

Abstract
Background/Aims
In a family with congenital hyperinsulinism (HI), first described in the 1950s by MacQuarrie, we examined the genetic locus and clinical phenotype of a novel form of dominant HI.
Methods
We surveyed 25 affected individuals, 7 of whom participated in tests of insulin dysregulation (24-hour fasting, oral glucose and protein tolerance tests). To identify the disease locus and potential disease-associated mutations we performed linkage analysis, whole transcriptome sequencing, whole genome sequencing, gene capture, and next generation sequencing.
Results
Most affecteds were diagnosed with HI before age one and 40% presented with a seizure. All affecteds responded well to diazoxide. Affecteds failed to adequately suppress insulin secretion following oral glucose tolerance test or prolonged fasting; none had protein-sensitive hypoglycemia. Linkage analysis mapped the HI locus to Chr10q21–22, a region containing 48 genes. Three novel non-coding variants were found in hexokinase 1 (HK1) and one missense variant in the coding region of DNA2.
Conclusion
Dominant, diazoxide-responsive HI in this family maps to a novel locus on Chr10q21–22. HK1 is the more attractive disease gene candidate since a mutation interfering with the normal suppression of HK1 expression in beta-cells could readily explain the hypoglycemia phenotype of this pedigree.
Key Terms: beta-cell, hypoglycemia, genetics, hyperinsulinism, insulin secretion
Introduction
Monogenic disorders of pancreatic beta-cell insulin secretion have become increasingly recognized to be important causes of hypoglycemia in infants and children [1]. Previously, such patients would have been classified as having “idiopathic hypoglycemia of infancy”, a term originated by McQuarrie in 1954 [2]. Dr. McQuarrie described a brother and sister with onset of hypoglycemia in early infancy, who were treated with pancreatectomy but continued to suffer from hypoglycemia afterwards.
We recently had the opportunity to investigate the family of the affected sibling pair. This pedigree now includes 25 affected individuals, spans four generations and follows an autosomal dominant inheritance pattern. There are now 8 genetic loci known to be associated with congenital HI and direct sequencing of genomic DNA from our cohort excluded all 8 genes [1, 3, 4]. Thus, we hypothesized that a novel genetic locus must be responsible for disease in this family. This report describes the clinical phenotype of this novel form of dominantly inherited HI and genetic tests demonstrating linkage of the disorder to an 8.2 Mb region of chromosome 10. Whole genome sequencing and gene capture of a candidate portion of this region identified variants in non-coding regions of hexokinase 1 (HK1) and in the coding sequence of DNA2 as potential candidate loci that may be responsible for this form of congenital HI.
Research Design and Methods
Patients
Family members donated saliva samples or blood for DNA isolation and participated in an interview about their hypoglycemic symptoms, treatment, and history. Diagnosis of HI was based on criteria including fasting hypoglycemia accompanied by inadequate suppression of plasma insulin, inappropriately low plasma free fatty acids (FFA) and plasma β-hydroxybutyrate (BOB) concentrations, and an inappropriate increase in serum glucose levels after administration of glucagon at the time of hypoglycemia [5–7].
Consent
Written informed consent was obtained from subjects or their parents for this study. This study was approved by The Children’s Hospital of Philadelphia (CHOP) Institutional Review Board.
Fasting tests
Fasting adaptation for 20–24 hours was evaluated by monitoring plasma concentrations of glucose, BOB, FFA, and insulin. Fasts were terminated at 20–24 hours or when plasma glucose fell to 2.8 mmol/L [8].
Oral protein tolerance tests
Subjects drank 1.5 g/kg protein (maximum 60 g) in water, 3–4 hours after a meal (Resource Instant Beneprotein Powder, Novartis Pharmaceuticals) [9, 10]. Blood samples were obtained for plasma glucose and insulin at times 0, 30, 60, 90, 120, 150, and 180 min. A fall in plasma glucose to below 3.9 mmol/L or a delta plasma glucose greater than 0.56 mmol/L was interpreted as abnormal [9, 11].
Oral glucose tolerance tests
Subjects drank 1.75 g/kg of glucose (maximum 75 g) as Glucola, 3 hr after a meal. Blood samples were obtained for plasma glucose and insulin at times 0, 30, 60, 90, 120,150,180, 210 and 240 min. Results were compared to published norms for children and adults [12].
Mutation Analysis
Genomic DNA was isolated from saliva samples using the Oragene DNA Self Collection Kit (DNA Genotek; Kanata, Ontario, Canada). Genomic DNA from one affected family member was used to sequence the coding regions and intron/exon splice junctions of candidate genes (SIRT1, NEUROG3, PCBD1, HK1, HKDC1) [10]. Resulting chromatograms were analyzed with Sequencher 4.9 (Gene Codes Corp; Ann Arbor, MI). Novel variants (absent in dbSNP v132 and/or 1000 genomes project) were sequenced across all family members to confirm tracking with affecteds and obligate carriers. These variants were tested against DNA from 100 healthy control individuals participating in the DNA Polymorphism Discovery Resource (Coriell Cell Depository; Camden, NJ) to exclude rare polymorphisms.
SNP genotyping and linkage analysis
DNA samples were genotyped on the Illumina HumanHap 550v1,v3 and 610v1 arrays at the CHOP Center for Applied Genomics. 24,602 SNPs in linkage equilibrium were included on the arrays and had genetic distances interpolated on the Rutgers second-generation combined linkage-physical map [13]. Thirty-one individuals were used for linkage analysis: 28 affecteds or obligate carriers and 3 unaffected individuals who married into the family. Linkage analysis was performed using Merlin [14], with a parametric rare dominant model in which the disease allele frequency was estimated at 0.0001 and the probability of being affected with 0, 1 and 2 alleles is 0.0001, 1.0 and 1.0, respectively.
Whole Transcriptome Sequencing
Total RNA extracted from lymphocyte cell lines from individuals 2a and 2b (Fig. 1) was used to create cDNA libraries followed by high throughput sequencing performed by Illumina Inc. (San Diego, CA). The data were analyzed by comparing the cDNA sequence to the UCSC genome browser [15] or by direct alignment to the published human genome sequence [16]. Both approaches generated quality analyzable data for 18 of 48 genes in the shared haplotype region. Genetic coordinates listed are based on GRCh37, hg19.
Figure 1.
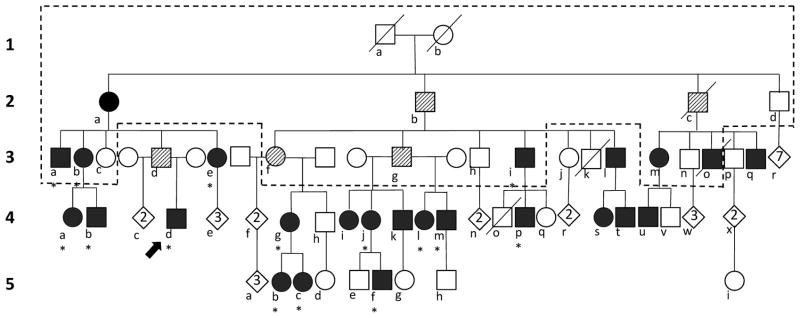
Pedigree of family with autosomal dominant congenital hyperinsulinism. Generation is indicated by number and family members are denoted with a letter. Males depicted as squares, females as circles, deceased individuals by a slashed line. Filled symbols are individuals diagnosed with hypoglycemia (n=25); hatched symbols are obligate carriers (n=5). Diamonds indicate number of unaffected descendants. Dashed lines outline original pedigree reported by McQuarrie (1). Asterisks indicate family members described in Table 1.
Whole Genome Sequencing
Genomic DNA from individual 2a (Fig. 1) was sent to Complete Genomics, Inc., (Mountain View, CA) for sequencing. Sequence data were analyzed by the CHOP Center for Applied Genomics.
Gene Capture and Next Generation Sequencing
Genomic DNA from individual 3d and a normal control was enriched for region specific extraction (RSE) encompassing a total of 205 kb (chr10: 70964058–71169635). Enriched samples were amplified with Qiagen Repli-g Midi amplification kit (Valencia, CA) sent to Illumina for library preparation and sequencing. Please see Supplemental Methods for details.
Results
The proband (4d; Fig. 1) was the product of an uncomplicated term pregnancy with a birth weight of 3405 gm. Shortly after birth his mother noted that he was a voracious eater and had spells of extreme irritability and inconsolable crying that resolved with feeding, despite being told his plasma glucose was normal in the newborn nursery. At 3 months of age he had a generalized tonic-clonic seizure and during evaluation at Cincinnati Children’s Hospital was diagnosed with HI. He was treated with diazoxide, which easily controlled his hypoglycemia and continued on diazoxide until age 10 when it was discontinued at his parents’ request. Evaluation off treatment at CHOP demonstrated an ability to maintain plasma glucose concentrations > 3.9 mmol/L for over 12 hours of fasting. At age 11 years he experienced another generalized tonic-clonic seizure and was restarted on diazoxide.
The proband’s father (3d) was never diagnosed with hypoglycemia. He denied having symptoms of hypoglycemia, apart from headaches and jitteriness during athletics, although he has completed marathons. He has two other sons who have no symptoms of hypoglycemia.
As shown in Fig. 1, the proband is the nephew of the brother and sister (3a and 3b) with “idiopathic hypoglycemia” described by McQuarrie. Three of their cousins (3i, 3m and 3o) were in McQuarrie’s original report and were diagnosed with hypoglycemia after seizures in infancy. Both 3a and 3b underwent pancreatectomy in unsuccessful attempts to control their hypoglycemia. All five original affecteds were treated with ACTH injections, with only partial effectiveness, until the 1960s when diazoxide was found to be effective in some forms of HI [17].
Currently, 25 members of this four-generation pedigree of northern European ancestry have been recognized as clinically affected with HI (Fig. 1). In addition, 5 individuals, including the father of our proband have been classified as obligate carriers based on an autosomal dominant inheritance pattern. Obligate carriers did not have recognized symptoms of hypoglycemia, but had children or grandchildren who were diagnosed and treated for HI. The mother of McQuarrie’s original proband (2a) was initially classified as an obligate carrier. However, at age 89 after a 10 lb weight loss from adult celiac disease, she suffered a hypoglycemic seizure (plasma glucose 1.7–2.2 mmol/L). She was diagnosed with HI, which improved greatly upon treatment with diazoxide.
Table 1 summarizes the features of 15 affected adults and children in the family. In general, their hyperinsulinism disorder was mild: most affecteds failed to show excessive prenatal growth and were not recognized to have hypoglycemia until late in the first year of life. Diazoxide therapy was uniformly effective in the 12 family members for whom it was prescribed and pancreatectomy was only performed in the original two cases. Of the 12 affected adults surveyed, all continued to have symptoms of hypoglycemia, although only case 2a is currently on treatment. Adult diabetes has developed only in the two cases treated with pancreatectomy. Apart from 3a, who suffered permanent brain damage from repeated episodes of hypoglycemia, family members have normal intellectual development; several have advanced degrees.
Table 1.
Clinical characteristics of 15 affected family members
| Current age | 21 mos-89 yrs |
| LGA birth weight | 5 of 13 |
| History of seizures | 6 of 16 |
| Age at diagnosis | 3–17 mos* |
| ACTH Treatment | 5 of 15 |
| Diazoxide Treatment | 12 of 16 |
| Pancreatectomy | 2 of 15 |
| Education - HS diploma or above | 15 of 16 |
| Adult with symptomatic hypoglycemia | 12 of 12 |
| Adult BMI – mean (range) | 23 (19.9–26.4) |
| Diabetes Mellitus | 2 of 12** |
Excluding patient 2a diagnosed at age 89;
Both adults with diabetes had pancreatectomy in infancy
In order to define the biochemical phenotype of this family’s form of congenital hyperinsulinism, 7 of the affected family members participated in tests of fasting and of oral glucose and protein tolerance (Table 2). The results showed a pattern of failure to adequately suppress insulin secretion during prolonged fasting and following stimulation with oral glucose. Plasma glucose levels fell to 3.9 mmol/L or below in 5 of the 7 affecteds within 4 hr after oral glucose loading; 4 dropped their plasma glucose to approximately 3.1 mmol/L and developed symptoms. The peak glucose and insulin responses to oral glucose were not unusual in most cases; 3 of the adults showed signs of insulin resistance (peak insulin >510 pmol/L), despite having a BMI of 25 or less. Protein sensitive hypoglycemia was not apparent in any of the 5 individuals tested (data not shown). After 20–24 hr of fasting, 6 of the 7 affected individuals dropped their plasma glucose to 3.9 mmol/L or below, with little or no activation of ketogenesis; 5 developed symptoms of hypoglycemia. One of the affected children (5b) gradually developed hypoglycemia with inadequate suppression of insulin and delayed and inadequate ketogenesis by the 23rd hour of fasting.
Table 2.
Responses to oral glucose tolerance tests and fasting tests in 7 affecteds and obligate carriers
| Patient | Age (yrs) | Sex | OGTT | Fasting Test | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Glucose (mmol/L) | Insulin (pmol/L) | Durations (hrs) | Glucose (mmol/L) | Insulin (pmol/L) | BOB (mmol/L) | ||||||
| basal | peak | nadir | basal | peak | nadir | final | final | ||||
| 5b | 4 | F | 4.3 | 7.9 | 3.1 | 90 | 234 | 24 | 3.1 | 89 | 1.3 |
| 4d | 6 | M | 3.8 | 7.4 | 3.1 | * | * | 20 | 3.6 | * | 0.2 |
| 4a | 28 | F | 5.3 | 12.4 | 7.5 | 72 | 774 | 23 | 3.8 | 34 | 0.1 |
| 4b | 30 | M | 4.1 | 8.4 | 4.1 | 24 | 174 | 24 | 3.9 | 26 | 0.1 |
| 4g | 32 | F | 5.4 | 9.2 | 3.0 | 48 | 528 | 24 | 3.9 | 25 | 0.6 |
| 3e | 45 | F | 3.6 | 5.2 | 2.8 | 24 | 234 | 20 | 3.5 | 29 | 0.3 |
| 3d | 48 | M | 6.2 | 10.7 | 4.1 | 78 | 540 | 20 | 5.2 | * | 0.2 |
Hemolyzed sample
Attempts to identify the genetic defect responsible for dominantly-transmitted HI in this family by direct sequencing of genomic DNA failed to detect a disease-causing mutation in the common genes for congenital HI (ABCC8, KCNJ11, GCK, GLUD1), as well as in the rarer genes (HADH, HNF4α, UCP2,)[1, 4]. Haplotype analysis excluded the gene for MCT1 (SLC16A1).
We employed positional cloning to map the location of the gene responsible for this novel form of HI by linkage analysis using Illumina SNP arrays. As shown in Fig. 2a, the only significant LOD score was identified on chromosome 10q21–22, which does not contain any known HI locus. The region of highest LOD score was 6.82 and the shared haplotype was between SNP markers rs10995440 and rs6480513 (genetic distance =11.1 cM; physical distance = 8.2 Mb).
Figure 2.


(a) Linkage analysis showing LOD score of 6.82 at chromosome 10q21–22. Shaded bar indicates the 11.1 cM shared haplotype region spanning 8.2 Mb and containing 48 genes. (b) Location of 3 novel disease-associated variants in non-coding regions of HK1. The promoters of the three HK1 isoforms are identified: PT (testes specific), PE (erythrocyte specific), and PU (ubiquitous). Tissue specific exons are designated T1-T6 and E1. Exons 2–18 are shared by all 3 isoforms. Transcription start sites are indicated with arrows.
Direct Sequencing of Candidate Genes
The region of shared haplotype contains 48 genes (Table 3). Several of are potential candidate genes for HI, including hexokinase 1 (HK1), which encodes a glucose phosphorylating enzyme with greater affinity for glucose than glucokinase, the hexokinase expressed in beta-cells. Direct sequencing of the coding exons and intron/exon boundaries of HK1 and 4 other candidate genes (SIRT1, NEUROG3, PCBD1, HKDC1) did not identify any coding mutations in affected family members. Coding mutations in candidate gene SLC25A16 were excluded by analysis of the whole genome sequencing data.
Table 3.
Tests of genes in the shared haplotype region
| Gene | Direct Sequencing | Transcriptome sequencing | Whole genome sequencing | Gene Capture |
|---|---|---|---|---|
| NRBF2 | + | + | ||
| JMJD1C | + | + | ||
| REEP3 | + | + | ||
| CTNNA3 | + | |||
| LRRTM3 | + | |||
| DNAJC12 | + | |||
| SIRT1 | + | + | ||
| HERC4 | + | + | ||
| MYPN | + | |||
| ATOH7 | + | |||
| PBLD | + | |||
| HNRPH3 | + | + | ||
| RUFY2 | + | |||
| DNA2 | + | |||
| SLC25A16 | + | |||
| TET1 | + | |||
| CCAR1 | + | + | ||
| STOX1 | + | |||
| DDX50 | + | + | ||
| DDX21 | + | + | ||
| KIAA1279 | + | + | ||
| SRGN | + | + | ||
| VPS26A | + | + | ||
| SUPV3L1 | + | + | ||
| HKDC1 | + | + | + | |
| HK1 | + | + | + | + |
| TACR2 | + | |||
| TSPAN15 | + | |||
| NEUROG3 | + | + | ||
| C10orf35 | + | |||
| COL13A1 | + | |||
| H2AFY2 | + | |||
| AIFM2 | + | + | ||
| TYSND1 | + | |||
| SAR1A | + | + | ||
| PPA1 | + | + | ||
| NPFFR1 | + | |||
| LRRC20 | + | |||
| EIF4EBP2 | + | + | ||
| NODAL | + | |||
| KIAA1274 | + | |||
| PRF1 | + | |||
| ADAMTS14 | + | |||
| C10orf27 | + | |||
| SGPL1 | + | |||
| PCBD1 | + | + | + | |
| UNC5B | + | |||
| CR617227 | + |
Whole Transcriptome Sequencing
We next used whole transcriptome sequencing of lymphoblast cDNA to search for exonic mutations among the 48 candidate genes. Good quality sequences were obtained for 18 of the 48 genes, but failed to detect any novel variants and, therefore, excluded these as candidate disease genes (Table 3).
Whole Genome Sequencing
Whole genome sequencing (Complete Genomics, Inc., Mountain View, CA 94043) was performed on one affected (2a) to search for possible disease variants within the entire shared haplotype region. This analysis detected the presence of 3 novel, non-coding variants in HK1, all of which tracked with all affected individuals and obligate carriers in the family. None of the three were reported in db137 or in the 1000 genomes project (4,362 alleles). As shown in Fig. 2b, HK1 variants #1 and #2 were found separately at ≤ 0.5% frequency in 100 healthy controls; however, variant #3 was not present in any healthy controls. The whole genome sequence analysis also detected only one exonic mutation in one of the other 47 genes within the shared haplotype region: a non-synonymous Q991K missense mutation in DNA2 (Chr10: 70179634 G>T). No intronic, coding or promoter mutations were detected in other candidate genes within the shared haplotype region (NEUROG3, SIRT1 and SLC25A16). The DNA2 variant was present in all of the affecteds and obligate carriers, but was predicted to be tolerated and benign by both SIFT and PolyPhen software programs. It was not found in dbSNPv137 or in 100 healthy controls, but was reported in the ESP database (Exome Sequencing Project-(http://evs.gs.washington.edu/EVS/) [April, 2013]) in in 82/11,274 European and African American alleles (75/8083 European alleles and 7/3641 African American alleles) and it was also reported in the 1000 genomes project in 6/4362 alleles (4/1512 European alleles) [18, 19]. Therefore, the DNA2 Q991K variant was not considered to be disease causing.
Gene Capture and Next Generation Sequencing
We hypothesized that one of the 3 non-coding variants in HK1, identified through whole genome sequencing, or a combination of these variants, could be disease-causing by leading to inappropriate expression of this high affinity hexokinase in beta-cells. In order to confirm the detection of the HK1 sequence variants, genomic DNA was used to capture the entire 205 kb HK1 region, as well as the adjacent HK1 pseudogene, HKDC1 (chr10: 70964058–71169635) for next generation sequencing. Homozygous variants and variants present in normal controls or reported in dbSNPv137 were eliminated as polymorphisms [18]. Only the same 3 variants detected by whole genome sequencing were detected by gene capture and next generation sequencing. As shown in Fig. 2b, the three HK1 variants included: (variant 1) chr10: 71046674 C>T, located in intron 1 of the testis specific HK1 isoform; (variant 2) chr10: 71095271 C>G, located in intron 1 of the ubiquitous HK1 isoform; and (variant 3) chr10: 71108666 dup G, located in intron 2 of the ubiquitous HK1 isoform. Analysis of DNA from 100 healthy controls did not identify a single individual that was heterozygous for more than one the three HK1 variants.
Discussion
The results of these studies show that the hypoglycemia in this family is a form of congenital HI which is autosomal dominant and diazoxide-responsive. Linkage studies mapped this disorder to a novel locus for congenital HI within an 8.2 Mb region on chromosome 10q21–22. The phenotype of the disorder, including hypoglycemia both with prolonged fasting and 3–4 hours following a glucose load, indicates a defect in suppression, rather than in stimulation of insulin secretion; protein sensitive hypoglycemia was not part of the phenotype. Although the hypoglycemia in this family appears to be less severe than several of the other genetic forms of congenital HI, some affecteds suffered profound hypoglycemic brain injury and many of the affecteds continue to suffer episodes of symptomatic hypoglycemia as adults. Genetic analysis of the 10q21–22 region shared by affected family members identified 3 non-coding variants in HK1 and an amino acid substitution in DNA2. As described below, HK1 is an attractive candidate disease locus, but DNA2, an endonuclease which functions in mitochondrial DNA replication and repair, has no obvious connection to regulation of insulin secretion [20–23].
The phenotype of HI in this family indicates that the appropriate suppression of insulin release at low plasma glucose concentrations is impaired, based on responses of affected children and adults to fasting or oral glucose challenge. A lack of protein sensitive hypoglycemia distinguishes this disorder from HI due to mutations of the KATP channel genes or GLUD1 or HADH [1, 4]. This suggests that the defect involves the glucose threshold for insulin secretion. Similar to a defect in the beta cell glucose threshold without protein sensitive hypoglycemia a similar, albeit more severe, defect in beta-cell glucose threshold occurs in HI due to glucokinase activating mutations. The escape of ketogenesis from suppression by insulin observed in patient 5b during fasting (Fig. 1; Table 2) has also been observed in patients with glucokinase HI, consistent with a lowered glucose threshold for insulin release [24]. Unlike patients with glucokinase HI, birthweight was not increased in our family. This is consistent with the milder course of the disorder in this family compared to that of glucokinase HI in which many patients cannot achieve normoglycemia with diazoxide treatment [1].
The clinical features of this novel form of HI bear some resemblance to those seen in diazoxide-responsive dominant KATP channel HI [10, 25, 26]. In both conditions, presentation of hypoglycemia is usually within the first year of life, often with the occurrence of a hypoglycemic seizure, sometimes leading to permanent brain injury. In addition, some affected individuals present with severe symptoms at birth while other family members carrying the same mutation have minimal symptoms, never seeking treatment. In some of the other milder forms of dominant HI (e.g., due to mutations in GDH) affected carriers also often escape recognition. Family members of both dominant KATP channel HI and this novel form of HI report that episodes of hypoglycemia seem to become less severe with age and that adults often stop treatment; however, our patient 2a first became symptomatic at age 89, indicating that the dysregulation of insulin secretion persists. In both the diazoxide-responsive forms of dominant KATP HI [1] and this novel form of HI, there does not appear to be an association with the later development of diabetes, with the exceptions of 3a and 3b who had pancreatectomies.
Since it did not appear that a coding region mutation was likely to explain the hyperinsulinism in our family, we extended our search for mutations to non-coding regions of HK1 that might affect its expression in beta-cells. In beta-cells, glucose phosphorylation is normally carried out by glucokinase, a hexokinase whose low affinity for glucose as substrate, is responsible for setting the beta-cell threshold for insulin release at a glucose concentrations of ~5mM [27–29]. The importance of glucokinase in controlling blood glucose levels is illustrated by the fact that inactivating mutations cause a monogenic form of diabetes (MODY2), while activating mutations of glucokinase lead to HI [1]. In addition, glucose-stimulated insulin release is enhanced by a group of glucokinase activator drugs [30–33]. In contrast to glucokinase, HK1 has a high affinity for glucose (Km < 1 mM) and lacks cooperativity with glucose as substrate [27, 28]. Expression of HK1 is normally “disallowed” in beta-cells, since the presence of this enzyme with high activity at very low levels of glucose would result in hypoglycemia [34]. This has been demonstrated by experiments in which expression of HK1 in MIN6 cells and isolated rat islets leads to elevated basal glycolysis and insulin release and by observations of hypoglycemia in transgenic mice expressing yeast hexokinase in beta-cells [35–37]. It is interesting that Henquin, et al. recently described increased HK1 immunostaining in islets of 5 of 6 infants with atypical HI defined by a mosaic pattern of hyperfunctional and hypofunctional islets within the pancreas[38]. These authors suggested that the hyperinsulinism in their 5 cases might be related to the apparent increase of HK1 in the abnormal islets, although they did not have sufficient material to confirm this by showing altered levels of HK1 activity or HK1 sequence changes in affected islets. The mechanism(s) for preventing expression of disallowed genes, such as HK1, in beta-cells are not known, but could involve transcription factor regulation, epigenetic mechanisms, or short noncoding RNAs[34, 39]. It is noteworthy that recent genome wide association studies have linked the HK1 locus to increased hemoglobin A1C and Type 2 diabetes, although specific mutations affecting HK1 expression or activity have not been reported [40–42].
Precedent for a genetic form of HI due to aberrant beta-cell expression of a disallowed gene is provided by the exercise-induced HI disorder (EIHI) associated with mutations of SLC16A1, which encodes MCT1, a plasma membrane monocarboxylic acid transporter for pyruvate and lactate. MCT1 is not expressed in beta-cells and, therefore, pyruvate and lactate normally do not stimulate insulin secretion. In patients with EIHI, Otonkoski, et al. showed that elevations of plasma pyruvate by anaerobic exercise induced a large release of insulin followed by an episode of hypoglycemia. They also demonstrated that an infusion of pyruvate could provoke an abnormal acute insulin response [43, 44]. The EIHI patients had no coding sequence mutations of SLC16A1, but mutations were identified in the upstream promoter region, which, presumably, interfered with the process by which MCT1 expression is normally silenced in beta-cells. We propose that a similar mechanism, leading to a failure in the normal silencing of HK1 expression in beta-cells, may be the basis for HI in our family.
Our whole genome sequencing and gene capture experiments identified three HK1 non-coding region variants as potential candidates for causing disease (Fig. 2b). Variant 1 was in intron 1 of the testicular isoform of HK1, upstream of the start-site of the ubiquitous form of HK1. Despite the fact that it occurred with a 0.5% frequency in normal control alleles (although it was not in any of the standard SNP databases), this variant is an attractive disease-causing candidate, since it disrupts a consensus sequence for binding of LBP-1 (Fig. 2b) [45]. LBP-1 is a transcription factor known to be a negative regulator of the HIV-1 promoter by preventing binding of the TFII-1 transcriptional activator, while also recruiting histone deacetylases [46–48]. The second candidate disease-causing variant was in intron 1 of the ubiquitous isoform of HK1 and also occurred at 0.5% frequency in normals, although it was also not listed in any SNP database. This region of intron 1 of the ubiquitous HK1 isoform appears to be enriched for activating histone modifications in cells that express HK1 (UCSC Browser ENCODE data), whereas it shows little enrichment for activating histone modifications in normal islets[49]. The third candidate disease-causing variant was located in intron 2 of the ubiquitous isoform of HK1; it was not found either in normal control alleles or in the SNP databases. Although intron 2 of the ubiquitous HK1 isoform shows histone marks consistent with a role in expression regulation, there do not appear to be marked differences in histone modifications in this region between human islets and tissues which normally express HK1[15, 49]. An examination of the species conservation of the nucleotides at the HK1 variant sites shows that there is conservation among primate species and cow at all 3 sites; the third HK1 variant site has nucleotide-specific conservation through additional species (Fig. 2b).
After an exhaustive search for the specific mutation causing dominant hyperinsulinism in this pedigree, we confidently report that this family represents a novel form of the disease. The previously described HI loci have clearly been excluded and the linkage analysis convincingly points to a novel region on chromosome 10 for this form of HI. The various sequencing experiments narrow the possible genes responsible to HK1 and DNA2 and the potential role that the HK1 variants and possibly the missense mutation found in DNA2 play in disrupting the normal silencing of HK1 in beta-cells remains speculative at present. Direct proof of the functional consequences of the sequence variants in HK1 and DNA2 would require islets from affected patients and, since the disorder is successfully treated with diazoxide, there are no affected islets available for study. In addition, an approach such as using a reporter construct to test the effects of each variant in clonal rodent beta-cells, similar to that taken with MCT1 variants by Otonkoski, et al, is not feasible due to great distance of the HK1 variants from their promoters (Fig. 2b) [43, 44]. Study of additional families with this condition would be helpful in determining the causal variant.
In summary, the hypoglycemia disorder in this historically important family represents a novel congenital HI disorder which maps to chromosome 10q21–22. The disorder has an autosomal dominant inheritance pattern and results in a diazoxide-responsive form of HI. Disease-associated variants were found in non-coding regions of HK1 and exon 18 of DNA2. HK1 appears to be the stronger candidate disease gene since mutations leading to impaired silencing of HK1 expression in beta-cells could lower the glucose threshold for insulin secretion and readily explain the clinical phenotype of this family.
Supplementary Material
Acknowledgments
This work was supported in part by: CAS: R37 DK056268, R01 DK053012; SEP: K08 DK090302, PES Research Fellowship, K12 HD043245; and P30-DK19525 and UL1RR024134.
The authors would like to thank Philippe Backeljauw, MD and his colleagues in the Division of Endocrinology at Cincinnati Children’s Hospital for referring their patient to the Hyperinsulinism Center at CHOP.
Footnotes
DISCLOSURE STATEMENT: The authors have nothing to disclose.
References
- 1.Stanley CA, De Leon DD, editors. Monogenic hyperinsulinemic hypoglycemia disorders. Basal, Switzerland: Karger; 2012. p. 21. [Google Scholar]
- 2.McQuarrie I. Idiopathic spontaneously occurring hypoglycemia in infants: Clinical significance of problem and treatment. American Journal of Diseases of Childhood. 1954;87:399. [PubMed] [Google Scholar]
- 3.De Leon DD, Stanley CA. Mechanisms of disease: Advances in diagnosis and treatment of hyperinsulinism in neonates. Nat Clin Pract Endocrinol Metab. 2007;3:57–68. doi: 10.1038/ncpendmet0368. [DOI] [PubMed] [Google Scholar]
- 4.Monogenic disorders of insulin secretion: Congenital hyperinsulinism and neonatal diabetes. Pediatric Diabetes. 2012;13:344–368. [Google Scholar]
- 5.Finegold DN, Stanley CA, Baker L. Glycemic response to glucagon during fasting hypoglycemia: An aid in the diagnosis of hyperinsulinism. J Pediatr. 1980;96:257–259. doi: 10.1016/s0022-3476(80)80817-1. [DOI] [PubMed] [Google Scholar]
- 6.Stanley CA, Baker L. Hyperinsulinism in infancy: Diagnosis by demonstration of abnormal response to fasting hypoglycemia. Pediatrics. 1976;57:702–711. [PubMed] [Google Scholar]
- 7.Stanley CA, Baker L. Hyperinsulinism in infants and children: Diagnosis and therapy. Adv Pediatr. 1976;23:315–355. [PubMed] [Google Scholar]
- 8.Stanley CA, Thornton PS, Ganguly A, MacMullen C, Underwood P, Bhatia P, Steinkrauss L, Wanner L, Kaye R, Ruchelli E, Suchi M, Adzick NS. Preoperative evaluation of infants with focal or diffuse congenital hyperinsulinism by intravenous acute insulin response tests and selective pancreatic arterial calcium stimulation. J Clin Endocrinol Metab. 2004;89:288–296. doi: 10.1210/jc.2003-030965. [DOI] [PubMed] [Google Scholar]
- 9.Hsu BY, Kelly A, Thornton PS, Greenberg CR, Dilling LA, Stanley CA. Protein-sensitive and fasting hypoglycemia in children with the hyperinsulinism/hyperammonemia syndrome. J Pediatr. 2001;138:383–389. doi: 10.1067/mpd.2001.111818. [DOI] [PubMed] [Google Scholar]
- 10.Magge SN, Shyng SL, MacMullen C, Steinkrauss L, Ganguly A, Katz LE, Stanley CA. Familial leucine-sensitive hypoglycemia of infancy due to a dominant mutation of the beta-cell sulfonylurea receptor. J Clin Endocrinol Metab. 2004;89:4450–4456. doi: 10.1210/jc.2004-0441. [DOI] [PubMed] [Google Scholar]
- 11.Fourtner SH, Stanley CA, Kelly A. Protein-sensitive hypoglycemia without leucine sensitivity in hyperinsulinism caused by k(atp) channel mutations. J Pediatr. 2006;149:47–52. doi: 10.1016/j.jpeds.2006.02.033. [DOI] [PubMed] [Google Scholar]
- 12.Palladino AA, Sayed S, Levitt Katz LE, Gallagher PR, De Leon DD. Increased glucagon-like peptide-1 secretion and postprandial hypoglycemia in children after nissen fundoplication. The Journal of clinical endocrinology and metabolism. 2009;94:39–44. doi: 10.1210/jc.2008-1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matise TC, Chen F, Chen W, De La Vega FM, Hansen M, He C, Hyland FC, Kennedy GC, Kong X, Murray SS, Ziegle JS, Stewart WC, Buyske S. A second-generation combined linkage physical map of the human genome. Genome Res. 2007;17:1783–1786. doi: 10.1101/gr.7156307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abecasis GR, Cherny SS, Cookson WO, Cardon LR. Merlin--rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 15.Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman-Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ. The ucsc genome browser database: Update 2011. Nucleic Acids Res. 2011;39:D876–882. doi: 10.1093/nar/gkq963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann N, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blocker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowski J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 17.Drash A, Kenny F, Field J, Blizzard R, Langs H, Wolff F. The therapeutic application of diazoxide in pediatric hypoglycemic states. Ann N Y Acad Sci. 1968;150:337–355. doi: 10.1111/j.1749-6632.1968.tb19059.x. [DOI] [PubMed] [Google Scholar]
- 18.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. Dbsnp: The ncbi database of genetic variation. Nucleic Acids Res. 2001;29:308–311. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duxin JP, Dao B, Martinsson P, Rajala N, Guittat L, Campbell JL, Spelbrink JN, Stewart SA. Human dna2 is a nuclear and mitochondrial DNA maintenance protein. Mol Cell Biol. 2009;29:4274–4282. doi: 10.1128/MCB.01834-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng L, Zhou M, Guo Z, Lu H, Qian L, Dai H, Qiu J, Yakubovskaya E, Bogenhagen DF, Demple B, Shen B. Human dna2 is a mitochondrial nuclease/helicase for efficient processing of DNA replication and repair intermediates. Mol Cell. 2008;32:325–336. doi: 10.1016/j.molcel.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Budd ME, Campbell JL. A yeast gene required for DNA replication encodes a protein with homology to DNA helicases. Proc Natl Acad Sci U S A. 1995;92:7642–7646. doi: 10.1073/pnas.92.17.7642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Budd ME, Choe WC, Campbell JL. Dna2 encodes a DNA helicase essential for replication of eukaryotic chromosomes. J Biol Chem. 1995;270:26766–26769. doi: 10.1074/jbc.270.45.26766. [DOI] [PubMed] [Google Scholar]
- 24.Sayed S, Langdon DR, Odili S, Chen P, Buettger C, Schiffman AB, Suchi M, Taub R, Grimsby J, Matschinsky FM, Stanley CA. Extremes of clinical and enzymatic phenotypes in children with hyperinsulinism caused by glucokinase activating mutations. Diabetes. 2009;58:1419–1427. doi: 10.2337/db08-1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pinney SE, MacMullen C, Becker S, Lin YW, Hanna C, Thornton P, Ganguly A, Shyng SL, Stanley CA. Clinical characteristics and biochemical mechanisms of congenital hyperinsulinism associated with dominant katp channel mutations. J Clin Invest. 2008;118:2877–2886. doi: 10.1172/JCI35414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Thornton PS, MacMullen C, Ganguly A, Ruchelli E, Steinkrauss L, Crane A, Aguilar-Bryan L, Stanley CA. Clinical and molecular characterization of a dominant form of congenital hyperinsulinism caused by a mutation in the high-affinity sulfonylurea receptor. Diabetes. 2003;52:2403–2410. doi: 10.2337/diabetes.52.9.2403. [DOI] [PubMed] [Google Scholar]
- 27.Matschinsky F, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, Cohen D, Permutt MA, Tanizawa Y, Jetton TL, et al. Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J Clin Invest. 1993;92:2092–2098. doi: 10.1172/JCI116809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matschinsky FM. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes. 1990;39:647–652. doi: 10.2337/diab.39.6.647. [DOI] [PubMed] [Google Scholar]
- 29.Wilson JE. Isozymes of mammalian hexokinase: Structure, subcellular localization and metabolic function. J Exp Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- 30.Froguel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, Lesage S, Stoffel M, Takeda J, Passa P, et al. Familial hyperglycemia due to mutations in glucokinase. Definition of a subtype of diabetes mellitus. N Engl J Med. 1993;328:697–702. doi: 10.1056/NEJM199303113281005. [DOI] [PubMed] [Google Scholar]
- 31.Glaser B, Kesavan P, Heyman M, Davis E, Cuesta A, Buchs A, Stanley CA, Thornton PS, Permutt MA, Matschinsky FM, Herold KC. Familial hyperinsulinism caused by an activating glucokinase mutation. N Engl J Med. 1998;338:226–230. doi: 10.1056/NEJM199801223380404. [DOI] [PubMed] [Google Scholar]
- 32.Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, Guertin KR, Hilliard DW, Kester RF, Mahaney PE, Marcus L, Qi L, Spence CL, Tengi J, Magnuson MA, Chu CA, Dvorozniak MT, Matschinsky FM, Grippo JF. Allosteric activators of glucokinase: Potential role in diabetes therapy. Science. 2003;301:370–373. doi: 10.1126/science.1084073. [DOI] [PubMed] [Google Scholar]
- 33.Liang Y, Kesavan P, Wang LQ, Niswender K, Tanizawa Y, Permutt MA, Magnuson MA, Matschinsky FM. Variable effects of maturity-onset-diabetes-of-youth (mody)-associated glucokinase mutations on substrate interactions and stability of the enzyme. Biochem J. 1995;309 (Pt 1):167–173. doi: 10.1042/bj3090167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quintens R, Hendrickx N, Lemaire K, Schuit F. Why expression of some genes is disallowed in beta-cells. Biochem Soc Trans. 2008;36:300–305. doi: 10.1042/BST0360300. [DOI] [PubMed] [Google Scholar]
- 35.Becker TC, BeltrandelRio H, Noel RJ, Johnson JH, Newgard CB. Overexpression of hexokinase i in isolated islets of langerhans via recombinant adenovirus. Enhancement of glucose metabolism and insulin secretion at basal but not stimulatory glucose levels. J Biol Chem. 1994;269:21234–21238. [PubMed] [Google Scholar]
- 36.Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, Kikuchi M, Yazaki Y, Miyazaki J, Oka Y. Overexpression of hexokinase i but not glut1 glucose transporter alters concentration dependence of glucose-stimulated insulin secretion in pancreatic beta-cell line min6. J Biol Chem. 1994;269:3081–3087. [PubMed] [Google Scholar]
- 37.Epstein PN, Boschero AC, Atwater I, Cai X, Overbeek PA. Expression of yeast hexokinase in pancreatic beta cells of transgenic mice reduces blood glucose, enhances insulin secretion, and decreases diabetes. Proc Natl Acad Sci U S A. 1992;89:12038–12042. doi: 10.1073/pnas.89.24.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Henquin JC, Sempoux C, Marchandise J, Godecharles S, Guiot Y, Nenquin M, Rahier J. Congenital hyperinsulinism caused by hexokinase i expression or glucokinase-activating mutation in a subset of beta-cells. Diabetes. 2013;62:1689–1696. doi: 10.2337/db12-1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dayeh TA, Olsson AH, Volkov P, Almgren P, Ronn T, Ling C. Identification of cpg-snps associated with type 2 diabetes and differential DNA methylation in human pancreatic islets. Diabetologia. 2013;56:1036–1046. doi: 10.1007/s00125-012-2815-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pare G, Chasman DI, Parker AN, Nathan DM, Miletich JP, Zee RY, Ridker PM. Novel association of hk1 with glycated hemoglobin in a non-diabetic population: A genome-wide evaluation of 14,618 participants in the women’s genome health study. PLoS Genet. 2008;4:e1000312. doi: 10.1371/journal.pgen.1000312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bonnefond A, Vaxillaire M, Labrune Y, Lecoeur C, Chevre JC, Bouatia-Naji N, Cauchi S, Balkau B, Marre M, Tichet J, Riveline JP, Hadjadj S, Gallois Y, Czernichow S, Hercberg S, Kaakinen M, Wiesner S, Charpentier G, Levy-Marchal C, Elliott P, Jarvelin MR, Horber F, Dina C, Pedersen O, Sladek R, Meyre D, Froguel P. Genetic variant in hk1 is associated with a proanemic state and a1c but not other glycemic control-related traits. Diabetes. 2009;58:2687–2697. doi: 10.2337/db09-0652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gjesing AP, Nielsen AA, Brandslund I, Christensen C, Sandbaek A, Jorgensen T, Witte D, Bonnefond A, Froguel P, Hansen T, Pedersen O. Studies of a genetic variant in hk1 in relation to quantitative metabolic traits and to the prevalence of type 2 diabetes. BMC Med Genet. 2011;12:99. doi: 10.1186/1471-2350-12-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Otonkoski T, Kaminen N, Ustinov J, Lapatto R, Meissner T, Mayatepek E, Kere J, Sipila I. Physical exercise-induced hyperinsulinemic hypoglycemia is an autosomal-dominant trait characterized by abnormal pyruvate-induced insulin release. Diabetes. 2003;52:199–204. doi: 10.2337/diabetes.52.1.199. [DOI] [PubMed] [Google Scholar]
- 44.Otonkoski T, Jiao H, Kaminen-Ahola N, Tapia-Paez I, Ullah MS, Parton LE, Schuit F, Quintens R, Sipila I, Mayatepek E, Meissner T, Halestrap AP, Rutter GA, Kere J. Physical exercise-induced hypoglycemia caused by failed silencing of monocarboxylate transporter 1 in pancreatic beta cells. Am J Hum Genet. 2007;81:467–474. doi: 10.1086/520960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Veljkovic J, Hansen U. Lineage-specific and ubiquitous biological roles of the mammalian transcription factor lsf. Gene. 2004;343:23–40. doi: 10.1016/j.gene.2004.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Coull JJ, Romerio F, Sun JM, Volker JL, Galvin KM, Davie JR, Shi Y, Hansen U, Margolis DM. The human factors yy1 and lsf repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J Virol. 2000;74:6790–6799. doi: 10.1128/jvi.74.15.6790-6799.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.He G, Margolis DM. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (hiv-1) by the hiv-1 repressor yy1 and hiv-1 activator tat. Mol Cell Biol. 2002;22:2965–2973. doi: 10.1128/MCB.22.9.2965-2973.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kato H, Horikoshi M, Roeder RG. Repression of hiv-1 transcription by a cellular protein. Science. 1991;251:1476–1479. doi: 10.1126/science.2006421. [DOI] [PubMed] [Google Scholar]
- 49.Bhandare R, Schug J, Le Lay J, Fox A, Smirnova O, Liu C, Naji A, Kaestner KH. Genome-wide analysis of histone modifications in human pancreatic islets. Genome Res. 2010;20:428–433. doi: 10.1101/gr.102038.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.