAT/PCET dichotomy, which normally typifies C-H vs. O-H/N-H bond activations. But here, the PCET propensity is highly amplified by the basic nature of 1–3 such that 1 and 2 show marked propensity for PCET. On the other hand, 3 traverses to the stepwise end of the mechanistic spectrum, reacting via PT/ET steps even with the C-H bond of S1. This mechanistic spectrum depends largely on the basicity/acidity relationships of the oxidants/X-H bonds of the substrates. Note that similar types of reactivity patterns have been observed for the corresponding MnIII-oxo and MnIV-oxo complexes with the [H3buea]3−ligand.17g
Diverse properties such as TS geometries, the electronic structures, the deformation energies in the TSs for HAT vs. PCET, and the corresponding reactivity patterns can all be distinguished in two ways: (a) by the nature of the singly occupied orbital in the H-abstraction moiety (O---H----X), and (b) by a plot of the deformation energy vs. the reaction barrier24 using the energy decomposition analysis approach to reactivity.25,26 Furthermore, VB conceptualization14,16 clearly illustrates how mixing of PT/ET states into HAT states brings about additional stabilization of TSs and influences the kinetic barriers. The VB modeling also explains that the deformation energy patterns provide a global behavior that is common to many iron-oxo H-abstraction reactivities (Figure 5b) and can serve as a physically-based classifier of the mechanistic dichotomy.
The insight into the underlying factors that govern H-abstraction mechanisms opens up new vistas in understanding the dichotomous behavior of HAT and PCET mechanisms that are operative in metalloenzymes and synthetic analogs. Furthermore, the distinct change in the nature of the H-abstraction mechanism (HAT or PCET) for X-H bonds with variable basicities gives clues for designing biomimetic catalysts that can exclusively activate C-H bonds over N-H/O-H bonds.1f
Supplementary Material
ACKNOWLEDGMENT
SS acknowledges support by the Israel Science Foundation (grant ISF 1183/13). ASB acknowledges grant GM50781 from the National Institute of Health USA.
Footnotes
Supporting information includes Tables S1–S21, Figures S1–S24, Cartesian coordinates of key species, and full reference of Gaussian 03 and Gaussian 09 (ref 32 and 37). This material is available at http://pubs.acs.org
References and Notes
- 1.(a) Sono M, Roach MP, Coulter ED, Dawson JH. Chem. Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]; (b) Costas M, Mehn MP, Jensen MP, Que L., Jr. Chem. Rev. 2004;104:939–986. doi: 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]; (c) Groves JT. J. Chem. Educ. 1985;62:928–931. [Google Scholar]; (d) Krebs C, Fujimori DG, Walsh CT, Bollinger JM., Jr. Acc. Chem. Res. 2007;40:484–492. doi: 10.1021/ar700066p. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Nam W, editor. Acc. Chem. Res. 2007;40:465–634. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (f) Borovik AS. Chem. Soc. Rev. 2011;40:1870–1874. doi: 10.1039/c0cs00165a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Schlichting I, Berendzen J, Chu K, Stock AM, Maves SA, Benson DE, Sweet RM, Ringe D, Petsko GA, Sligar SG. Science. 2000;287:1615–1622. doi: 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]; (b) Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]; (c) Chakrabarty S, Austin RN, Deng D, Groves JT, Lipscomb JD. J. Am. Chem. Soc. 2007;129:3514–3515. doi: 10.1021/ja068188v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Derat E, Shaik S. J. Am. Chem. Soc. 2006;128:13940–13949. doi: 10.1021/ja065058d. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Yang C, Wang H, Han K, Shaik S. Chem. Bio. Chem. 2007;8:277–81. doi: 10.1002/cbic.200600510. [DOI] [PubMed] [Google Scholar]; (c) Schyman P, Lai WZ, Chen H, Wang Y, Shaik S. J. Am. Chem. Soc. 2011;133:7977–7984. doi: 10.1021/ja201665x. [DOI] [PubMed] [Google Scholar]; (d) Hsuanyu Y, Dunford HB. Arch. Biochem. Biophys. 1992;292:213–220. doi: 10.1016/0003-9861(92)90070-d. [DOI] [PubMed] [Google Scholar]
- 4.(a) Goodwin DC, Grover TA, Aust SD. Biochemistry. 1997;36:139–147. doi: 10.1021/bi961465y. [DOI] [PubMed] [Google Scholar]; (b) Kretschmer R, Zhang X, Schlangen M, Schwarz H. Chem. Eur. J. 2011;17:3886–3892. doi: 10.1002/chem.201003620. [DOI] [PubMed] [Google Scholar]; (c) Hirao H, Chuanprasit P, Cheong YY, Wang X. Chem. Eur. J. 2013;19:7361–7369. doi: 10.1002/chem.201300689. [DOI] [PubMed] [Google Scholar]; (d) Koymans L, Lenthe JHV, Kelder GMDD, Vermeulen NPE. Mol. Pharm. 1990;37:452–460. [PubMed] [Google Scholar]
- 5.(a) Que L., Jr. Acc. Chem. Res. 2007;40:493–500. doi: 10.1021/ar700024g. [DOI] [PubMed] [Google Scholar]; (b) Nam W. Acc. Chem. Res. 2007;40:522–531. doi: 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]; (c) Hohenberger J, Ray K, Meyer K. Nature Commun. doi: 10.1038/ncomms1718. DOI:10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]; (d) Maiti D, Lee D-H, Gaoutchenova K, Würtele C, Holthausen MC, Sarjeant AAN, Sundermeyer J, Schindler S, Karlin KD. Angew. Chem. Int. Ed. 2008;47:82–85. doi: 10.1002/anie.200704389. [DOI] [PubMed] [Google Scholar]; (e) Maiti D, Fry C, Woertink JS, Vance MA, Solomon EI, Karlin KD. J. Am. Chem. Soc. 2007;129:264–265. doi: 10.1021/ja067411l. [DOI] [PubMed] [Google Scholar]; (f) Cho J, Woo J, Han JE, Kubo M, Ogura T, Nam W. Chem. Sci. 2011;2:2057–2062. [Google Scholar]; (g) Company A, Yao S, Ray K, Driess M. Chem. Eur. J. 2010;16:9669–9675. doi: 10.1002/chem.201001138. [DOI] [PubMed] [Google Scholar]; (h) Kundu S, Pfaff FF, Miceli E, Zaharieva I, Herwig C, Yao S, Farquhar ER, Kuhlmann U, Bill E, Hildebrandt P, Dau H, Driess M, Limberg C, Ray K. Angew. Chem. Int. Ed. 2013;52:5622–5626. doi: 10.1002/anie.201300861. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Itoh S, Fukuzumi S. Acc. Chem. Res. 2007;40:592–600. 13234–13242. doi: 10.1021/ar6000395. [DOI] [PubMed] [Google Scholar]; (j) Company A, Prat I, Frisch JR, Mas-Ballesté R, Güell M, Juhász G, Ribas X, Münck E, Luis JM, Que L, Jr., Costas M. Chem. Eur. J. 2011;17:1622–1634. doi: 10.1002/chem.201002297. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Shook RL, Borovik AS. Chem. Comm. 2008;2:6095–6107. doi: 10.1039/b810957e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Bell RP. Proc. R. Soc. London, Ser. A. 1936;154:414–429. [Google Scholar]; (b) Evans MG, Polanyi M. Trans. Faraday Soc. 1938;34:11–24. [Google Scholar]
- 7.(a) Mayer JM. Acc. Chem. Res. 2011;44:36–46. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mayer JM. Acc. Chem. Res. 1998;31:441–450. [Google Scholar]; (c) Warren JJ, Tronic TA, Mayer JM. Chem. Rev. 2010;110:6961–7001. doi: 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Mayer JM. J. Phys. Chem. Lett. 2011;2:1481–1489. doi: 10.1021/jz200021y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chung LW, Li X, Hirao H, Morokuma K. J. Am. Chem. Soc. 2011;133:20076–20079. doi: 10.1021/ja2084898. [DOI] [PubMed] [Google Scholar]
- 9.(a) Cowley RE, Eckert NA, Vaddadi S, Figg TM, Cundari TR, Holland PL. J. Am. Chem. Soc. 2011;133:9796–9811. doi: 10.1021/ja2005303. [DOI] [PubMed] [Google Scholar]; (b) Holland PL. Acc. Chem. Res. 2008;41:905–914. doi: 10.1021/ar700267b. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Goldsmith CR, Stack TDP. Inorg. Chem. 2006;45:6048–6055. doi: 10.1021/ic060621e. [DOI] [PubMed] [Google Scholar]
- 10.(a) Klinker EJ. PhD Thesis. Chapter 3. University of Minnesota; 2007. p. 20. [Google Scholar]; (b) Fiedler AT, Que L., Jr. Inorg. Chem. 2009;48:11038–11047. doi: 10.1021/ic901391y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sastri CV, Lee J, Oh K, Lee YJ, Lee J, Jackson TA, Ray K, Hirao H, Shin W, Halfen JA, Kim J, Que L, Jr., Shaik S, Nam W. Proc. Nat. Acad. Sci. USA. 2007;104:19181–19186. doi: 10.1073/pnas.0709471104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang D, Farquhar ER, Stubna A, Münck E, Que L., Jr. Nat. Chem. 2009;1:145–150. doi: 10.1038/nchem.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For some general references on PCET mechanisms, Huynh MHV, Meyer TJ. Chem. Rev. 2007;107:5004–5064. doi: 10.1021/cr0500030.. Mayer JM. Annu. Rev. Phys. Chem. 2004;55:363–390. doi: 10.1146/annurev.physchem.55.091602.094446.. Cukier RI, Nocera DG. Annu. Rev. Phys. Chem. 1998;49:337–369. doi: 10.1146/annurev.physchem.49.1.337.. Hammes-Schiffer S. Acc. Chem. Res. 2009;42:1881–1889. doi: 10.1021/ar9001284.. Weinberg DR, Gagliardi CJ, Hull JF, Murphy CF, Ken CA, Westlake BC, Paul A, Ess DH, McCraffety DG, Meyer TJ. Chem. Rev. 2012;112:4016–4093. doi: 10.1021/cr200177j.. For PCET in PS II, see: Siegbahn PEM. Acc. Chem. Res. 2009;42:1871–1880. doi: 10.1021/ar900117k. Costentin C, Robert M, Savéant J-M. Acc. Chem. Res. 2010;43:1019–1029. doi: 10.1021/ar9002812.. Wenger OS. Chem.-Eur. J. 2011;17:11692–11702. doi: 10.1002/chem.201102011.. Liu S, Ess DH, Schauer CK. J. Phys. Chem. A. 2011;115:4738–4742. doi: 10.1021/jp112319d..
- 14.(a) Lai WZ, Li C, Chen H, Shaik S. Angew. Chem. Int. Ed. 2012;51:5556–5578. doi: 10.1002/anie.201108398. [DOI] [PubMed] [Google Scholar]; (b) Li C, Danovich D, Shaik S. Chem. Sci. 2012;3:1903–1918. [Google Scholar]
- 15.For PCET/HAT relationships see: Skone JH, Soudackov AV, Hammes-Schiffer S. J. Am. Chem. Soc. 2006;128:16655–16663. doi: 10.1021/ja0656548.; Hammes-Schiffer S. Acc. Chem. Res. 2001;34:273–281. doi: 10.1021/ar9901117.. Tishchenko O, Truhlar DG, Ceulemans A, Nguyen MT. J. Am. Chem. Soc. 2008;130:7000–7010. doi: 10.1021/ja7102907..
- 16.Cembran A, Provorse MR, Wang C, Wu W, Gao J. J. Chem. Theory. Comput. 2012;8:4347–4358. doi: 10.1021/ct3004595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lacy DC, Gupta R, Stone KL, Greaves J, Ziller JW, Hendrich MP, Borovik AS. J. Am. Chem. Soc. 2010;132:12188–2190. doi: 10.1021/ja1047818.. Gupta R, Lacy DC, Bominaar EL, Borovik AS, Hendrich MP. J. Am. Chem. Soc. 2012;134:9775–9784. doi: 10.1021/ja303224p.. Lacy DC. Ph.D. Thesis. University of California-Irvine; Irvine, CA: 2012. . MacBeth CE, Golombek AP, Young VG, Jr., Yang C, Kuczera K, Hendrich MP, Borovik AS. Science. 2000;289:938–941. doi: 10.1126/science.289.5481.938.. Gupta R, Borovik AS. J. Am. Chem. Soc. 2003;125:13234–13242. doi: 10.1021/ja030149l.. Dey A, Hocking RK, Larsen P, Borovik AS, Hodgson KO, Hedman B, Solomon EI. J. Am. Chem. Soc. 2006;128:9825–9833. doi: 10.1021/ja061618x.. See also, Parsell TH, Yang M-Y, Borovik AS. J. Am. Chem. Soc. 2009;131:2762–2763. doi: 10.1021/ja8100825..
- 18.(a) Bernasconi L, Louwerse MJ, Baerends EJ. Eur. J. Inorg. Chem. 2007:3023–3033. [Google Scholar]; (b) Solomon EI, Wong SD, Liu LV, Decker A, Chow MS. Curr. Opin. Chem. Biol. 2009;13:99–113. doi: 10.1016/j.cbpa.2009.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Johansson AJ, Blomberg MRA, Siegbahn PEM. J. Phys. Chem. C. 2007;111:12397–12406. [Google Scholar]; (d) Ye S, Neese F. Proc. Nat. Acad. Sci. USA. 2011;108:1228–1233. doi: 10.1073/pnas.1008411108. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) McDonald AR, Guo Y, Vu VV, Bominaar EL, Münck E, Que L., Jr. Chem. Sci. 2012;3:1680–1693. doi: 10.1039/C2SC01044E. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) England J, Guo Y, Van Heuvelen KM, Cranswick MA, Rohde GT, Bominaar EL, Münck E, Que L., Jr. J. Am. Chem. Soc. 2011;133:11880–11883. doi: 10.1021/ja2040909. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Seo SM, Kim NH, Cho K-B, So JE, Park SK, Clémancey M, Garcia-Serres R, Latour J-M, Shaik S, Nam W. Chem. Sci. 2011;2:1039–1045. [Google Scholar]; (h) Xue G, DeHont R, Münck E, Que L., Jr. Nat. Chem. 2010;2:400–405. doi: 10.1038/nchem.586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Latifi R, Sainna MA, Rybak-Akimova EV, de Visser SP. Chem.-Eur. J. 2013;19:4058–4068. doi: 10.1002/chem.201202811. [DOI] [PubMed] [Google Scholar]
- 20.Kaizer J, Klinker EJ, Oh NY, Rohde J-U, Song WJ, Stubna A, Kim J, Münck E, Nam W, Que L., Jr. J. Am. Chem. Soc. 2004;126:472–473. doi: 10.1021/ja037288n. [DOI] [PubMed] [Google Scholar]
- 21.Mayer JM, Hrovat DA, Thomas JL, Borden WT. J. Am. Chem. Soc. 2002;124:11142–11147. doi: 10.1021/ja012732c. [DOI] [PubMed] [Google Scholar]
- 22.Lingwood M, Hammond JR, Hrovat DA, Mayer JM, Borden WT. J. Chem. Theory Comput. 2006;2:740–745. doi: 10.1021/ct050282z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Gao J, Cembran A, Mo Y. J. Chem. Theory Comput. 2010;6:2402–2410. doi: 10.1021/ct100292g. [DOI] [PubMed] [Google Scholar]; (b) Cembran A, Song L, Mo Y, Gao J. J. Chem. Theory Comput. 2009;5:2702–2716. doi: 10.1021/ct9002898. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mo Y, Song L, Lin Y. J. Phys. Chem. A. 2007;111:8291–8301. doi: 10.1021/jp0724065. [DOI] [PubMed] [Google Scholar]; (d) Song L, Mo Y, Zhang Q, Wu W. J. Comput. Chem. 2005;26:514–521. doi: 10.1002/jcc.20187. [DOI] [PubMed] [Google Scholar]
- 24.Usharani D, Janardanan D, Li C, Shaik S. Acc. Chem. Res. 2013;46:471–482. doi: 10.1021/ar300204y. [DOI] [PubMed] [Google Scholar]
- 25.For development of the method see Morokuma K. J. Chem. Phys. 1971;55:1236–1244.. Kitaura K, Morokuma K. Int. J. Quantum Chem. 1976;10:325–340.. For usage of the term distortion energy see: Ziegler T, Rauk A. Theor. Chim. Acta. 1977;46:1–10.. Ziegler T, Rauk A. Inorg. Chem. 1979;18:1558–1565.. For usage of the terms distortion/deformation energies see: Strozier RW, Caramella P, Houk KN. J. Am. Chem. Soc. 1979;101:1340–1343.. Mitchell DJ, Schlegel HB, Shaik S, Wolfe S. Can. J. Chem. 1985;63:1642–1649.. Guthrie JP. Chem. Phys. Chem. 2003;4:809–816. doi: 10.1002/cphc.200200327.. Ess DH, Houk KN. J. Am. Chem. Soc. 2007;129:10646–10646. doi: 10.1021/ja0734086.. Legault CY, Garcia Y, Merlic CA, Houk KN. J. Am. Chem. Soc. 2007;129:12664–12665. doi: 10.1021/ja075785o.. Ess DH, Houk KN. J. Am. Chem. Soc. 2008;130:10187–10198. doi: 10.1021/ja800009z..
- 26.For a similar development and usage of the term `activation strain' see van Zeist W-J, Bickelhaupt FM. Org. Biomol. Chem. 2010;8:3118–3127. doi: 10.1039/b926828f.. Diefenbach A, Bickelhaupt FM. J. Phys. Chem. A. 2004;108:8460–8466..
- 27.Johansson AJ, Blomberg MR, Siegbahn PEM. J. Chem. Phys. 2008;129:154301–13. doi: 10.1063/1.2991180. [DOI] [PubMed] [Google Scholar]
- 28.Janardanan D, Usharani D, Chen H, Shaik S. J. Phys. Chem. Lett. 2011;2:2610–2617. [Google Scholar]
- 29.(a) Roelfes G, Lubben M, Chen K, Ho YNR, Meetsma A, Genseberger S, Hermant RM, Hage R, Mandal SK, Young VG, Jr., Zang Y, Kooijman H, Spek AL, Que L, Jr., Feringa BL. Inorg. Chem. 1999;38:1929–1936. doi: 10.1021/ic980983p. [DOI] [PubMed] [Google Scholar]; (b) Klinker EJ, Kaizer J, Brennessel WW, Woodrum NL, Cramer CJ, Que L., Jr. Angew. Chem. Int. Ed. 2005;44:3690–3694. doi: 10.1002/anie.200500485. [DOI] [PubMed] [Google Scholar]
- 30.(a) Becke AD. J. Chem. Phys. 1992;96:2155–2160. [Google Scholar]; (b) Becke AD. J. Chem. Phys. 1992;97:9173–9177. [Google Scholar]; (c) Becke AD. J. Chem. Phys. 1993;98:5648–5652. [Google Scholar]; (d) Lee C, Yang W, Parr RG. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 31.(a) Hay PJ, Wadt WR. J. Chem. Phys. 1985;82:299–310. [Google Scholar]; (b) Friesner RA, Murphy RB, Beachy MD, Ringnalda MN, Pollard WT, Dunietz BD, Cao Y. J. Phys. Chem. A. 1999;103:1913–1928. [Google Scholar]
- 32.Frisch MJ, et al. Gaussian 03. Revision E. 01 Gaussian, Inc.; Wallingford, CT: 2004. [Google Scholar]
- 33.(a) Tannor DJ, Marten B, Murphy R, Friesner RA, Sitkoff D, Nicholls A, Ringnalda M, Goddard WA, III, Honig B. J. Am. Chem. Soc. 1994;116:11875–11882. [Google Scholar]; (b) Marten B, Kim K, Cortis C, Friesner RA, Murphy RB, Ringnalda MN, Sitkoff D, Honig B. J. Phys. Chem. 1996;100:11775–11788. [Google Scholar]
- 34.Jaguar . Version 7.6 Schrödinger, LLC; New York, NY: 2008. [Google Scholar]
- 35.Grimme S, Antony J, Ehrlich S, Krieg H. J. Chem. Phys. 2010;132:154104–19. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 36.(a) Weigend F, Häser M, Patzelt H, Ahlrichs R. Chem. Phys. Lett. 1998;294:143–152. [Google Scholar]; (b) Weigend F, Ahlrichs R. Phys. Chem. Chem. Phys. 2005;7:3297–3305. doi: 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- 37.Frisch MJ, et al. Gaussian 09. Revision B1 Gaussian, Inc.; Wallingford CT: 2009. [Google Scholar]
- 38.Marenich AV, Cramer CJ, Truhlar DG. J. Phys. Chem. B. 2009;113:6378–6396. doi: 10.1021/jp810292n. [DOI] [PubMed] [Google Scholar]
- 39.Lynch BJ, Fast PL, Harris M, Truhlar DG. J. Phys. Chem. A. 2000;104:4811–4815. [Google Scholar]
- 40.(a) Adamo C, Barone V. J. Chem. Phys. 1999;110:6158–6170. [Google Scholar]; (b) Perdew JP, Burke K, Ernzerhof M. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]; Phys. Rev. Lett. (Erratum) 1997;78:1386. [Google Scholar]
- 41.Zhao Y, Truhlar DG. J. Chem. Phys. 2006;125:194101–18. doi: 10.1063/1.2370993. [DOI] [PubMed] [Google Scholar]
- 42.Zhao Y, Truhlar DG. Theor. Chem. Acc. 2008;120:215–241. [Google Scholar]
- 43.Dolg M, Wedig U, Stoll H, Preuss H. J. Chem. Phys. 1987;86:866–872. [Google Scholar]
- 44.Martin JML, Sundermann A. J. Chem. Phys. 2001;114:3408–3420. [Google Scholar]
- 45.(a) England J, Martinho M, Farquhar ER, Frisch JR, Bominaar EL, Münck E, Que L., Jr. Angew. Chem. Int. Ed. 2009;48:3622–3626. doi: 10.1002/anie.200900863. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Janardanan D, Wang Y, Schyman P, Que L, Jr., Shaik S. Angew. Chem. Int. Ed. 2010;49:3342–3345. doi: 10.1002/anie.201000004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shaik S, Chen H, Janardanan D. Nat. Chem. 2011;3:19–27. doi: 10.1038/nchem.943. [DOI] [PubMed] [Google Scholar]
- 47.Chen H, Lai WZ, Shaik S. J. Phys. Chem. Lett. 2010;1:1533–1540. [Google Scholar]
- 48.(a) Ye S, Geng C-Y, Shaik S, Neese F. Phys. Chem. Chem. Phys. 2013;15:8017–8030. doi: 10.1039/c3cp00080j. [DOI] [PubMed] [Google Scholar]; (b) Geng C, Ye S, Neese F. Angew. Chem. Int. Ed. 2010;49:5717–5720. doi: 10.1002/anie.201001850. [DOI] [PubMed] [Google Scholar]
- 49.For the importance of oxyl character, see: Dietl N, Schlangen M, Schwarz H. Angew. Chem. Int. Ed. 2012;51:5544–5555. doi: 10.1002/anie.201108363.. Groves JT, Nemo TE. J. Am. Chem. Soc. 1983;105:6243–6248..
- 50.Maitre P, Hiberty PC, Ohanessian G, Shaik S. J. Phys. Chem. 1990;94:4089–4093.. Shaik S, Hibery PC. A Chemist's Guide to Valence Bond Theory. Chapter 6. John Wiley & Sons Inc.; New York: 2008. . For importance of ET-type states in radical reactions, see: Shurki A, Shaik S. Angew. Chem. Int. Ed. 1999;38:586–625. doi: 10.1002/(SICI)1521-3773(19990301)38:5<586::AID-ANIE586>3.0.CO;2-T..
- 51.For importance of ET-type states in radical reactions, see: Fischer H, Radom L. Angew. Chem. Int. Ed. 2001;40:1340–1371. doi: 10.1002/1521-3773(20010417)40:8<1340::aid-anie1340>3.0.co;2-#.. Pross A. Theoretical and Physical Principles of Organic Reactivity. Wiley Interscience; New York: 1995. pp. 263–266..
- 52.Lai WZ, Chen H, Shaik S. J. Phys. Chem. B. 2009;113:7912–7917. doi: 10.1021/jp902288q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.