

Abstract
Alzheimer's disease (AD) is one of the most prevalent severe neurological disorders afflicting our aged population. Cognitive decline, a major symptom exhibited by AD patients, is associated with neuritic dystrophy, a degenerative growth state of neurites. The molecular mechanisms governing neuritic dystrophy remain unclear. Mounting evidence indicates that the AD-causative agent, β-amyloid protein (Aβ), induces neuritic dystrophy. Indeed, neuritic dystrophy is commonly found decorating Aβ-rich amyloid plaques (APs) in the AD brain. Furthermore, disruption and degeneration of the neuronal microtubule system in neurons forming dystrophic neurites may occur as a consequence of Aβ-mediated downstream signaling. This review defines potential molecular pathways, which may be modulated subsequent to Aβ-dependent interactions with the neuronal membrane as a consequence of increasing amyloid burden in the brain.
1. Introduction
Several neurodegenerative disorders share common characteristics including aggregation of misfolded mutant proteins in neurons leading to their deafferentation or loss with resultant structural or functional deficits in specific regions of the central nervous system (CNS) [1]. The most prevalent symptoms of age-related neurodegenerative disease are cognitive decline and movement disorders, along with brainstem and cerebellar signs. Such age-dependent disorders include Alzheimer's disease (AD), Huntington's disease (HD), Parkinson's disease (PD), and Spinal Cerebellar Ataxias (SCAs) [1]. There exists complexity in identifying fundamental molecular mechanisms precipitating neurodegeneration in these age-related brain diseases. However, common molecular signalling pathways have been defined in the specific neuronal populations associated with pathology [2]. Although the initiators of neuronal dysfunction may differ for each neurodegenerative disorder, there may be common molecular pathways which when being dysregulated, drive and exacerbate neurodegeneration. For example, the degeneration seen in AD is a result of amyloid plaques and phosphorylated tau deposition in the cerebral cortex and specific subcortical regions, leading to degeneration in the temporal lobe and parietal lobe, along with parts of the frontal cortex and cingulate gyrus [2]. AD also displays dysregulation in kinase and phosphatase mechanisms along with microtubule motor proteins during the degeneration phase [3, 4]. Therefore, a major question that remains unresolved is whether the dysregulation in specific kinases/phosphatases and vesicular transport mechanisms are aetiological contributors to AD pathology.
2. Neurodegeneration and Alzheimer's Disease (AD)
Over the past century, the ageing of our population (≥65 years) in industrialised countries has exceeded that of the population as a whole. It is predicted that in subsequent generations, the proportion of the elderly population will double and so will the proportion of persons suffering from neurodegenerative disorders [5]. Diagnosis of neurodegenerative disease is usually based on clinical symptoms as there are no suitable noninvasive tests that can specifically predict onset of these conditions. However, with the advent of specialised magnetic resonance imaging (MRI) techniques, it is now possible to detect early pathological changes in the brain [6], providing clinicians with a unique window for early therapeutic intervention. Nevertheless, it is imperative that biomarker(s) of neurodegeneration are identified to assist in the early detection of these idiopathic cognitive disorders. Such biomarkers may take the form of modified proteins or peptides that are released into the circulation or alternatively sequestered intrathecally [1, 2].
Biomarkers of neurodegeneration may well be derived from dysfunctional/modified proteins that form the basis of pathological signal transduction cascades [1]. The dysregulation of signalling molecules central for maintaining neuronal function may stimulate the onset of neurodegeneration. For example, while Rho kinase (mainly ROCKII), glycogen synthase kinase-3β (GSK-3β), cyclin-dependent kinase-5 (Cdk5), and phosphatases are all essential for normal neuronal development [7], they may all be involved in a plethora of neurodegenerative disorders through a central pathogenic mechanism.
3. Amyloid Beta (Aβ) and Amyloid Plaque Pathology
It is well documented that the aging process is the major determinant of developing amyloid plaques with or without disease [8]. These extracellular senile plaques are composed of accumulated Aβ protein aggregating as β-pleated sheets and are derived from the aberrant cleavage of the transmembrane protein, APP [9–11]. Under normal physiological conditions, APP is a cell surface protein that is thought to be involved in signal transduction, axonal elongation, and cell migration [12–20]. It was also demonstrated that the C-terminus of APP plays a central role in gene expression and neuronal cell survival [21]. Such physiological mechanisms are only effective when APP is cleaved by various enzymes which can include intramembranous degradation by beta-site AβPP-cleaving enzyme (BACE1) to form the β C-terminal fragment (βCTF) [22, 23], subsequently followed by gamma-secretase which forms the small 4 kilodalton (kDa) amyloid-β (Aβ) peptides Aβ1-40 and Aβ1-42, which are released at the synapse (Figure 1) [22, 24, 25]. It has been demonstrated that the extent of APP cleavage is amplified in AD brains and that Aβ treatment further enhances this cleavage [21]. It has also been established that APP and its degradation products localise to neuritic vesicles [26] in the axons of AD brains, along with other neurodegenerative diseases, suggesting that APP accumulation may represent a hallmark of axonal injury [27, 28]. For instance, in APP transgenic mice, it has been demonstrated that elevated Aβ levels result in the loss of synapses and neuronal transmission along with behavioural abnormalities, before the formation of amyloid plaques [21]. Accumulation of Aβ, mainly Aβ1-42, results in the rare early-onset familial AD (EOFAD) which is caused by mutations in the enzymes that cleave APP, leading to rapid and aberrant cleavage with resultant overproduction of Aβ [29]. On the other hand, the common late-onset AD (LOAD) is thought to result from either the failure of Aβ to be cleared from the brain [30, 31] by microglial cells, lower expression of Aβ degrading proteases such as insulysin (insulin degrading enzyme IDE), a decline in the availability of Aβ chaperone low density lipoprotein receptor-related protein (LRP1) to transport Aβ out of the brain, reduced vascular and perivascular drainage, or a combination of the above [32]. Although Aβ monomers are relatively nonpathogenic, accumulating soluble Aβ oligomeric forms have been shown to be synaptotoxic and can prune dendritic spines, disconnecting the memory-encoding neuronal network in the entorhinal cortex, the parahippocampal gyrus, and the hippocampus [33]. These oligomers eventually form large insoluble fibrillar aggregates or plaques that by themselves do not directly induce neuronal death but rather attract microglia and astrocytes that produce cytotoxic proinflammatory cytokines and reactive oxygen species that may indirectly cause neuronal death [34].
Figure 1.
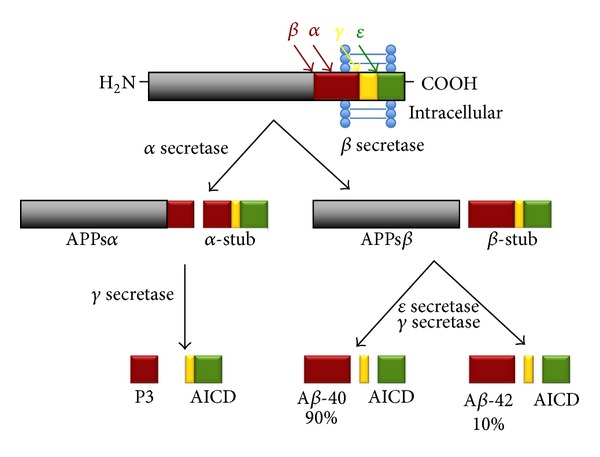
The processing of APP through the beta-site AβPP-cleaving enzyme BACE1, followed by presenilin-1 (PS1). Sequential beta and gamma-secretase cleavage of APP generates the synaptotoxic amyloid-β (Aβ) peptide species, Aβ1-40 and Aβ1-42.
Additionally, other proposed mechanisms that contribute to neuronal damage include the vulnerability of cells to secondary insults, tau hyperphosphorylation, induction of the apoptosome and lysosomal protease activity, changes in calcium influx, and direct damage (peroxidation) of membranes [35].
Although the plaques are found extracellularly, it is thought that production, oligomerisation, and accumulation of Aβ occur within neuronal processes with the possibility that the incorporation of aggregates into plaques occurs after the neurites are dissolved [36]. Certainly, studies performed in the well-established mouse models of AD have identified Aβ in several neuronal compartments such as the Golgi apparatus, the endoplasmic reticulum, the secretory vesicles, endosomes, and autophagic vacuoles, suggesting intraneuronal aggregation and pathology [36]. However, recent evidence supports the extracellular deposition of Aβ as the initiating pathogenic mechanism in the AD brain [37], with a direct correlation with the inhibition of anterograde axonal transport [38]. Despite direct evidence of Aβ-dependent neurodegeneration, Aβ pathology occurs prior to the appearance of clinical symptoms [37]. Accordingly, determining the level of amyloid deposition in an AD patient's brain (Aβ load) in a time-dependent manner would be informative in evaluating the progression of the disease and monitoring patient's response to antiamyloid therapies. Interestingly, through amyloid imaging, recent studies have demonstrated binding of the PET Pittsburgh compound B (PiB-PET) to Aβ peptides [39]. In this study, PET amyloid imaging with Pittsburgh compound B (PiB) showed increased cortical PiB binding in AD patients when compared to control subjects and intermediate binding levels in patients with mild cognitive impairment (MCI) [39]. This compound could be beneficial in the early detection of AD and evaluation of disease progression.
Recently, it was demonstrated by a combination of in vivo and in vitro studies that Aβ binding to the cellular prion protein (PrPc), an oligomer-specific high-affinity binding site for Aβ, can play a central role in Aβ-induced memory deficits, axon degeneration, synapse loss, and neuronal death in the AD brain through Fyn kinase activation [40]. The activation of this kinase results in alterations in N-methyl-D-aspartate receptor (NMDAR) function by increasing surface NMDAR expression along with its phosphorylation, and eventually leading to dendritic spine, in association with surface receptor loss [40]. The data suggests that by inhibiting PrPc in the APPswe/PSEN1-M146L double transgenic mouse, reversal of memory deficits and restoration of synaptic density could be achieved [40]. It has been demonstrated that Fyn kinase associates with the tau protein and that abnormal Fyn-tau interactions sensitise synapses to glutamate excitoxicity [40]. Together, these data suggest that PrPc-Fyn signalling may contribute to Aβ and tau pathologies and thus its downregulation may be a potential therapeutic approach.
4. Tau Protein Pathology
The tau protein is an integral component of the neuronal cytoskeleton [41] with a molecular weight ranging from 45 kDa to 65 kDa [41] and is responsible for the promotion of microtubule assembly in the normal brain [42]. Microtubule assembly is tightly regulated by a combination of protein kinases and phosphatases that balance the amount of tau phosphorylation [43, 44]. The most common tau pathology is seen in AD, but it is manifest in other diseases such as frontotemporal dementias and Parkinson's disease [45, 46]. In the AD brain, tau exists in a hyperphosphorylated state, which leads to aberrant secondary structures and loss of function, resulting in a reduced ability to bind to microtubules and to promote their assembly [47]. The abnormal translocation of tau from axonal microtubules to neuropil thread inclusions, cell bodies and dendritic processes, where tau aggregates and accumulates, are pther prominent cytopathological hallmarks observed within AD brain sections [48]. The tau protein is initially synthesised as a single chain polypeptide and then targeted by posttranslational modifications that alter its conformation, promoting tau dimerisation in an antiparallel manner [49]. Stable tau dimers subsequently form tau oligomers, which aggregate at an increasing rate to form subunits of filaments, called protomers. Two protomers twisted around each other with a crossover repeat of 80 nm, constitute the width varying between w10 and w22 nm to form paired helical filaments (PHFs), a characteristic of AD neuronal pathology [49, 50]. Assembly of PHFs finally establishes the neurofibrillary tangles (NFTs), which can be observed microscopically (Figure 2) [51]. Hyperphosphorylated tau sequesters normal tau and other neuronal microtubule associated proteins (MAPs), such as MAP1A, MAP1B, and MAP2, contributing further to disassembled microtubules, disruption of the axonal cytoskeleton, and transport, culminating as damaged neurons [52]. After neuronal death, tau oligomers are released into the extracellular environment which leads to microglial cell activation and, as a consequence, further progressive bystander neuronal degeneration [53]. It has been suggested that tau pathology results from elevated protein kinase activity, a reduction in the activity of protein phosphatase, or both [45]. Analysis of phosphorylated tau isolated from AD brains has identified numerous target serine or threonine residues [45]. It has been demonstrated that MAP-kinase, GSK-3β, and/or Cdk5 are the main kinases involved in tau phosphorylation. However, in AD not all tau phosphorylation events can be attributed to these kinases [45].
Figure 2.
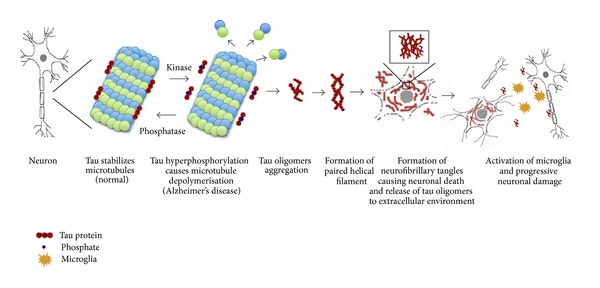
Stabilisation of microtubules by the tau protein is regulated by kinases and phosphatases. Abnormal hyperphosphorylation of tau proteins causes catastrophic microtubule depolymerisation and the formation of insoluble cytoplasmic tau oligomers, which aggregate to form protomers. Two protomers twisted around each other to form paired helical filaments (PHFs), which assemble to produce neurofibrillary tangles (NFTs).
The mechanism by which tau exerts its neuronal toxicity is still controversial [54]. It has been suggested that a series of degenerative signals such as Aβ aggregation, iron overload [55], oxygen free radicals [56], cholesterol levels in neuronal rafts, LDL species [57], and homocysteine can activate the innate immune response [53]. The activation of microglial cells, for instance, results in the subsequent release of pro-inflammatory cytokines that modify neuronal behaviour through anomalous signalling cascades, with the end result being the promotion of tau hyperphosphorylation [53]. However, numerous cellular and transgenic animal models indicate that tau is crucial for Aβ-induced neurotoxicity [54]. For instance, cultured hippocampal neurons from tau deficient mice are protected against Aβ pathology [54]. Furthermore, in cultured hippocampal neurons from wild-type mice, the silencing of tau by siRNA has demonstrated that tau is required for prefibrillar Aβ-induced microtubule disassembly. Furthermore, it was demonstrated that a reduction in soluble Aβ and tau but not Aβ alone causes cognitive decline in the triple transgenic AD mouse model with plaques and tangles [58]. These data suggest that although Aβ is the initial trigger, tau accumulation plays a central role in neurodegeneration. Finally, in the AD-like transgenic model that expresses human APP with familial mutations, suppression of endogenous tau prevented Aβ-dependent water maze learning and memory deficits without reversing the amyloid pathology [58]. Collectively, these data suggest a link between Aβ and tau that drive the neural pathologies and the manifestations of clinical symptoms. Preliminary data on the inhibition of tau aggregation by methylene blue chloride (MTC) has indicated a lower rate of cognitive decline in treated patients compared with those sporadic AD patients on alternate therapies, implicating tau as the key initiator of cognitive deficits [50]. However, the exact role of Aβ dependent in signal transduction cascades that are associated with pathogenic tau modifications and the contribution to the progression of neuronal death require further investigation [54].
5. Signalling Molecules Linked with Neuronal Cytoskeleton Disassembly
5.1. Rho Kinase (ROCK)
The Rho-associated coiled-coil forming protein kinases (ROCKs) include the ROCK-1 and ROCK-2 isoforms. These two kinases contain highly conserved aminoterminal but different carboxy-terminal domains [59]. Both ROCK-1 and ROCK-2 were originally shown to be involved in cell differentiation, essential for the regulation of myogenesis from embryonic fibroblasts along with skeletal muscle maturation and differentiation [60]. Both Rho kinase (ROCKs) and p21-activated kinase (PAKs) are members of the serine/threonine class of protein kinases. However, they are known to have antagonistic effects on the actin cytoskeleton and therefore on the plasticity of synapses. PAK also has two major isoforms, PAK1 and PAK3, and they have downstream signalling effects on Rho/Rac (for review see [61]). PAK can stimulate actin polymerisation [62], axon outgrowth, and the formation of dendritic spines [63] through LIM kinase stimulation [62]. PAK can also inhibit the myosin light chain kinase (MLCK) which diminishes actomyosin contractility [64, 65].
It has been reported that 13-month-old AD-like mice (PDAPP) displayed a substantial decrease in PAK 1-3 activity compared to normal controls [21]. Furthermore, the hippocampi of patients exhibiting the early clinical signs of AD have displayed high PAK 1-3 activity which was then shown to decline in the late stages of AD pathology [21]. It was further suggested that C-terminal cleavage of APP at the Asp664 site mediates PAK abnormalities and that an Asp664 mutation may potentially prevent these abnormalities [21]. On the other hand, ROCKs stimulate the retraction of axonal and dendritic growth cones by activating MLCK through the phosphorylation of myosin light chain proteins to promote an interaction with actin [66]. In addition, ROCK2 can phosphorylate collapsin response mediator protein 2 (CRMP2), another microtubule associated protein, to induce growth cone collapse [67].
Moreover, many developmentally or pathologically regulated molecules can also activate the RhoA/ROCK pathway to inhibit axonal growth including semaphorins, ephrins, and myelin inhibitory factors, such as Nogo and myelin-associated glycoprotein (MAG). On the other hand, there are some signalling molecules such as Sema4D/plexin-B1 that activate the RhoA/ROCK pathway especially in hippocampal neurons that may induce dendritic spine formation. It has been speculated that this may be due to the activation of LIM kinase and the PAK-type response via actin-depolymerising factor ADF/cofilin [68, 69].
In AD, dendritic spine defects play a major role in cognitive impairments [61]. It has been reported that dendritic postsynaptic proteins are excessively distorted with disease progression [70]. For instance, neuronal loss in the hippocampi of AD patients is approximately 5–40% while the loss of postsynaptic proteins such as the developmentally regulated actin-regulating brain protein (drebrin), which is targeted by Aβ oligomers, reaches 70–95% [71]. This study in particular suggested that Aβ-induced alteration in postsynaptic PAK may have a central role in the massive drebrin loss and cognitive deficits found in AD, which could be prevented by an antibody to Aβ and/or by in vivo or in vitro overexpression of wild-type PAK [71].
Cognitive defects and eventually dementia are important clinical features of AD (for review see [2]). It has been reported that there exists a relationship between the cognitive-decline occurring in AD along with genetic mental retardation syndromes and synaptic dysfunction, primarily since the postsynaptic maintenance of dendritic spines is lost. To maintain synaptic balance, both ROCK1 and 2 transduce signals to retract the growth cones and dendritic spines (for review see [72]). It has been shown that ROCK may provoke APP breakdown to the toxic β-amyloid 1-42 peptide. For example, ROCK inhibitors, such as Y27632, inhibit the toxic processing of APP [73]. An intriguing conundrum is that the binding of Aβ on neurons may activate RhoA and ROCK2 to potentiate the phosphorylation of its substrates [74, 75]. One of the specific substrates that our group has recently defined is CRMP-2 (Figure 3). It has been shown that CRMP-2 exhibits hyperphosphorylation in the cortex of AD postmortem brains [76]. Experimentally, it has been illustrated that other kinases such as GSK-3 and Cdk5 can also phosphorylate CRMP-2 and produce growth cone collapse in neurons [77] (Figure 3). Our data suggest that β-amyloid can increase the RhoA-GTP level in differentiated SH-SY5Y cells increasing CRMP-2 phosphorylation and reducing the neurite lengths in cultured neuroblastoma cells. Additionally, RhoA and CRMP-2 levels are elevated in neurons surrounding amyloid plaques in the cerebral cortex of the APP (Swe) Tg2576 AD mouse model. Our work indicates that Aβ induces Rho GTPase activity and ROCK2 to promote CRMP-2 phosphorylation which can lead to the inhibition of neurite outgrowth [78] (Figure 3). However, a direct link with the reduction of ROCK2-dependent CRMP-2 phosphorylation and the limitation of cognitive decline is yet to be established in the context of Aβ-dependent neurodegeneration.
Figure 3.
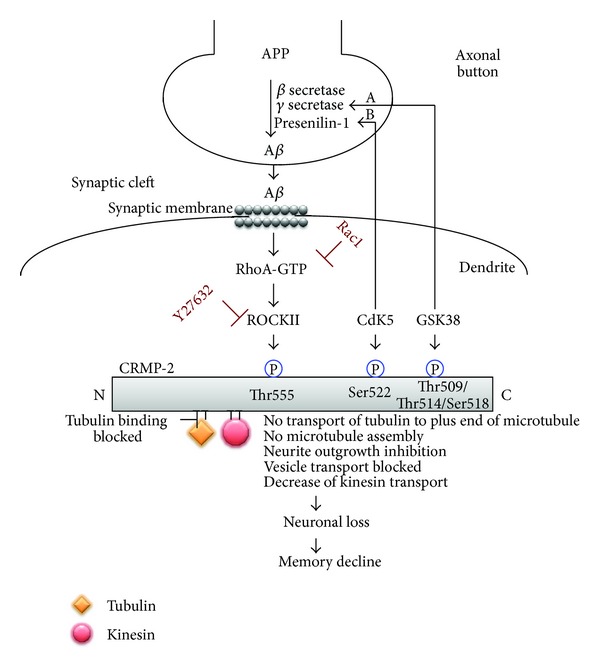
Model of Aβ-mediated neurite outgrowth inhibition. Aβ (oligomeric) activates the small GTPase, RhoA, which inhibits the proneurite outgrowth GTPase Rac1. RhoA-GTP activates Rho Kinase (ROCK II) to effect microfilament rearrangement and also potentiate microtubule disassembly. Microtubule disassembly occurs when ROCK II directly phosphorylates CRMP-2 at the Thr555 position preventing the association of CRMP-2 with tubulin heterodimers, thereby affecting neurite outgrowth inhibition. Neurite outgrowth is further impeded by CRMP-2 phosphorylation since this prevents the microtubule motor protein, kinesin, to associate with CRMP-2 and transport growth-related vesicular cargo, such as BDNF, antergradely to the distal end of the neurite. It is demonstrated that CRMP-2 is also phosphorylated by GSK-3β and Cdk-5. (A) Studies have demonstrated that GSK-3β activity can also regulate the processing of APP resulting in the production of Aβ, which in turn can further increase GSK-3β activity through PI3K inhibition, illustrating as a potential feedback loop. (B) Additionally, it has been suggested that Cdk5 may phosphorylate presenilin-1 at Thr354 destabilising its carboxy-terminal fragment, leading to increased APP processing.
5.2. Glycogen Synthase Kinase-3β (GSK-3β)
The proline-directed serine/threonine kinase, glycogen synthase kinase-3 (GSK-3), is important for several cellular processes such as metabolism, cell structure, and apoptosis and in the regulation of gene expression (for review see [79]). The GSK-3 family contains two members, GSK-3α and GSK-3β, that are highly expressed in the brain and spinal cord with GSK-3β playing a central role in neuronal differentiation and the maintenance of neurons (for review see [80]). Activation of GSK-3 requires prephosphorylation by other priming kinases such as Cdk5 at serine or threonine sites located 4 residues, C-terminal to the site phosphorylated by GSK-3 (for review see [81]) (Figure 3). Abnormal GSK-3 function has been implicated in different brain pathologies indicating its fundamental role in controlling basic mechanisms of neuronal function, modulation of neuronal polarity, migration, proliferation, and survival, not to mention the establishment of neuronal circuits (for review see [82]). It has been demonstrated that phosphorylation of GSK-3 may influence cytoskeletal proteins altering neuronal plasticity (for review see [83]). Neuronal cytoskeletal changes occur due to an altered rate in the stabilisation/destabilisation of microtubules (MT), thereby altering the dynamics of dendrites, spines, axons, and synapses. Intensified efforts in the identification of enzymes involved in regulating tau phosphorylation in vivo have revealed GSK-3β as a candidate kinase for therapeutic targeting [79] during AD pathology.
It has been hypothesised that GSK-3 overactivity may potentiate sporadic and familial forms of AD by enhancing tau hyperphosphorylation [84] and APP processing and possibly through the phosphorylation of CRMP-2 leading to profound memory impairment [81] (Figures 2 and 3). It has been established that the expression of full-length unmodified or unphosphorylated CRMP-2, in primary hippocampal neurons or SH-SY5Y neuroblastoma cells, promotes axon elongation. Moreover, cultured neurons expressing CRMP-2 with mutant GSK-3 phosphorylation sites (T509A, S518A) display significantly reduced axon elongation [81]. On the other hand, studies have demonstrated that GSK-3β phosphorylation of the CRMP-2 T509 site can play a crucial role in mediating the repulsive action of Sema3A [85] and promoting growth cone collapse [77]. Recently, Cole et al. have demonstrated that dephosphorylation of CRMP-2 at the GSK-3β-dependent sites (Ser-518/Thr-514/Thr-509) can be carried out by a protein phosphatase 1 (PP1) in vitro, observed in neuroblastoma cells and primary cortical neurons, and that the inhibition of GSK-3β by insulin-like growth factor-1 or the highly selective inhibitor CT99021 results in dephosphorylation of CRMP-2 at these sites [86]. How this may be translated to real therapeutic outcomes during AD pathology is yet to be demonstrated, even within animal models of disease.
5.3. Cyclin-Dependent Kinase-5 (Cdk5)
The other proline-directed serine/threonine kinase, identified as a major priming enzyme for tau phosphorylation, is cyclin-dependent kinase-5 (Cdk5) [87]. Although Cdk5 is ubiquitously expressed in most tissues, it is not directly involved in mediating progression through the cell cycle as it requires prior activation by p35 and p39, which are expressed almost exclusively in the CNS [88]. Cdk5 plays an important role in CNS development possibly by mediating interactions between neurons and glia during radial migration, which is essential for developing appropriate cortical laminar architecture [89, 90]. Furthermore, Cdk5 has been reported to also play a role in neuronal differentiation, axonal guidance, synaptic plasticity, cellular motility, cellular adhesion, and neurodegeneration (for review see [91]).
Studies have shown that inhibition of Cdk5 reduces Aβ-induced neurodegeneration in cortical neurons [92] which highlights that targeting Cdk5 could be a future therapeutic strategy for neurodegenerative disorders. The critical microtubule associated protein, CRMP-2, has been also demonstrated to be a substrate for Cdk5 [77]. This study showed an orderly phosphorylation process of CRMP-2 by Cdk5 (defining it as the priming kinase) followed by GSK-3β as a consequence of Sema3A stimulation that inhibits axonal growth [77]. Alternatively, a non-phosphorylated form of CRMP-2 cannot respond to Sema3A signalling. This study also demonstrated that Sema3A promotes phosphorylation of CRMP-2 at Ser522, which is the established Cdk5 phosphorylation site [77]. Thus, targeted kinase inhibitors may possibly be therapeutically beneficial in AD to limit both tau and CRMP-2 phosphorylation. Deciphering which of the kinases precipitate neurodegeneration is still under investigation but when elucidated, the possibility exists that formulation of specific inhibitors to prevent cognitive decline associated with AD is achievable.
5.4. Phosphatases
Protein phosphatases provide unique endogenous signalling mechanisms for the dephosphorylation of proteins, reversing such posttranslational modifications, which may limit protein dysfunction. Protein phosphatase 2A (PP2A) is one of the most important serine/threonine phosphatases in the mammalian brain. It also exists in most tissues comprising up to 1% of total cellular protein. It has major roles in development, cell growth, transformation (for review see [3]), regulation of protein phosphorylation, and cell signalling pathways [93]. PP2A is composed of 3 subunits: subunit A (scaffolding/structural), subunit B (regulatory/targeting), and subunit C (catalytic) [94]. PP2A with PP1 collectively account for more than 80% of the total serine/threonine phosphatase activity in all mammalian cells [3, 95] making these enzymes integral to cellular physiology.
In situ, PP2A, PP1, PP5, and PP2B account for 71%, 11%, 10%, and 7%, respectively, of the total tau phosphatase activity in the human brain [96]. PP2A is the most prevalent phosphatase involved in tau dephosphorylation [97]. Knockdown of PP2A phosphatase activity was shown to lead to tau hyperphosphorylation [98]. Furthermore, when PP2A was inhibited in cultured cells and in transgenic mice with mutant PP2A, hyperphosphorylation of tau was observed [98]. Moreover, the naturally abundant SET protein, a potent PP2A inhibitor, is found to be elevated in AD brains [99], possibly illustrating reduced PP2A activity allowing for the hyperphosphorylation of cellular substrates to occur unabated and the potentiation of neurodegeneration. Interestingly, autopsy studies of brains from AD patients, non-AD dementia, and normal human brains demonstrate that there is loss in PP2A protein, mRNA, and enzymatic activity in areas of the brain affected by AD, the hippocampus and cortex, but not in the cerebellum [100]. In addition, the inhibition of PP2A activity mimics most of the phosphorylation events seen in AD, such as tau hyperphosphorylation [101].
Phosphorylation of APP by an array of kinases has been shown to influence its cleavage by β-secretase resulting in Aβ production [102]. It was demonstrated that PP2A has the ability to dephosphorylate APP at the Thr668 site and thus inhibit Aβ generation [103]. Studies of cells expressing the (APPswe) mutation, transgenic mice expressing both APPswe and presenilin mutations, and sections of hippocampus and entorhinal cortex from human AD patients, show that PP2A levels are decreased and Y307 levels (an inhibitor of PP2A) were increased [104] implying that the phosphatase affects the processing of APP and highlighting its importance in limiting AD pathology. In N2a cells, where PP2A was inhibited with okadaic acid (OA), the phosphorylation of APP and the secretion of both sAPPα and sAPPβ were all elevated [105]. In addition, inhibition of the protein phosphatases PP1 and PP2A in rat brain by OA results in the accumulation of hyperphosphorylated tau and Aβ species [45, 94]. Even though incubation of different types of cells with OA resulted in the stimulation of APP secretion, it was not proven that the effect was mediated by PP1 [106] and/or PP2A [107]. Moreover, it was demonstrated that demethylation of PP2A by nuclear phosphatase methylesterase-1 (PME-1) reduces its activity and thus leads to tau hyperphosphorylation along with APP phosphorylation, promoting APP cleavage and Aβ production [108–110]. Collectively, these results suggest that downregulation of PP2A may induce Aβ production and tau phosphorylation, precipitating AD pathology.
A direct link of PP2A activity with the progression of AD pathology has been affiliated to the fact that CRMP-2 phosphorylation may actually be a result of lowered PP2A activity [93]. Since CRMP-2 hyperphosphorylation was commonly observed to correspond with progressive neurodegeneration, decreased PP2A may well regulate such a disease-specific event. However, such a hypothesis would need to be substantiated beyond a causal link.
5.5. Collapsin Response Mediator Protein (CRMP)
The collapsin response mediator proteins (CRMPs) are members of the dihydropyrimidinase-related neuronal phosphoprotein family [111]. The CRMP family has five isoforms, CRMP1-5 [112]. The most well characterised of these, CRMP-2, is highly expressed in the adult mammalian CNS localising in the cytoplasm and neurites of postmitotic neurons [111]. CRMP-2 is also highly expressed in the areas of the adult brain of greatest plasticity such as the hippocampus, olfactory bulb, and cerebellum [113]. In neurons, CRMP-2 is concentrated within the distal portions of neurites, in synapses and in growth cones [114]. It regulates the polarity and differentiation of neurons through the assembly and trafficking of microtubules [115]. CRMP-2 has no known enzymatic activity by itself but through an interaction with other binding partners it can regulate neural differentiation, dendrite/axon fate specification, Ca2+ homeostasis, neurotransmitter release, regulation of cell surface receptor endocytosis, kinesin-dependent axonal transport, growth cone collapse, neurite outgrowth, and microtubule dynamics (for review see [78, 116]). The last three functions have been demonstrated to be regulated by phosphorylation near the C-terminus of CRMP-2 by kinases [117, 118] including cyclin-dependent kinase 5 (Cdk5), glycogen synthase kinase-3β (GSK-3β) [31, 76, 86, 119], Tau-tubulin kinase-1 (TTBK1) [120], and Rho kinase II (ROCKII) [78, 117, 121], all of which culminate in neurite retraction (for review see [78]). CRMP-2 hyperphosphorylation in AD was suggested to be a result of increased kinase activity, decreased phosphatase activity, or both [86]. All phosphorylation events can disrupt the association of mature full-length CRMP-2 with tubulin heterodimers possibly resulting in the destabilisation of the neuronal microtubule system rendering axonal retraction [67]. Moreover, disruption of the binding between CRMP-2 and tubulin due to the phosphorylation of CRMP-2 can block tubulin transport to the plus ends of microtubules for assembly (Figure 3) [78], blocking neurite outgrowth/elongation. In primary neurons and neuroblastoma cells, it has been demonstrated that overexpression of CRMP-2 results in axon elongation [114] while overexpression of truncated CRMP-2, lacking the C-terminus tubulin binding domain, inhibits axon growth. These data implicate this region of CRMP-2 to play a central role in axonal growth [114]. Both the Cdk5 and GSK-3β phosphorylation of CRMP-2 have been shown to be increased in the cortex and hippocampus of the triple transgenic mouse (PS1/APP/Tau mutant), along with the double transgenic mouse (PS1/APP mutant), that develop AD-like plaques along with NFTs. However, in transgenic mice, which display only mutant tau (P301L) that develop tangles but do not develop amyloid plaques, Cdk5 phosphorylation of CRMP-2 does not occur. These results indicate that hyperphosphorylation of CRMP-2 might be induced by APP overexpression and/or its enhanced processing, thereby generating a high amyloid load within the brain of these transgenic mice [76].
Our laboratory has recently demonstrated that, in human neuroblastoma SH-SY5Y cells and in the Tg2576 mouse model of AD, Aβ can reduce the length of neurites by inactivating the neurite outgrowth-signalling molecule Rac1 [78]. Furthermore, the data suggested that Aβ-mediated reduction in neurite length could be reversed by the Rho Kinase inhibitor (Y27632). Additionally, the Aβ-mediated decrease in neurite length was linked to the promotion of a threonine phosphorylation of CRMP-2 (unrelated to GSK-3β-dependant phosphorylation), conferring a reduced binding capacity to tubulin, both of which can be reversed by inhibiting RhoA activity [78]. These data suggested that Aβ-mediated neurite outgrowth inhibition results from the activity of RhoA-GTP and the dysregulation of CRMP-2 to bind tubulin for neurite outgrowth [78] (Figure 3).
Studies using transgenic mouse models expressing the Swedish familial AD mutant (APP/TTBK1) demonstrated that the induced upregulation of tau tubulin kinase-1 (TTBK1) can promote axonal degeneration via phosphorylation of CRMP-2 and tau within the entorhinal cortex and hippocampus, implicating TTBK1 as a potential therapeutic target for AD [120].
Despite the profound link to CRMP-2-dependent degeneration through kinase-mediated phosphorylation, another function of CRMP2 is mediated through its known association with kinesin, facilitating the anterograde molecular transport of growth promoting vesicles along axonal microtubules [122]. The exact mechanism of binding and transport and its contribution to AD will be discussed in detail below.
6. CRMP2-Tubulin Binding
The microtubule and actin cytoskeleton orchestrates axonal growth cone dynamics by a process of signal transduction leading to either depolymerisation or polymerisation events, for directional growth [119]. As already discussed above, the binding of CRMP2 to tubulin heterodimers can enhance microtubule assembly leading to axon outgrowth [123, 124]. Semaphorin-3A (Sema3A) is an extracellular protein that can block axonal outgrowth [77] through the activation of Cdk5, with downstream phosphorylation of both tau and CRMP-2 [31, 77]. Such phosphorylation can disrupt their tubulin association limiting axonal growth. Following the Cdk5 phosphorylation of CRMP-2, the latter may potentiate a conformational change leading to subsequent phosphorylation by GSK-3β [31, 77]. However, it has been demonstrated that in GSK-3β overexpressing mice, no hyperphosphorylation of CRMP-2 can be identified at the GSK-3β phosphorylation sites and furthermore phosphorylation of tau does not increase [125]. This may explain the finding that activation of GSK-3β alone can not induce growth cone collapse (for review see [119]). Interestingly, protein lysates from human AD cortex and animal models of AD show hyperphosphorylation of CRMP-2 at residues Thr509, Thr514, and Ser518 which are known to be the GSK-3β phosphorylation sites as well as Ser522, the well-known Cdk5 phosphorylation site (for review see [78]). These findings indicate that Sema3A signalling may regulate microtubule polymerisation through the physiological actions of tau and CRMP-2, which regulate the dynamics of microtubules and tubulin dimers, respectively [126]. Phosphorylation of CRMP-2 by Rho kinase at the Thr555 site, however, can also reduce the CRMP-2 association with tubulin heterodimers and induce growth cone collapse unrelated to Sema3A signalling and quite possibly be the result of Aβ-dependent signalling [31, 77]. The phosphorylation of CRMP-2 by Cdk5, GSK-3β, and Rho kinase may therefore play a central role in coordinating cytoskeletal activities in response to multiple axon guidance cues [31, 77].
The plausible hypothesis exists that activation of all thre kinases Cdk5/GSK-3b/ROCK2, contribute to the destabilisation of the neuronal microtubule system in AD. Consequently, tau and CRMP-2 have some similarities in that both control microtubule polymerisation and stability and they both respond to the growth cone guidance molecule Sema3A [77]. Therefore, it can be theorised that a balanced treatment which may successfully decrease CRMP-2 phosphorylation could also be effective in regard to tau aggregation and vice versa in AD (for review see [31]).
7. Microtubules (MT)
One of the most important physiological features of the multipolar neuron is to have a polarised axon, that can extend to more than 1 meter in the human CNS [127]. For the neuron to function normally, it should be able to transport vital molecular cargo from its body to synaptic terminals and vice versa in a timely manner through the axon via anterograde and retrograde transport mechanisms, respectively [127, 128]. Therefore, it stands to reason that the integrity of the microtubule transport system is crucial for axonal transport [129]. The microtubule system facilitates ATP driven transport through molecular motors of the cell's vital components which include vesicles, proteins, mitochondria, chromosomes, and large macromolecules such as microtubule heterodimers themselves [128, 130]. The transport machinery directly interacts with microtubules and includes two families of proteins categorised according to their directional movement. These proteins include either microtubule plus end-directed kinesins or the microtubule minus end-directed cytoplasmic dynein [127].
Many neurodegenerative diseases, such as AD, display a blockade in microtubule transport, emphasising its significance in normal physiology and highlighting abnormal neuronal vesicle trafficking as a potential pathogenic mechanism [130–132]. It is believed that Aβ may cause mitochondrial dysfunction and, therefore, axonal transport defects [132]. It has been demonstrated that APP processing and Aβ overproduction in the mitochondria lead to mitochondrial dysfunction and therefore reduction of mitochondrial energy supply and inhibition of axonal transport [133]. Enhancing energy supply of neurons could be critical to compensate for the Aβ-dependent loss of energy and thus facilitate axonal transport.
Microtubule depolymerisation has been touted as a contributing factor in the gross loss of memory, as it is necessary to stabilise newly formed microtubules in spines for long-lasting memory [134, 135]. There exists evidence implicating tubulin sequestration [136] and blockade in microtubule assembly as a pathogenic mechanism of AD [129]. It has been recently demonstrated that in vitro, microtubules can be assembled from the cytosol of normal autopsy brain obtained within five hours postmortem, while this is not possible from identically treated AD postmortem brain tissue [129]. Furthermore, it has been documented that axonal transport is defective in neurons from AD postmortem brains indicating the destruction of the microtubule cytoskeleton in axons of diseased neurons [134]. There also exist data suggesting that the abnormality in axonal transport might stimulate the formation of, or enhance the accumulation of, Aβ [134, 137], through autophagocytosis of mitochondria without normal lysosomal degradation [137].
One of the main physiological functions of tau is to stimulate microtubule assembly by polymerising with tubulin, maintaining the microtubule structure and stability through its capacity to anchor polymerised microtubules to the internal axolemma [129]. Evidence for the role of tau and microtubule destabilisation arises from tau transgenic mice which show spinal cord tau inclusions [131]. In this animal model, an inability of tau to stabilise microtubules can be compensated with the MT-stabilising agent paclitaxel resulting in increased MT density and marked improvement in motor function [131]. However, paclitaxel is thought to have poor blood-brain barrier permeability and thus is an unlikely candidate for human therapy during neurodegeneration [131].
In the early stages of AD pathogenesis, observations within the neuropil demonstrate that there exists an abnormal aggregation of the activated actin-associated protein cofilin, a protein that modulates actin-rich dendritic spine architecture, which is important for learning and memory [43]. Those neuropil threads can disrupt the cytoskeletal network by blocking cargo trafficking to synapses, resulting in memory and cognition impairment [43]. It is also suggested that abnormal activation of cofilin may trigger the accumulation of phosphorylated tau in neuropil threads [43]. The activities of cofilin and the protein actin-depolymerising factor (ADF) are regulated by phosphorylation and dephosphorylation through LIM and other kinases, along with chronophin phosphatases, respectively [43]. Heredia et al. found that β-amyloid may activate LIMK1 and thus stimulate ADF/cofilin phosphorylation in cultured neurons [69]. Moreover, they demonstrated, in the AD brain, that the number of P-LIMK1-positive neurons was extensively increased in the affected regions [69]. A recent study of AD transgenic mice demonstrated that neuronal cell bodies are viable although the neurites are damaged [138]. Taken together, these studies highlighted that the development of in vivo methods to disrupt LIMK1 activation, the formation of the cofilin-actin rods, and/or the interaction between cofilin and pMAP, may be a plausible way to stop the disease early in its presentation.
8. Kinesin
The microtubule motor protein complex, kinesin-1, has a fundamental role in the vesicular transport from the neuronal cell body, along the axon and anterograde, to the synapse (for review see [139]). The motor protein complex consists of two kinesin heavy chains (KHC) that have both an ATP and the microtubule binding motif which are essential for vesicle transport [140]. Two kinesin light chains (KLC) that associate with the heavy chain and vesicular cargo membranes [140] complete the structure of the transport protein. APP is one of the molecular candidates for receptors that attach kinesin-1 to vesicular cargo [139]. The carboxy terminus of APP binds directly to the light-chain subunits of kinesin-1 [140] and thus plays a major role in the recruitment of kinesin-1 to axonal vesicles [141]. Moreover, the level of axonal APP is suggested to play a central role in determining expression levels of kinesin-1 decorating vesicles, providing the ability to determine the anterograde movement behaviour of APP-containing vesicles [141]. It has been reported that kinesin blockade and axonal swellings are involved in the pathogenesis of the early stages of AD even before the formation of amyloid plaques and neurofibrillary tangles, although the initiating events are not clear [142]. Moreover, in animal models, β-amyloid formation and its subsequent transport are enhanced when kinesin transport is abrogated or impaired [38, 141]. Axonal transport damage results in the development of axonal swellings where APP is processed into smaller Aβ species. APP axonal transport is mediated by direct binding to KLC1 [143]. Genetic manipulation designed to damage APP axonal transport in AD mouse models, such as Tg-swAPPPrp, demonstrated the enhancement in the incidence of axonal swellings, elevated Aβ levels, and potentiated the production of amyloid deposition [142]. In particular, APP directly interacts with KLC1 (the microtubule transport machinery) through its carboxy terminus, suggesting that impaired interaction of APP and KLC1 might play a central role in the AD pathogenesis [144]. Decreased KLC1 transport may also stimulate tau hyperphosphorylation and formation of NFTs as well as axonal swellings producing catastrophic damage to axons. Such damage may arise from increased Aβ levels and tau hyperphosphorylation, further disrupting axonal transport [145].
It is now well established that CRMP-2 plays a central role in negotiating fast axonal transport by acting as an adaptor protein to the microtubule motor kinesin-1, for propagation of anterograde vesicle transport of key traffic molecules such as the high affinity neurotrophin receptor, tyrosine kinase (TrkB). Following distal localisation of this receptor, TrkB is inserted into the cell membrane and activated by its cognate ligand brain-derived neurotrophic factor (BDNF), resulting in axonal growth through signalling within the growth cone, thereby establishing the accumulation and polymerisation of F-actin and tubulin. In AD, phosphorylated CRMP-2 releases kinesin-1, inhibiting TrkB function and limiting the structural integrity of the actin-based cytoskeleton in distal axons, growth cones, and synapses [146]. Inhibiting CRMP-2 phosphorylation could be beneficial to restore tubulin and kinesin-1 binding to CRMP-2 and thus promoting axonal outgrowth and transport of important molecular cargo.
9. Conclusion
Alzheimer's disease (AD) is an age-related progressive neurodegenerative disorder and is the most common form of dementia in the elderly. The hallmarks of AD pathology are the extracellular deposition of a 4 kDa amyloid beta (Aβ) polypeptide and the formation of intracellular neurofibrillary tangles (NFTs) along with dystrophic neurites, degenerating neurons, and activated astrocytes and microglia, a part of the reactive pathology observed around senile plaques. Neuritic plaques result from the aggregation of the amyloid β protein (Aβ) which is a consequence of amyloid precursor protein (APP) aberrant processing. The corresponding accumulation of filamentous inclusions within the CNS as neurofibrillary tangles (NFTs), resulting from the hyperphosphorylation of the microtubule-associated protein, tau and amyloid deposition, are both pathognomonic to sporadic AD. There is an impressive list of genes and proteins involved in AD pathologies including APP, presenilins, secretases, kinases, and phosphatases all touted as being responsible for either increasing the production of the neurotoxic Aβ protein or promoting the hyperphosphorylation of CRMP-2 or tau, leading to the devastating neurodegenerative sequelae. The understanding of the major gene players cooperating with key environmental factors that contribute to the manifestation of AD pathology is fundamental in the derivation of a more comprehensive understanding of AD pathogenesis and for the development of specific and more effective treatments of this devastating age-dependent disease.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
Acknowledgments
Sara Mokhtar was supported by King Abdul-Aziz University postgraduate scholarship; Steven Petratos was supported by National Multiple Sclerosis Society (USA) Project Grant ID no. RG43981/1.
References
- 1.Kozlowski H, Luczkowski M, Remelli M, Valensin D. Copper, zinc and iron in neurodegenerative diseases (Alzheimer’s, Parkinson’s and prion diseases) Coordination Chemistry Reviews. 2012;256(19-20):2129–2141. [Google Scholar]
- 2.Kumar P, Pradhan K, Karunya R, Ambasta RK, Querfurth HW. Cross-functional E3 ligases Parkin and C-terminus Hsp70-interacting protein in neurodegenerative disorders. Journal of Neurochemistry. 2012;120(3):350–370. doi: 10.1111/j.1471-4159.2011.07588.x. [DOI] [PubMed] [Google Scholar]
- 3.Liu R, Wang J. Protein phosphatase 2A in Alzheimer’s disease. Pathophysiology. 2009;16(4):273–277. doi: 10.1016/j.pathophys.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 4.Crews L, Rockenstein E, Masliah E. APP transgenic modeling of Alzheimer’s disease: mechanisms of neurodegeneration and aberrant neurogenesis. Brain Structure and Function. 2010;214(2-3):111–126. doi: 10.1007/s00429-009-0232-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Przedborski S, Vila M, Jackson-Lewis V. Neurodegeneration: what is it and where are we? Journal of Clinical Investigation. 2003;111(1):3–10. doi: 10.1172/JCI17522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chan KC, Cai KX, Su HX, et al. Early detection of neurodegeneration in brain ischemia by manganese-enhanced MRI. Proceedings of the IEEE Conference on Engineering in Medicine and Biology Society; 2008; pp. 3884–3887. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe D, Mandelkow E, Holtzman D. Deciphering Alzheimer disease. Cold Spring Harbor Laboratory Press. 2012;2(1) doi: 10.1101/cshperspect.a011460.a011460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morimatsu M, Hirai S, Muramatsu A, Yoshikawa M. Senile degenerative brain lesions and dementia. Journal of the American Geriatrics Society. 1975;23(9):390–406. doi: 10.1111/j.1532-5415.1975.tb00425.x. [DOI] [PubMed] [Google Scholar]
- 9.Sisodia SS, Koo EH, Beyreuther K, Unterbeck A, Price DL. Evidence that β-amyloid protein in Alzheimer’s disease is not derived by normal processing. Science. 1990;248(4954):492–495. doi: 10.1126/science.1691865. [DOI] [PubMed] [Google Scholar]
- 10.Kirschner DA, Abraham C, Selkoe DJ. X-ray diffraction from intraneuronal paired helical filaments and extraneuronal amyloid fibers in Alzheimer disease indicates cross-β conformation. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(2):503–507. doi: 10.1073/pnas.83.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esch FS, Keim PS, Beattie EC, et al. Cleavage of amyloid β peptide during constitutive processing of its precursor. Science. 1990;248(4959):1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 12.Williamson TG, Mok SS, Henry A, et al. Secreted glypican binds to the amyloid precursor protein of Alzheimer’s disease (APP) and inhibits APP-induced neurite outgrowth. Journal of Biological Chemistry. 1996;271(49):31215–31221. doi: 10.1074/jbc.271.49.31215. [DOI] [PubMed] [Google Scholar]
- 13.Small DH, Williamson T, Reed G, et al. The role of heparan sulfate proteoglycans in the pathogenesis of Alzheimer’s disease. Annals of the New York Academy of Sciences. 1996;777:316–321. doi: 10.1111/j.1749-6632.1996.tb34439.x. [DOI] [PubMed] [Google Scholar]
- 14.Small DH, Nurcombe V, Reed G, et al. A heparin-binding domain in the amyloid protein precursor of Alzheimer’s disease is involved in the regulation of neurite outgrowth. Journal of Neuroscience. 1994;14(4):2117–2127. doi: 10.1523/JNEUROSCI.14-04-02117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wei WQ, Ferreira A, Miller C, Koo EH, Selkoe DJ. Cell-surface β-amyloid precursor protein stimulates neurite outgrowth of hippocampal neurons in an isoform-dependent manner. Journal of Neuroscience. 1995;15(3):2157–2167. doi: 10.1523/JNEUROSCI.15-03-02157.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ohsawa I, Hirose Y, Ishiguro M, Imai Y, Ishiura S, Kohsaka S. Expression, purification, and neurotrophic activity of amyloid precursor protein-secreted forms produced by yeast. Biochemical and Biophysical Research Communications. 1995;213(1):52–58. doi: 10.1006/bbrc.1995.2097. [DOI] [PubMed] [Google Scholar]
- 17.Milward EA, Papadopoulos R, Fuller SJ, et al. The amyloid protein precursor of Alzheimer’s disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9(1):129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- 18.Jin LW, Ninomiya H, Roch J, et al. Peptides containing the RERMS sequence of amyloid β/A4 protein precursor bind cell surface and promote neurite extension. Journal of Neuroscience. 1994;14(9):5461–5470. doi: 10.1523/JNEUROSCI.14-09-05461.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breen KC, Bruce M, Anderton BH. Beta amyloid precursor protein mediates neuronal cell-cell and cell-surface adhesion. Journal of Neuroscience Research. 1991;28(1):90–100. doi: 10.1002/jnr.490280109. [DOI] [PubMed] [Google Scholar]
- 20.Allinquant B, Hantraye P, Mailleux P, Moya K, Bouillot C, Prochiantz A. Downregulation of amyloid precursor protein inhibits neurite outgrowth in vitro. Journal of Cell Biology. 1995;128(5):919–927. doi: 10.1083/jcb.128.5.919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen TVV, Galvan V, Huang W, et al. Signal transduction in Alzheimer disease: p21-activated kinase signaling requires C-terminal cleavage of APP at Asp664. Journal of Neurochemistry. 2008;104(4):1065–1080. doi: 10.1111/j.1471-4159.2007.05031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinha S, Lieberburg I. Cellular mechanisms of β-amyloid production and secretion. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(20):11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anderson JP, Chen Y, Kim KS, Robakis NK. An alternative secretase cleavage produces soluble Alzheimer amyloid precursor protein containing a potentially amyloidogenic sequence. Journal of Neurochemistry. 1992;59(6):2328–2331. doi: 10.1111/j.1471-4159.1992.tb10128.x. [DOI] [PubMed] [Google Scholar]
- 24.Snyder EM, Nong Y, Almeida CG, et al. Regulation of NMDA receptor trafficking by amyloid-β . Nature Neuroscience. 2005;8(8):1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 25.Mucke L, Masliah E, Yu G, et al. High-level neuronal expression of Aβ(1-42) in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. Journal of Neuroscience. 2000;20(11):4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muresan V, Varvel NH, Lamb BT, Muresan Z. The cleavage products of amyloid-β precursor protein are sorted to distinct carrier vesicles that are independently transported within neurites. Journal of Neuroscience. 2009;29(11):3565–3578. doi: 10.1523/JNEUROSCI.2558-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahlgren S, Li GL, Olsson Y. Accumulation of β-amyloid precursor protein and ubiquitin in axons after spinal cord trauma in humans: immunohistochemical observations on autopsy material. Acta Neuropathologica. 1996;92(1):48–55. doi: 10.1007/s004010050488. [DOI] [PubMed] [Google Scholar]
- 28.Cras P, Kawai M, Lowery D, Gonzalez-DeWhitt P, Greenberg B, Perry G. Senile plaque neurites in Alzheimer disease accumulate amyloid precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(17):7552–7556. doi: 10.1073/pnas.88.17.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sisodia SS, Tanzi RE. Alzheimer's Disease. Advances in Genetics and Cellular Biology. New York, NY, USA: Springer Science+Business Media LLC; 2007. [Google Scholar]
- 30.Chakravarthy B, Gaudet C, Ménard M, et al. Amyloid-β peptides stimulate the expression of the p75NTR neurotrophin receptor in SHSY5Y human neuroblastoma cells and AD transgenic mice. Journal of Alzheimer’s Disease. 2010;19(3):915–925. doi: 10.3233/JAD-2010-1288. [DOI] [PubMed] [Google Scholar]
- 31.Hensley K, Venkova K, Christov A, Gunning W, Park J. Collapsin response mediator protein-2: an emerging pathologic feature and therapeutic target for neurodisease indications. Molecular Neurobiology. 2011;43(3):180–191. doi: 10.1007/s12035-011-8166-4. [DOI] [PubMed] [Google Scholar]
- 32.Donahue JE, Flaherty SL, Johanson CE, et al. RAGE, LRP-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathologica. 2006;112(4):405–415. doi: 10.1007/s00401-006-0115-3. [DOI] [PubMed] [Google Scholar]
- 33.Shankar GM, Li S, Mehta TH, et al. Amyloid-β protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature Medicine. 2008;14(8):837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiarini A, Pra ID, Whitfield JF, Armato U. The killing of neurons by β-amyloid peptides, prions, and pro-inflammatory cytokines. Italian Journal of Anatomy and Embryology. 2006;111(4):221–246. [PubMed] [Google Scholar]
- 35.Behl C, Davis JB, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77(6):817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 36.Muresan Z, Muresan V. Neuritic deposits of amyloid-β peptide in a subpopulation of central nervous system-derived neuronal cells. Molecular and Cellular Biology. 2006;26(13):4982–4997. doi: 10.1128/MCB.00371-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jack CR, Jr., Knopman DS, Jagust WJ, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. The Lancet Neurology. 2010;9(1):119–128. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rodrigues E, Weissmiller A, Golstein L. Enhanced β-secretase processing alters APP axonal transport and leads to axonal defects. Human Molecular Genetics. 2012;21(21):4587–4601. doi: 10.1093/hmg/dds297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Devanand DP, Schupf N, Stern Y, et al. Plasma A β and PET PiB binding are inversely related in mild cognitive impairment. Neurology. 2011;77(2):125–131. doi: 10.1212/WNL.0b013e318224afb7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Um JW, Nygaard HB, Heiss JK, et al. Alzheimer amyloid-β oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nature Neuroscience. 2012;15(9):1227–1235. doi: 10.1038/nn.3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hirokawa N, Shiomura Y, Okabe S. Tau proteins: the molecular structure and mode of binding on microtubules. Journal of Cell Biology. 1988;107(4):1449–1459. doi: 10.1083/jcb.107.4.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Scott CW, Klika AB, Lo MMS, Norris TE, Caputo CB. Tau protein induces bundling of microtubules in vitro: comparison of different tau isoforms and a tau protein fragment. Journal of Neuroscience Research. 1992;33(1):19–29. doi: 10.1002/jnr.490330104. [DOI] [PubMed] [Google Scholar]
- 43.Whiteman IT, Gervasio OL, Cullen KM, et al. Activated actin-depolymerizing factor/cofilin sequesters phosphorylated microtubule-associated protein during the assembly of Alzheimer-like neuritic cytoskeletal striations. Journal of Neuroscience. 2009;29(41):12994–13005. doi: 10.1523/JNEUROSCI.3531-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li L, Liu Z, Liu J, et al. Ginsenoside Rd attenuates beta-amyloid-induced tau phosphorylation by altering the functional balance of glycogen synthase kinase 3beta and protein phosphatase 2A. Neurobiology of Disease. 2013;54:320–328. doi: 10.1016/j.nbd.2013.01.002. [DOI] [PubMed] [Google Scholar]
- 45.Arendt T, Holzer M, Fruth R, Brückner MK, Gärtner U. Phosphorylation of tau, aβ-formation, and apoptosis after in vivo inhibition of PP-1 and PP-2A. Neurobiology of Aging. 1998;19(1):3–13. doi: 10.1016/s0197-4580(98)00003-7. [DOI] [PubMed] [Google Scholar]
- 46.Ittner LM, Fath T, Ke YD, et al. Parkinsonism and impaired axonal transport in a mouse model of frontotemporal dementia. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(41):15997–16002. doi: 10.1073/pnas.0808084105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Terry RD. The cytoskeleton in Alzheimer disease. Journal of Neural Transmission. 1998;(53):141–145. doi: 10.1007/978-3-7091-6467-9_12. [DOI] [PubMed] [Google Scholar]
- 48.Velasco ME, Smith MA, Siedlak SL, Nunomura A, Perry G. Striation is the characteristic neuritic abnormality in Alzheimer disease. Brain Research. 1998;813(2):329–333. doi: 10.1016/s0006-8993(98)01034-8. [DOI] [PubMed] [Google Scholar]
- 49.Martin L, Latypova X, Terro F. Post-translational modifications of tau protein: implications for Alzheimer’s disease. Neurochemistry International. 2011;58(4):458–471. doi: 10.1016/j.neuint.2010.12.023. [DOI] [PubMed] [Google Scholar]
- 50.Bulic B, Pickhardt M, Mandelkow E, Mandelkow E. Tau protein and tau aggregation inhibitors. Neuropharmacology. 2010;59(4-5):276–289. doi: 10.1016/j.neuropharm.2010.01.016. [DOI] [PubMed] [Google Scholar]
- 51.Shelton SB, Johnson GVW. Cyclin-dependent kinase-5 in neurodegeneration. Journal of Neurochemistry. 2004;88(6):1313–1326. doi: 10.1111/j.1471-4159.2003.02328.x. [DOI] [PubMed] [Google Scholar]
- 52.Iqbal K, Alonso ADC, Grundke-Iqbal I. Cytosolic abnormally hyperphosphorylated tau but not paired helical filaments sequester normal MAPs and inhibit microtubule assembly. Journal of Alzheimer’s Disease. 2008;14(4):365–370. doi: 10.3233/jad-2008-14402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Maccioni RB, Farías G, Morales I, Navarrete L. The revitalized tau hypothesis on Alzheimer’s disease. Archives of Medical Research. 2010;41(3):226–231. doi: 10.1016/j.arcmed.2010.03.007. [DOI] [PubMed] [Google Scholar]
- 54.Amadoro G, Corsetti V, Ciotti MT, et al. Endogenous Aβ causes cell death via early tau hyperphosphorylation. Neurobiology of Aging. 2011;32(6):969–990. doi: 10.1016/j.neurobiolaging.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 55.Lavados M, Guillón M, Mujica MC, Rojo LE, Fuentes P, Maccioni RB. Mild cognitive impairment and Alzheimer patients display different levels of redox-active CSF iron. Journal of Alzheimer’s Disease. 2008;13(2):225–232. doi: 10.3233/jad-2008-13211. [DOI] [PubMed] [Google Scholar]
- 56.Zambrano CA, Egaña JT, Núñez MT, Maccioni RB, González-Billault C. Oxidative stress promotes τ dephosphorylation in neuronal cells: the roles of cdk5 and PP1. Free Radical Biology and Medicine. 2004;36(11):1393–1402. doi: 10.1016/j.freeradbiomed.2004.03.007. [DOI] [PubMed] [Google Scholar]
- 57.Neumann KF, Rojo L, Navarrete LP, Farías G, Reyes P, Maccioni RB. Insulin resistance and Alzheimer’s disease: molecular links amp; clinical implications. Current Alzheimer Research. 2008;5(5):438–447. doi: 10.2174/156720508785908919. [DOI] [PubMed] [Google Scholar]
- 58.Roberson ED, Scearce-Levie K, Palop JJ, et al. Reducing endogenous tau ameliorates amyloid β-induced deficits in an Alzheimer’s disease mouse model. Science. 2007;316(5825):750–754. doi: 10.1126/science.1141736. [DOI] [PubMed] [Google Scholar]
- 59.Nakagawa O, Fujisawa K, Ishizaki T, Saito Y, Nakao K, Narumiya S. ROCK-I and ROCK-II, two isoforms of Rho-associated coiled-coil forming protein serine/threonine kinase in mice. FEBS Letters. 1996;392(2):189–193. doi: 10.1016/0014-5793(96)00811-3. [DOI] [PubMed] [Google Scholar]
- 60.Sordella R, Jiang W, Chen G, Curto M, Settleman J. Modulation of Rho GTPase signaling regulates a switch between adipogenesis and myogenesis. Cell. 2003;113(2):147–158. doi: 10.1016/s0092-8674(03)00271-x. [DOI] [PubMed] [Google Scholar]
- 61.Zhao L, Ma Q, Calon F, et al. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nature Neuroscience. 2006;9(2):234–242. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]
- 62.Gorovoy M, Niu J, Bernard O, et al. LIM kinase 1 coordinates microtubule stability and actin polymerization in human endothelial cells. Journal of Biological Chemistry. 2005;280(28):26533–26542. doi: 10.1074/jbc.M502921200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Daniels RH, Hall PS, Bokoch GM. Membrane targeting of p21-activated kinase 1 (PAK1) induces neurite outgrowth from PC12 cells. EMBO Journal. 1998;17(3):754–764. doi: 10.1093/emboj/17.3.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Goeckeler ZM, Masaracchia RA, Zeng Q, Chew T, Gallagher P, Wysolmerski RB. Phosphorylation of myosin light chain kinase by p21-activated kinase PAK2. Journal of Biological Chemistry. 2000;275(24):18366–18374. doi: 10.1074/jbc.M001339200. [DOI] [PubMed] [Google Scholar]
- 65.Chew T, Masaracchia RA, Goeckeler ZM, Wysolmerski RB. Phosphorylation of non-muscle myosin II regulatory light chain by p21-activated kinase (γ-PAK) Journal of Muscle Research and Cell Motility. 1998;19(8):839–854. doi: 10.1023/a:1005417926585. [DOI] [PubMed] [Google Scholar]
- 66.Gallo G. Myosin II activity is required for severing-induced axon retraction in vitro. Experimental Neurology. 2004;189(1):112–121. doi: 10.1016/j.expneurol.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 67.Arimura N, Inagaki N, Chihara K, et al. Phosphorylation of collapsin response mediator protein-2 by Rho-kinase: evidence for two separate signaling pathways for growth cone collapse. Journal of Biological Chemistry. 2000;275(31):23973–23980. doi: 10.1074/jbc.M001032200. [DOI] [PubMed] [Google Scholar]
- 68.Niederöst B, Oertle T, Fritsche J, McKinney RA, Bandtlow CE. Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. Journal of Neuroscience. 2002;22(23):10368–10376. doi: 10.1523/JNEUROSCI.22-23-10368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Heredia L, Helguera P, De Olmos S, et al. Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid β-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer’s disease. Journal of Neuroscience. 2006;26(24):6533–6542. doi: 10.1523/JNEUROSCI.5567-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gylys KH, Fein JA, Yang F, Wiley DJ, Miller CA, Cole GM. Snaptic changes in alzheimer’s disease: increased amyloid-β and gliosis in surviving terminals is accompanied by decreased PSD-95 fluorescence. American Journal of Pathology. 2004;165(5):1809–1817. doi: 10.1016/s0002-9440(10)63436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ma QL, Yang F, Calon F, et al. p21-activated kinase-aberrant activation and translocation in Alzheimer disease pathogenesis. Journal of Biological Chemistry. 2008;283(20):14132–14143. doi: 10.1074/jbc.M708034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salminen A, Suuronen T, Kaarniranta K. ROCK, PAK, and Toll of synapses in Alzheimer’s disease. Biochemical and Biophysical Research Communications. 2008;371(4):587–590. doi: 10.1016/j.bbrc.2008.04.148. [DOI] [PubMed] [Google Scholar]
- 73.Leuchtenberger S, Kummer MP, Kukar T, et al. Inhibitors of Rho-kinase modulate amyloid-β (Aβ) secretion but lack selectivity for Aβ42. Journal of Neurochemistry. 2006;96(2):355–365. doi: 10.1111/j.1471-4159.2005.03553.x. [DOI] [PubMed] [Google Scholar]
- 74.Del Pozo MA, Alderson NB, Kiosses WB, Chiang H, Anderson RGW, Schwartz MA. Integrins regulate rac targeting by internalization of membrane domains. Science. 2004;303(5659):839–842. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
- 75.Guirland C, Suzuki S, Kojima M, Lu B, Zheng JQ. Lipid rafts mediate chemotropic guidance of nerve growth cones. Neuron. 2004;42(1):51–62. doi: 10.1016/s0896-6273(04)00157-6. [DOI] [PubMed] [Google Scholar]
- 76.Cole AR, Noble W, Aalten LV, et al. Collapsin response mediator protein-2 hyperphosphorylation is an early event in Alzheimer’s disease progression. Journal of Neurochemistry. 2007;103(3):1132–1144. doi: 10.1111/j.1471-4159.2007.04829.x. [DOI] [PubMed] [Google Scholar]
- 77.Uchida Y, Ohshima T, Sasaki Y, et al. Semaphorin3A signalling is mediated via sequential Cdk5 and GSK3β phosphorylation of CRMP2: implication of common phosphorylating mechanism underlying axon guidance and Alzheimer’s disease. Genes to Cells. 2005;10(2):165–179. doi: 10.1111/j.1365-2443.2005.00827.x. [DOI] [PubMed] [Google Scholar]
- 78.Petratos S, Li Q, George AJ, et al. The β-amyloid protein of Alzheimer’s disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain. 2008;131(1):90–108. doi: 10.1093/brain/awm260. [DOI] [PubMed] [Google Scholar]
- 79.Proctor CJ, Gray DA. GSK3 and p53-is there a link in Alzheimer’s disease? Molecular Neurodegeneration. 2010;5(1):p. 7. doi: 10.1186/1750-1326-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kim W-Y, Snider WD. Functions of GSK-3 signaling in development of the nervous system. Frontiers in Molecular Neuroscience. 2011;4:p. 44. doi: 10.3389/fnmol.2011.00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cole AR, Knebel A, Morrice NA, et al. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. Journal of Biological Chemistry. 2004;279(48):50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochemical Journal. 2001;359(1):1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Salcedo-Tello P, Ortiz-Matamoros A, Arias C. GSK3 function in the brain during development, neuronal plasticity, and neurodegeneration. International Journal of Alzheimer’s Disease. 2011;2011:12 pages. doi: 10.4061/2011/189728.189728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Qian W, Shi J, Yin X, et al. PP2A regulates tau phosphorylation directly and also indirectly via activating GSK-3β . Journal of Alzheimer’s Disease. 2010;19(4):1221–1229. doi: 10.3233/JAD-2010-1317. [DOI] [PubMed] [Google Scholar]
- 85.Ryan KA, Pimplikar SW. Activation of GSK-3 and phosphorylation of CRMP2 in transgenic mice expressing APP intracellular domain. Journal of Cell Biology. 2005;171(2):327–335. doi: 10.1083/jcb.200505078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cole AR, Soutar MPM, Rembutsu M, et al. Relative resistance of Cdk5-phosphorylated CRMP2 to dephosphorylation. Journal of Biological Chemistry. 2008;283(26):18227–18237. doi: 10.1074/jbc.M801645200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Piedrahita D, Hernández I, López-Tobón A, et al. Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s mice. Journal of Neuroscience. 2010;30(42):13966–13976. doi: 10.1523/JNEUROSCI.3637-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Peterson DW, Ando DM, Taketa DA, Zhou H, Dahlquist FW, Lew J. No difference in kinetics of tau or histone phosphorylation by CDK5/p25 versus CDK5/p35 in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(7):2884–2889. doi: 10.1073/pnas.0912718107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Chae T, Kwon YT, Bronson R, Dikkes P, En L, Tsai L. Mice lacking p35, a neuronal specific activator of Cdk5, display cortical lamination defects, seizures, and adult lethality. Neuron. 1997;18(1):29–42. doi: 10.1016/s0896-6273(01)80044-1. [DOI] [PubMed] [Google Scholar]
- 90.Ohshima T, Ward JM, Huh C, et al. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology and perinatal death. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(20):11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cheung ZH, Ip NY. Cdk5: a multifaceted kinase in neurodegenerative diseases. Trends in Cell Biology. 2012;22(3):169–175. doi: 10.1016/j.tcb.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 92.Wen Y, Planel E, Herman M, et al. Interplay between cyclin-dependent kinase 5 and glycogen synthase kinase 3β mediated by neuregulin signaling leads to differential effects on tau phosphorylation and amyloid precursor protein processing. Journal of Neuroscience. 2008;28(10):2624–2632. doi: 10.1523/JNEUROSCI.5245-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hill JJ, Callaghan DA, Ding W, Kelly JF, Chakravarthy BR. Identification of okadaic acid-induced phosphorylation events by a mass spectrometry approach. Biochemical and Biophysical Research Communications. 2006;342(3):791–799. doi: 10.1016/j.bbrc.2006.02.029. [DOI] [PubMed] [Google Scholar]
- 94.Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC. Protein phosphatases and Alzheimer’s Disease. Progress in Molecular Biology and Translational Science. 2012;106:343–379. doi: 10.1016/B978-0-12-396456-4.00012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Martin L, Latypova X, Wilson CM, Magnaudeix A, Perrin ML, Terro F. Tau protein phosphatases in Alzheimer's disease: the leading role of PP2A. Ageing Research Reviews. 2013;12(1):39–49. doi: 10.1016/j.arr.2012.06.008. [DOI] [PubMed] [Google Scholar]
- 96.Liu F, Grundke-Iqbal I, Iqbal K, Gong C. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. European Journal of Neuroscience. 2005;22(8):1942–1950. doi: 10.1111/j.1460-9568.2005.04391.x. [DOI] [PubMed] [Google Scholar]
- 97.Xu Y, Chen Y, Zhang P, Jeffrey PD, Shi Y. Structure of a protein phosphatase 2A holoenzyme: insights into B55-mediated tau dephosphorylation. Molecular Cell. 2008;31(6):873–885. doi: 10.1016/j.molcel.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kins S, Crameri A, Evans DRH, Hemmings BA, Nitsch RM, Götz J. Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. Journal of Biological Chemistry. 2001;276(41):38193–38200. doi: 10.1074/jbc.M102621200. [DOI] [PubMed] [Google Scholar]
- 99.Tanimukai H, Grundke-Iqbal I, Iqbal K. Up-regulation of inhibitors of protein phosphatase-2A in Alzheimer’s disease. American Journal of Pathology. 2005;166(6):1761–1771. doi: 10.1016/S0002-9440(10)62486-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sontag E, Luangpirom A, Hladik C, et al. Altered expression levels of the protein phosphatase 2A ABαC enzyme are associated with Alzheimer disease pathology. Journal of Neuropathology and Experimental Neurology. 2004;63(4):287–301. doi: 10.1093/jnen/63.4.287. [DOI] [PubMed] [Google Scholar]
- 101.Gong CX, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K. Phosphorylation of microtubule-associated protein tau is regulated by protein phosphatase 2A in mammalian brain. Implications for neurofibrillary degeneration in Alzheimer’s disease. Journal of Biological Chemistry. 2000;275(8):5535–5544. doi: 10.1074/jbc.275.8.5535. [DOI] [PubMed] [Google Scholar]
- 102.Ando K, Iijima K, Elliott JI, Kirino Y, Suzuki T. Phosphorylation-dependent regulation of the interaction of amyloid precursor protein with Fe65 affects the production of β-amyloid. Journal of Biological Chemistry. 2001;276(43):40353–40361. doi: 10.1074/jbc.M104059200. [DOI] [PubMed] [Google Scholar]
- 103.Iijima K, Ando K, Takeda S, et al. Neuron-specific phosphorylation of Alzheimer’s β-amyloid precursor protein by cyclin-dependent kinase 5. Journal of Neurochemistry. 2000;75(3):1085–1091. doi: 10.1046/j.1471-4159.2000.0751085.x. [DOI] [PubMed] [Google Scholar]
- 104.Liu R, Zhou X-W, Tanila H, et al. Phosphorylated PP2A (tyrosine 307) is associated with Alzheimer neurofibrillary pathology: in focus. Journal of Cellular and Molecular Medicine. 2008;12(1):241–257. doi: 10.1111/j.1582-4934.2008.00249.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Sontag E, Nunbhakdi-Craig V, Sontag J, et al. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. Journal of Neuroscience. 2007;27(11):2751–2759. doi: 10.1523/JNEUROSCI.3316-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Da Cruz Silva EEF, Da Cruz Silva EOA, Zaia CT, Greengard P. Inhibition of protein phosphatase 1 stimulates secretion of Alzheimer amyloid precursor protein. Molecular Medicine. 1995;1(5):535–541. [PMC free article] [PubMed] [Google Scholar]
- 107.Holzer M, Brückner MK, Beck M, Bigl V, Arendt T. Modulation of APP processing and secretion by okadaic acid in primary guinea pig neurons. Journal of Neural Transmission. 2000;107(4):451–461. doi: 10.1007/s007020070087. [DOI] [PubMed] [Google Scholar]
- 108.De Baere I, Derua R, Janssens V, et al. Purification of porcine brain protein phosphatase 2A leucine carboxyl methyltransferase and cloning of the human homologue. Biochemistry. 1999;38(50):16539–16547. doi: 10.1021/bi991646a. [DOI] [PubMed] [Google Scholar]
- 109.Sontag JM, Nunbhakdi-Craig V, Montgomery L, Arning E, Bottiglieri T, Sontag E. Folate deficiency induces in vitro and mouse brain region-specific downregulation of leucine carboxyl methyltransferase-1 and protein phosphatase 2A Bα subunit expression that correlate with enhanced tau phosphorylation. Journal of Neuroscience. 2008;28(45):11477–11487. doi: 10.1523/JNEUROSCI.2816-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Vogelsberg-Ragaglia V, Schuck T, Trojanowski JQ, Lee VM-Y. PP2A mRNA expression is quantitatively decreased in Alzheimer’s disease hippocampus. Experimental Neurology. 2001;168(2):402–412. doi: 10.1006/exnr.2001.7630. [DOI] [PubMed] [Google Scholar]
- 111.Wang LH, Strittmatter SM. Brain CRMP forms heterotetramers similar to liver dihydropyrimidinase. Journal of Neurochemistry. 1997;69(6):2261–2269. doi: 10.1046/j.1471-4159.1997.69062261.x. [DOI] [PubMed] [Google Scholar]
- 112.Hamajima N, Matsuda K, Sakata S, Tamaki N, Sasaki M, Nonaka M. A novel gene family defined by human dihydropyrimidinase and three related proteins with differential tissue distribution. Gene. 1996;180(1-2):157–163. doi: 10.1016/s0378-1119(96)00445-3. [DOI] [PubMed] [Google Scholar]
- 113.Wang LH, Strittmatter SM. A family of rat CRMP genes is differentially expressed in the nervous system. Journal of Neuroscience. 1996;16(19):6197–6207. doi: 10.1523/JNEUROSCI.16-19-06197.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Inagaki N, Chihara K, Arimura N, et al. CRMP-2 induces axons in cultured hippocampal neurons. Nature Neuroscience. 2001;4(8):781–782. doi: 10.1038/90476. [DOI] [PubMed] [Google Scholar]
- 115.Morita T, Sobuě K. Specification of neuronal polarity regulated by local translation of CRMP2 and tau via the mTOR-p70S6K pathway. Journal of Biological Chemistry. 2009;284(40):27734–27745. doi: 10.1074/jbc.M109.008177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Arimura N, Menager C, Fukata Y, Kaibuchi K. Role of CRMP-2 in neuronal polarity. Journal of Neurobiology. 2004;58(1):34–47. doi: 10.1002/neu.10269. [DOI] [PubMed] [Google Scholar]
- 117.Arimura N, Ménager C, Kawano Y, et al. Phosphorylation by Rho kinase regulates CRMP-2 activity in growth cones. Molecular and Cellular Biology. 2005;25(22):9973–9984. doi: 10.1128/MCB.25.22.9973-9984.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K. GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell. 2005;120(1):137–149. doi: 10.1016/j.cell.2004.11.012. [DOI] [PubMed] [Google Scholar]
- 119.Soutar MPM, Thornhill P, Cole AR, Sutherland C. Increased CRMP2 phosphorylation is observed in Alzheimer’s disease; does this tell us anything about disease development? Current Alzheimer Research. 2009;6(3):269–278. doi: 10.2174/156720509788486572. [DOI] [PubMed] [Google Scholar]
- 120.Asai H, Ikezu S, Varnum M, Ikezu T. Phosphorylation of collapsin response mediator protein-2 and axonal degeneration in transgenic mice expressing a familial Alzheimer's disease mutant of APP and tau-tubulin kinase 1. Alzheimer's & Dementia. 2012;8(4):649–650. [Google Scholar]
- 121.Yoneda A, Morgan-Fisher M, Wait R, Couchman JR, Wewer UM. A collapsin response mediator protein 2 isoform controls myosin II-mediated cell migration and matrix assembly by trapping ROCK II. Molecular and Cellular Biology. 2012;32(10):1788–1804. doi: 10.1128/MCB.06235-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kimura T, Arimura N, Fukata Y, Watanabe H, Iwamatsu A, Kaibuchi K. Tubulin and CRMP-2 complex is transported via Kinesin-1. Journal of Neurochemistry. 2005;93(6):1371–1382. doi: 10.1111/j.1471-4159.2005.03063.x. [DOI] [PubMed] [Google Scholar]
- 123.Kawano Y, Yoshimura T, Tsuboi D, et al. CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Molecular and Cellular Biology. 2005;25(22):9920–9935. doi: 10.1128/MCB.25.22.9920-9935.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Nishimura T, Fukata Y, Kato K, et al. CRMP-2 regulates polarized Numb-mediated endocytosis for axon growth. Nature Cell Biology. 2003;5(9):819–826. doi: 10.1038/ncb1039. [DOI] [PubMed] [Google Scholar]
- 125.Tan M, Ma S, Huang Q, Hu K, Song B, Li M. GSK-3α/β-mediated phosphorylation of CRMP-2 regulates activity-dependent dendritic growth. Journal of Neurochemistry. 2013;125(5):685–697. doi: 10.1111/jnc.12230. [DOI] [PubMed] [Google Scholar]
- 126.Fukata Y, Itoh TJ, Kimura T, et al. CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nature Cell Biology. 2002;4(8):583–591. doi: 10.1038/ncb825. [DOI] [PubMed] [Google Scholar]
- 127.Reis GF, Yang G, Szpankowski L, et al. Molecular motor function in axonal transport in vivo probed by genetic and computational analysis in Drosophila. Molecular Biology of the Cell. 2012;23(9):1700–1714. doi: 10.1091/mbc.E11-11-0938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Falzone TL, Stokin GB. Imaging amyloid precursor protein in vivo: an axonal transport assay. Methods in Molecular Biology. 2012;846:295–303. doi: 10.1007/978-1-61779-536-7_25. [DOI] [PubMed] [Google Scholar]
- 129.Iqbal K, Grundke-Iqbal I. Alzheimer abnormally phosphorylated tau is more hyperphosphorylated than the fetal tau and causes the disruption of microtubules. Neurobiology of Aging. 1995;16(3):375–379. [Google Scholar]
- 130.Potter H, Borysov S, Ari C, et al. Aβ inhibits specific kinesin motors involved in both mitosis and neuronal function; potential implications for neurogenesis and neuroplasticity in Alzheimer's disease and Down syndrome. Alzheimer's & Dementia. 2011;7(4):p. 24. [Google Scholar]
- 131.Brunden KR, Yao Y, Potuzak JS, et al. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacological Research. 2011;63(4):341–351. doi: 10.1016/j.phrs.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ye X, Tai W, Zhang D. The early events of Alzheimer’s disease pathology: from mitochondrial dysfunction to BDNF axonal transport deficits. Neurobiology of Aging. 2012;33(6):1122.e1–1122.e10. doi: 10.1016/j.neurobiolaging.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 133.Mao P, Reddy PH. Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in Alzheimer’s disease: implications for early intervention and therapeutics. Biochimica et Biophysica Acta. 2011;1812(11):1359–1370. doi: 10.1016/j.bbadis.2011.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Goldstein LSB. Axonal transport and Alzheimer’s diseasein. In: Larry RS, editor. Encyclopedia of Neuroscience. Oxford, UK: Academic Press; 2009. pp. 1189–1194. [Google Scholar]
- 135.Mitsuyama F, Futatsugi Y, Okuya M, et al. Stimulation-dependent intraspinal microtubules and synaptic failure in Alzheimer’s disease: a review. International Journal of Alzheimer’s Disease. 2012;2012:7 pages. doi: 10.1155/2012/519682.519682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Paula-Barbosa M, Tavares MA, Cadete-Leite A. A quantitative study of frontal cortex dendritic microtubules in patients with Alzheimer’s disease. Brain Research. 1987;417(1):139–142. doi: 10.1016/0006-8993(87)90188-0. [DOI] [PubMed] [Google Scholar]
- 137.Fiala JC. Mechanisms of amyloid plaque pathogenesis. Acta Neuropathologica. 2007;114(6):551–571. doi: 10.1007/s00401-007-0284-8. [DOI] [PubMed] [Google Scholar]
- 138.Adalbert R, Nogradi A, Babetto E, et al. Severely dystrophic axons at amyloid plaques remain continuous and connected to viable cell bodies. Brain. 2009;132(2):402–416. doi: 10.1093/brain/awn312. [DOI] [PubMed] [Google Scholar]
- 139.Goldstein LSB. Kinesin molecular motors: transport pathways, receptors, and human disease. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(13):6999–7003. doi: 10.1073/pnas.111145298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dhaenens C, Van Brussel E, Schraen-Maschke S, Pasquier F, Delacourte A, Sablonnire B. Association study of three polymorphisms of kinesin light-chain 1 gene with Alzheimer’s disease. Neuroscience Letters. 2004;368(3):290–292. doi: 10.1016/j.neulet.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 141.Szpankowski L, Encalada SE, Goldstein LSB. Subpixel colocalization reveals amyloid precursor protein-dependent kinesin-1 and dynein association with axonal vesicles. Proceedings of the National Academy of Sciences of the United States of America. 2012;109(22):8582–8587. doi: 10.1073/pnas.1120510109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stokin GB, Lillo C, Falzone TL, et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s diseases. Science. 2005;307(5713):1282–1288. doi: 10.1126/science.1105681. [DOI] [PubMed] [Google Scholar]
- 143.Kamal A, Stokin GB, Yang Z, Xia C, Goldstein LSB. Axonal transport of amyloid precursor protein is mediated by direct binding to the kinesin light chain subunit of kinesin-I. Neuron. 2000;28(2):449–459. doi: 10.1016/s0896-6273(00)00124-0. [DOI] [PubMed] [Google Scholar]
- 144.Traina G, Federighi G, Brunelli M. Up-regulation of kinesin light-chain 1 gene expression by acetyl-l-carnitine: therapeutic possibility in Alzheimer’s disease. Neurochemistry International. 2008;53(6–8):244–247. doi: 10.1016/j.neuint.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 145.Falzone TL, Gunawardena S, McCleary D, Reis GF, Goldstein LSB. Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Human Molecular Genetics. 2010;19(22):4399–4408. doi: 10.1093/hmg/ddq363.ddq363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Quach TT, Duchemin A, Rogemond V, et al. Involvement of collapsin response mediator proteins in the neurite extension induced by neurotrophins in dorsal root ganglion neurons. Molecular and Cellular Neuroscience. 2004;25(3):433–443. doi: 10.1016/j.mcn.2003.11.006. [DOI] [PubMed] [Google Scholar]