

Abstract
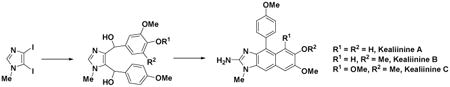
Short total syntheses of the Leucetta-derived alkaloids, kealiinines A-C, have been accomplished using an intramolecular Friedel-Crafts-dehydration sequence of a bis benzylic diol. The precursor diol was obtained through a series of position specific Grignard reactions from 1-methyl-4,5-diiodoimidazole. C2-Azidation and hydrogenation of the azide then provided the reported structures of kealiinines A-C. While the 1H NMR data did not completely match for these materials, the HPLC data were consistent with the assigned structure of these materials.
The Leucetta alkaloids are a group of 60 or so 2-aminoimidazole alkaloids1 isolated from sponges of the Leucetta and Clathrina families.2-4 Structurally, all of these alkaloids contain at least one oxygenated benzyl group (Figures 1 and 2), e.g., clathridine A (1),5 and in many cases there are two benzyl groups e.g., naamidine G (2).6 Within this collection of alkaloids a subgroup 6-8 containing a naphthimidazole moiety has been reported that still retains the two benzylic groups,7-9 but these are now embedded within the heterocyclic framework (Figure 2). Kealiinines A-C (8a-c)7 were isolated by the Proksch group in 2004, along with naamine G and naamidine H (3); the former are structurally related to the more highly oxygenated kealiiquinone (6)9 and 2-deoxy-2-aminokealiiquinone (7).8 It has been suggested that kealiinine C (8c) may serve as a biosynthetic precursor to 6 and 7,7 but there is no experimental evidence to support this hypothesis.10 No bioactivity was reported by Proksch for 8b and 8c,7 presumably as a consequence of the modest amounts of material isolated, but kealiinine A (8a) was shown to be active in the brine shrimp toxicity assay.7 To date, only two published synthetic studies towards this subgroup of alkaloids have appeared resulting in a total synthesis of kealiiquinone (6)11 and the non-natural 7′-desmethyl derivative (4).12 Furthermore, since the biological activity of the kealiinines has not been extensively investigated, it would appear that a program directed towards their synthesis is warranted.13 Over the past few years, our laboratory has developed several synthetic methods for the total synthesis of 2-aminoimidazole alkaloids using site selective functionalization of polyhaloimidazoles. Using this strategy, we have reported total syntheses of several Leucetta alkaloids including clathridine A (1),14 naamidine G (2),15 naamidine H (3)16 and calcaridine A (5).17 We have more recently turned our attention to members of this class containing a naphthimidazole framework such as kealiinines A-C (8a-c), kealiiquinone (6)12 and the 2-amino congener 7. While the details of the biosynthesis of these natural products remain to be elucidated experimentally, hypothetical biosynthetic relationships between both the simple systems and the more highly oxidized derivatives are evident. It is thought that naamine type systems 9 may serve as precursors for other family members through net oxidative functionalization (Figure 2). Some of these potential relationships are outlined in Figure 2 and while they are purely hypothetical, they have provided a framework for our synthetic programs. Indeed, we have used such a biomimetic strategy en route to calcaridine A (5) via 9 (R = X = Y = H, Z = OMe).17 It has been speculated that the naphthimidazole framework arises from a net oxidative coupling between C12 and C13 (kealiinine numbering) and further oxidation to lead to the natural product. Circumstantial evidence to support this idea can be found in reports of C12-oxygenated naamidine derivatives,6 but there is no experimental data to confirm this hypothesis. From a synthetic perspective, one can envision that a similar outcome might be obtained through a Friedel-Crafts type of strategy to form the C12-C13 bond followed by dehydration.11 Such a strategy in principle would provide access to all family members with only C2-amination and deprotection necessary. Oxidation of the C-ring would provide an entry to kealiiquinone and congeners.
Figure 1. Structures of the Leucetta-family of alkaloids.

Figure 2. Putative biosynthetic relationships among the Leucetta alkaloids.

Scheme 2.

Completion of kealiinine A (8a).
In order to establish the viability of the strategy we selected kealiinine C (8c) as our first target, thereby avoiding potential regioselectivity issues in the cyclization step forming the naphthimidazole system. In a forward sense, metallation of 12 with EtMgBr in CH2Cl2 provides the 5-imidazolyl Grignard, which upon reaction with the trimethoxybenzaldehyde 13c results in the formation of the corresponding alcohol 14c. Treatment of alcohol 14c with excess EtMgBr leads to the formation of the 4-imidazolyl Grignard, which reacts with N-methyl formanilide (15), producing aldehyde 16c. Treatment of aldehyde 16c with excess p-methoxyphenylmagnesium bromide affords the diol 17c as an inconsequential diastereomeric mixture in good yield. Attempts to introduce the benzyl moiety directly, i.e. 14c→17c via metalation and reaction with p-anisaldehyde, were not successful leading to the two step sequence described. Gratifyingly, treatment of unpurified 17c with HCl resulted in an intramolecular Friedel-Crafts cyclization-dehydration sequence to provide the naphthimidazole 18c, a result that was confirmed by X-ray crystallography. C2-Metalation with n-BuLi, followed by exposure to TrisN3, produced the 2-azido compound 19c. Subsequent treatment with Pd-C/H2 led to the reduction of the azide to the amine and the completion of the synthesis of the kealiinine C (8c).
The 1H NMR spectroscopic data of synthetic kealiinine C in DMSO-d6 were not in agreement with the data reported for the natural product and 13C NMR data were not obtained for the natural product.7 For example, the aromatic proton at C6 on the naphthimidazole framework appeared at δ 7.33 in the synthetic material compared to δ 7.66 in the natural material. Similarly, there were some inconsistencies with the signals due to the methyl substituents. In particular we noted methyl signals at 3.49 (N-Me) and 3.06 ppm (MeO at C11) in the synthetic material, whereas in the naturally-occurring material the highest field signal was at 3.40 ppm. We had prepared substantially more material than was isolated from the sponge, and thus we hypothesized that the differences simply may be due to concentration effects. However, while dilution of NMR samples of synthetic material resulted in some minor shift changes (Δδ ∼ 0.05 ppm), the spectroscopic data were still not in agreement with that reported for the natural product. Attemps to reproduce the “as isolated” sample by the addition of water and/or TFA also failed to cause substantial changes in the chemical shifts. While the connectivity of the naphthimidazole framework and the relative location of the p-methoxyphenyl and methyl groups was confirmed through the X-ray analysis of 18c, it is conceivable that metalation with n-BuLi may have occurred at sites other than the imidazole C2. Fortunately, we were able to obtain an X-ray crystal structure of our synthetic final product 8c, which unequivocally indicated that the required substitution pattern corresponding to the reported structure was obtained. Interestingly, the X-ray structure indicated that we had obtained the amino tautomer, rather than the imino tautomer described for the natural product. While extrapolating from the solid state to the solution phase must be done with caution, one possible explanation for the discrepancy in the spectroscopic data between the isolated natural product and the synthetic material may lie in the extent of the equilibrium. Since only a very small amount of the natural material was isolated, the presence of impurities in the natural sample may substantially influence the position of the tautomeric equilibrium, which in turn may affect the chemical shifts of this material. A related issue with tautomers has been observed with kealiiquinone which was isolated as this enol tautomer,9 but the synthetic material was prepared as the keto form.11 To partially address this issue, the Proksch lab was kind enough to perform comparative HPLC analysis of our synthetic material with a natural product sample. This analysis showed that the two samples co-eluted suggesting that they were in fact chromatographically identical.18
Given that there was some uncertainty in the structure of kealiinine C (8c), we decided to prepare the other two family members, kealiinine A (8a) and B (8b). Their syntheses follow largely the same strategy, simply substituting the appropriate benzaldehyde for 8a and 8b providing the targets in 7 steps (11% overall yield) and 6 steps (21% overall yield) respectively from 12. In the case of kealiinine A (8a) deprotection of the O-benzyl group was accomplished prior to C2-azidation and reduction as metallation was more effective with a free phenolic OH (Scheme 2). X-ray crystallographic analysis of 20 unequivocally confirmed the connectivity of the Friedel-Crafts product. Puzzlingly however, the spectroscopic data for both synthetic materials also did not coincide with the data reported for by the Proksch lab. On the other hand the chromatographic properties of the synthetic and natural materials were identical based on HPLC data. In the case of kealiinine A (8a) the 13C NMR data were reported, but our data did not appear to match well either. However, the major disagreements are largely confined to carbons assigned to C4 and C5, which are consistent with the hypothesis of the different tautomeric identities of the isolated and synthetic materials.
In summary, we have developed high yielding, protecting group-free synthesis of the Leucetta alkaloids kealiinine A-C (8a-c) using sequential and chemoselective halogen-magnesium exchange reactions. Subsequent Friedel-Crafts cyclization and lithiation at C2 with electrophilic trapping leads to introduction of the 2-amino moiety. While the structures of the synthetic materials are secure and their chromatographic properties match the natural materials, the spectroscopic data do not match. One explanation for these observations may be related to the preparation of the amino form rather than the isolated imino forms. Attempts to reproduce the spectroscopic characteristics of the as isolated material through changing the sample concentration and introduction of additives have been unsuccessful.
Supplementary Material
Scheme 1. Assembly of the benzimidazole framework (for yields see Table 1).

Table 1. Yields for synthesis of the kealiinines A-C (8a-c).
Confirmed by X-ray analysis
Acknowledgments
We would like to thank Prof. Peter Proksch (Universität Düsseldorf) for providing copies of the original spectra of natural kealiinine C, for performing the comparative HPLC analyses and for valuable discussion pertaining to the the discrepancies in the data. We would also like to thank Prof. Ryan Looper (University of Utah) for valuable discussion and exchange of data prior to publication. This work was supported by the Robert A. Welch Foundation (Y-1362) and in part by the NIH (GM065503). The NSF (CHE-0234811 and CHE-0840509) is thanked for partial funding of the purchases of the NMR spectrometers used in this work.
Footnotes
Supporting Information Available Detailed experimental procedures and copies of 1H and 13C NMR spectra for all new compounds. CIFs for compounds 8c, 18c, and 20. This information is free of charge from the internet at http://pubs.acs.org.
References
- 1.Jin Z. J Nat Prod. 2011;28:1143. doi: 10.1039/c0np00074d. [DOI] [PubMed] [Google Scholar]
- 2.Koswatta PB, Lovely CJ. Nat Prod Rep. 2011;28:511. doi: 10.1039/c0np00001a. [DOI] [PubMed] [Google Scholar]
- 3.Sullivan JD, Giles RL, Looper RE. Curr Bioact Cpds. 2009;5:39. [Google Scholar]
- 4.Roue M, Quevrain E, Domart-Coulon I, Bourguet-Kondracki ML. Nat Prod Rep. 2012;29:739. doi: 10.1039/c2np20040f. [DOI] [PubMed] [Google Scholar]
- 5.Ciminiello P, Fattorusso E, Magno S, Mangoni A. Tetrahedron. 1989;45:3873. [Google Scholar]
- 6.Mancini I, Guella G, Debitus C, Pietra F. Helv Chim Acta. 1995;78:1178. [Google Scholar]
- 7.Hassan W, Edrada R, Ebel R, Wray V, Berg A, Van Soest R, Wiryowidagdo S, Proksch P. J Nat Prod. 2004;67:817. doi: 10.1021/np0305223. [DOI] [PubMed] [Google Scholar]
- 8.Fu X, Barnes JR, Do T, Schmitz FJ. J Nat Prod. 1997;60:497. [Google Scholar]
- 9.Akee RK, Carroll TR, Yoshida WY, Scheuer PJ, Stout TJ, Clardy J. J Org Chem. 1990;55:1944. [Google Scholar]
- 10.We have demonstrated the feasibility of such a transformation in the conversion of a kealiinine-type intermediate into kealiiquinone and 2-deoxy-2-aminokealiiquinone.
- 11.Kawasaki I, Taguchi N, Yamamoto T, Yamashita M, Ohta S. Tetrahedron Lett. 1995;36:8251. [Google Scholar]
- 12.Lima HM, Sivappa R, Yousufuddin M, Lovely CJ. Org Lett. 2012;14:2274. doi: 10.1021/ol300704w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.While our manuscript was in preparation the total synthesis of kealiinines A and B were reported see: Gibbons JB, Gligorich KM, Welm BE, Looper RE. Org Lett. 2012;14:4734. doi: 10.1021/ol3019242.
- 14.Koswatta PB, Lovely CJ. Tetrahedron Lett. 2009;50:4998. doi: 10.1016/j.tetlet.2009.10.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koswatta PB, Lovely CJ. Tetrahedron Lett. 2010;51:164. doi: 10.1016/j.tetlet.2009.10.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koswatta PB, Lovely CJ. Chem Comm. 2010;46:2148. doi: 10.1039/b926285g. [DOI] [PubMed] [Google Scholar]
- 17.Koswatta PB, Sivappa R, Dias HVR, Lovely CJ. Org Lett. 2008;10:5055. doi: 10.1021/ol802018r. and references cited therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
18.An alternative interpretation of these observations is that the assigned structure is in fact incorrect. To address this possibility, we prepared an isomeric congener through similar chemistry and found that although the NMR data were a better match (but not exact), the chromatographic properties were substantially different from the naturally occurring material.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.