

Abstract
We used oriented solid-state NMR spectroscopy and biochemical cross-linking experiments to demonstrate that the ligand-free membrane protein transporter EmrE forms anti-parallel dimers with different monomer tilt angles relative to the lipid bilayer. Our results also show the subtle conformational changes efflux pumps experience in response to drug binding and emphasize the importance of studying membrane proteins in a fluid bilayer environment.
Keywords: EmrE, oriented solid-state NMR, membrane proteins, multidrug resistance, molecular recognition
Multidrug resistance (MDR) is a major public health problem that reduces the efficacy of antibiotics in the treatment of infections.[2] One of the most common mechanisms in bacteria for conferring MDR is the coupling of drug efflux with the proton motive force.[3] A prototype for studying ion-coupled active transport is the polytopic E. coli membrane protein EmrE, a member of the small multidrug resistance (SMR) family that has 110 residues and four transmembrane (TM) domains. EmrE oligomerizes to form a homodimer that effluxes a wide variety of cations such as ethidium, methyl viologen, and tetraphenylphosphonium (TPP+).[4] A combination of evidence from topology analyses,[5] crystallography,[6] spectroscopy,[7] and molecular modeling[1] all support EmrE forming an anti-parallel dimer and dual topologies in the cell membrane,[8] although controversy still remains in the field.[9] Recently, von Heijne and co-workers have demonstrated that the anti-parallel state has higher stability than the parallel form in vivo.[10] Low-resolution structural evidence has defined the general features of the anti-parallel quaternary arrangement, which includes cryo-electron microscopy images (7.5 Å x 16 Å)[6a] and an X-ray Cα model of TPP+ bound EmrE (4.5 Å).[6b] In addition, magic-angle-spinning data for E14[7b] and solution NMR experiments in isotropic bicelles also support the asymmetry for EmrE bound to TPP+.[7a] The latter study showed monomer interconversion within the dimer in agreement with an alternating access model of the transporter.[7, 11]
The objective of this Communication is to offer high-resolution structural insight into the ligand free or apo form of EmrE in lipid bilayers, since the plasticity of this conformation is responsible for adapting to the size and shape of the ligand.[12] Tate and co-workers have previously shown similarity between the apo and TPP+ asymmetric dimer structures with the latter crystals possessing a higher degree of order.[6a, 13] Using the microscopy images, biochemical restraints, and primary sequence conservation data, a backbone structural model was constructed for EmrE bound to TPP+ (PDB 2I68).[1] The X-ray structure was later reported and is also in qualitative agreement with this model of ligand-bound EmrE.[6b] However, as expected from the experimental resolution of the EmrE crystals, it was not possible to decipher atomic scale details of the structure. To date, no high-resolution structure exists for EmrE in the apo or ligand bound states.
Our structural studies are aimed at defining the features of the apo state that encode the molecular recognition mechanism. We first verified the functionality of our sample preparation using ITC in DMPC lipid vesicles. The dissociation constant for TPP+ of 46 nM corresponds to correctly folded and functional preparations (SI Figure 1).[14] Additionally, the fitted stoichiometry revealed a 1:2 TPP+:monomer ratio, which is consistent with the dimer as the functional oligomeric unit.[4b] To probe the possible parallel or antiparallel arrangement in the apo form, we carried out cross-linking experiments using a single Cys-mutant of EmrE (S107C) in DMPC liposomes with heterobifunctional (BMPS; amine- and thiol-reactive groups) and homofunctional reagents (o-PDM; thiol reactive groups). Consistent with previous results for TPP+ bound EmrE,[7a] we observed rapid cross-linking for BMPS between K22 (the only Lys in EmrE) and C107, which is only possible for the anti-parallel dimer arrangement (Figure 1). To the contrary, o-PDM showed a significant reduction in the cross-linking efficiency, and more importantly dimer formation decreased with increasing lipid to protein ratios (SI Figure 2). We found that dimer formation in the presence of BMPS did not significantly change (<5%) with the lipid to protein ratio (Figure 1). Our results support the conclusion that it is the interaction between anti-parallel dimers and not within the parallel dimer that leads to o-PDM cross-linking on the gel. These findings on the apo conformation are similar with the single molecule FRET experiments on the TPP+ bound form of EmrE that support the anti-parallel configuration of the dimer[7a].
Figure 1.

Cross-linking results for the apo form of EmrE in DMPC lipid vesicles. (A) SDS-PAGE gel of the BMPS cross-linking reaction with varying lipid:protein ratios having constant EmrE concentrations. (B) Normalized dimer formation relative to the 100:1 lipid:protein ratio. (C) Schematic of BMPS cross-linking between at K22 and C107, which is only possible for the antiparallel topology.
Does the apo form possess an asymmetric configuration that can be verified at atomic resolution?
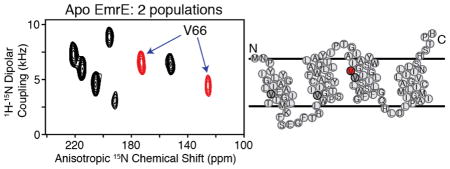
To answer this question, we utilized oriented solid-state NMR (O-SSNMR) spectroscopy, which is a direct method to probe the structure and tilt angles of membrane proteins relative to the lipid bilayer.[15] EmrE was reconstituted into magnetically aligned bicelles consisting of DMPC/DHPC in a 3.2/1 molar ratio (q=3.2), similar to the conditions for ITC and cross-linking experiments. The protein alignment in flipped bicelles was checked with 1D 1H/15N cross-polarization of [U-15N] labeled EmrE (SI Figure 3B). As expected for an aligned protein with short loops and no soluble domains, the 15N chemical shifts were clustered between 120–220 ppm. Due to overlapping resonances in this spectrum, we shifted our attention to selectively labeled samples ([15N-Val] and [15N-Met]) that gave local probes distributed in each of the TM domains (Figure 2): TM1 (V15, M21), TM2 (V34), TM3 (V66, V69), and TM4 (M91, M92, V98).
Figure 2.

PISEMA spectra of apo EmrE in flipped magnetically aligned bicelles (DMPC/DHPC, q=3.2). (A) [15N-Met] and (B) [15N-Val] wild-type EmrE PISEMA spectra. (C) [15N-Val] V98I EmrE PISEMA (black) showing two less peaks than in the wild-type spectrum (grey). The arrows indicate peaks corresponding to V98.
A sensitivity-enhanced version[16] of PISEMA[17] was recorded for each sample to correlate 15N anisotropic chemical shifts with 1H- 15N dipolar couplings. The PISEMA spectrum for [15N-Val] apo EmrE is shown in Figure 2B, and revealed 10 resolved peaks having chemical shifts corresponding to TM domain helices. Interestingly, this is twice the expected number of Val residues in the primary sequence and shows there are two populations with different angular dependencies with respect to the lipid bilayer normal. Similar to the Val spectrum, the experiment on [15N-Met] EmrE in Figure 2A also showed peak doubling for M21 and M91. The lack of doubling for M1 and M92 are due to the location at the dynamic N-terminus and the presence of overlapping populations, which is evident by the two-fold greater peak intensity of M92 with respect to M21 or M91. Taken together with our ITC and cross-linking results, the data strongly support the conclusion that the two asymmetric populations have different tilt angles with respect to the membrane and stem from the anti-parallel quaternary arrangement of the apo dimer.
In order to extract meaningful structural restraints, assignment of the PISEMA spectra was carried out. This is typically achieved by selective isotope labeling in conjunction with the assumption of periodic spectral patterns (PISA wheels)[18] and the use of spin diffusion techniques.[19] While somewhat straightforward for a single helix, polytopic membrane proteins present increased challenges due to overlapping PISA wheels. In fact, for the asymmetric EmrE dimer, this involves eight PISA wheels. Therefore, as an alternative, we implemented a mutagenesis approach to assign the spectra in Figure 2. All single-site mutations preserved the approximate size and hydrophobicity of the wild-type residue (Val and Met to Ile), and importantly did not disrupt the binding affinity or stoichiometry to TPP+ (SI Table I). An example of this assignment approach is displayed for [15N-Val] V98I EmrE, which shows eight resolved peaks that overlapped with eight of the ten peaks in the corresponding wild-type [15N-Val] spectrum (Figure 2C). This experiment with other mutant spectra shown in SI Figure 4 confirmed the two populations and provided an unambiguous way to assign the PISEMA spectra. While the mutagenesis approach to assign the O-SSNMR spectra did not directly distinguish between monomers A and B for each residue, these assignments were obtained by the qualitative agreement between the observed PISEMA profile and those calculated values from the computational model 2I68 (SI Figure 5). Future work will seek to assign the peaks to specific monomers without the assistance of the structural model.
What is the effect of ligand binding on the asymmetric apo EmrE conformation?
To provide insight into the tilt angle changes of the transporter, we carried out a titration with TPP+ and monitored the spectral changes using PISEMA spectroscopy. Upon addition of one-half molar equivalent TPP+ relative to the EmrE dimer concentration, we observed additional peak splitting in the slow chemical exchange timescale for several residues (Figure 3B). The slow exchange is consistent with the strong binding affinity to TPP+ and the presence of 50% free and 50% bound dimers. To the same sample, we then made a saturating addition of ligand and observed eight clearly distinguishable peaks, which was similar to the profile of the initial ligand-free spectrum. These data are evidence for a specific rearrangement of the transporter upon ligand binding and also validate the two asymmetric populations we observed in the apo form. The changes occurring upon TPP+ binding are also shown for Met residues in SI Figure 6. While we observed chemical shift perturbations in each TM domain (SI Figure 7A), one of the key features of the spectra is the differential behavior found for V66 and V69 within the same monomer (Figure 3 and SI Figure 8). The fact that one peak showed a chemical shift perturbation and the other did not is suggestive of a structural rearrangement involving helix bending around G67 in TM3 (sequence is G65-V66-G67-I68-V69). This conclusion is consistent with the computational model of EmrE having a discernible kink between V66 and V69 in TM3 of monomer B[1] and EPR paramagnetic accessibility experiments in liposomes.[12b] Note that all other helices in the computational model were ideal with no major deviations from uniform dihedral angles. In Figure 4, we present a depiction of changes in TM3 upon binding to TPP+. In this model, the N-terminal half of monomer A and the C-terminal side of monomer B tilt in response to ligand binding, which is consistent with our experimental PISEMA spectra.
Figure 3.

TPP+ binding titration of EmrE probed with PISEMA spectroscopy. (A) Ligand-free spectrum of [15N-Val] V98I, (B) 0.5 molar equivalents TPP+ relative to the dimer, and (C) a saturating addition of ligand to the same sample. 1D slices are shown in SI Figure 8.
Figure 4.

Bending motion of EmrE TM3. (A) Computational model of EmrE by Fleishman et al.[1] showing the kink in monomer A of 2I68. (B) Cartoon representation of TM3 bending motion consistent with the differential behavior of V66 and V69 in monomers A and B in the PISEMA spectra shown in Figure 3.
The combination of O-SSNMR and cross-linking experiments demonstrated that the ligand-free form of EmrE has an anti-parallel dimer and asymmetric tilt angles with respect to the lipid bilayer normal. This asymmetry is particularly unusual given the fact that nearly all homo-oligomeric proteins found in biology possess symmetry, which has been argued to enhance stability and allosteric control over assemblies.[21] Our atomic-resolution observables were possible with PISEMA spectroscopy and further validate the backbone structural models available from cryo-electron microscopy and X-ray crystallography.[6b, 13] Nevertheless, the PISEMA spectra showed quantitative differences with the structural models and spectral perturbations between the apo and TPP+ bound forms that were not easily discernible in the cryo-electron microscopy images (SI Figure 7).[13] One example is the fact that the fitted rotation angles from the PISEMA data for TM4 differed by ~33° and ~50° in monomers A and B with respect to those in PDB 2I68 (SI Figures 9 and 10). These subtle atomic-scale differences emphasize the need for a high-resolution structure of the apo and ligand-bound forms to fully understand the molecular recognition and transport mechanisms within the lipid membrane.[20] Since the MDR phenomenon hinges on the recognition of a wide variety of ligands, studies to give additional insight into the structural plasticity of apo EmrE is of paramount importance.
Experimental Section
Methods are given in the supporting information. Received: ((will be filled in by the editorial staff)) Published online on ((will be filled in by the editorial staff))
Supplementary Material
Acknowledgments
This work was supported by NIH grant K22AI083745 and start-up funds from New York University. We thank Prof. Ray Turner and Dr. Denice Bay for helpful discussions and Prof. Bobby Arora and Steve Joy for synthesis of BMPS.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Fleishman SJ, Harrington SE, Enosh A, Halperin D, Tate CG, Ben-Tal N. J Mol Biol. 2006;364:54–67. doi: 10.1016/j.jmb.2006.08.072. [DOI] [PubMed] [Google Scholar]
- 2.Nikaido H. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Higgins CF. Nature. 2007;446:749–757. doi: 10.1038/nature05630. [DOI] [PubMed] [Google Scholar]
- 4.a) Bay DC, Turner RJ. BMC Evolutionary Biology. 2009;9:140. doi: 10.1186/1471-2148-9-140. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Schuldiner S. Biochim Biophys Acta. 2009;1794:748–762. doi: 10.1016/j.bbapap.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 5.Rapp M, Seppala S, Granseth E, von Heijne G. Science. 2007;315:1282–1284. doi: 10.1126/science.1135406. [DOI] [PubMed] [Google Scholar]
- 6.a) Ubarretxena-Belandia I, Baldwin JM, Schuldiner S, Tate CG. EMBO J. 2003;22:6175–6181. doi: 10.1093/emboj/cdg611. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen YJ, Pornillos O, Lieu S, Ma C, Chen AP, Chang G. Proc Natl Acad Sci U S A. 2007;104:18999–19004. doi: 10.1073/pnas.0709387104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Morrison EA, DeKoster GT, Dutta S, Vafabakhsh R, Clarkson MW, Bahl A, Kern D, Ha T, Henzler-Wildman KA. Nature. 2012;481:45–50. doi: 10.1038/nature10703. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Lehner I, Basting D, Meyer B, Haase W, Manolikas T, Kaiser C, Karas M, Glaubitz C. J Biol Chem. 2008;283:3281–3288. doi: 10.1074/jbc.M707899200. [DOI] [PubMed] [Google Scholar]
- 8.Rapp M, Granseth E, Seppala S, von Heijne G. Nat Struc Mol Biol. 2006;13:112–116. doi: 10.1038/nsmb1057. [DOI] [PubMed] [Google Scholar]
- 9.Schuldiner S. Trends Biochem Sci. 2012;37:215–219. doi: 10.1016/j.tibs.2012.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lloris-Garcera P, Bianchi F, Slusky JS, Seppala S, Daley DO, von Heijne G. J Biol Chem. 2012;287:26052–26059. doi: 10.1074/jbc.M112.357590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yerushalmi H, Schuldiner S. Biochemistry. 2000;39:14711–14719. doi: 10.1021/bi001892i. [DOI] [PubMed] [Google Scholar]
- 12.a) Korkhov MV, Tate CG. J Mol Biol. 2008;377:1094–103. doi: 10.1016/j.jmb.2008.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Amadi ST, Koteiche HA, Mishra S, McHaourab HS. J Biol Chem. 2010;285:26710–26718. doi: 10.1074/jbc.M110.132621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tate CG, Kunji ER, Lebendiker M, Schuldiner S. EMBO J. 2001;20:77–81. doi: 10.1093/emboj/20.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tate CG. Curr Opin Struct Biol. 2006;16:457–464. doi: 10.1016/j.sbi.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 15.a) Sharma M, Yi M, Dong H, Qin H, Peterson E, Busath DD, Zhou HX, Cross TA. Science. 2010;330:509–512. doi: 10.1126/science.1191750. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) De Angelis AA, Howell SC, Nevzorov AA, Opella SJ. J Am Chem Soc. 2006;128:12256–12267. doi: 10.1021/ja063640w. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Traaseth NJ, Shi L, Verardi R, Mullen DG, Barany G, Veglia G. Proc Natl Acad Sci U S A. 2009;106:10165–10170. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gopinath T, Veglia G. J Am Chem Soc. 2009;131:5754–5756. doi: 10.1021/ja900096d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu CH, Ramamoorthy A, Opella SJ. J Magn Reson. 1994;109:270–272. [Google Scholar]
- 18.a) Marassi FM, Opella SJ. J Magn Reson. 2000;144:150–155. doi: 10.1006/jmre.2000.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang J, Denny J, Tian C, Kim S, Mo Y, Kovacs F, Song Z, Nishimura K, Gan Z, Fu R, Quine JR, Cross TA. J Magn Reson. 2000;144:162–167. doi: 10.1006/jmre.2000.2037. [DOI] [PubMed] [Google Scholar]
- 19.a) Knox RW, Lu GJ, Opella SJ, Nevzorov AA. J Am Chem Soc. 2010;132:8255–8257. doi: 10.1021/ja102932n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mote KR, Gopinath T, Traaseth NJ, Kitchen J, Gor’kov PL, Brey WW, Veglia G. J Biomolec NMR. 2011;51:339–346. doi: 10.1007/s10858-011-9571-8. [DOI] [PubMed] [Google Scholar]; c) Xu J, Struppe J, Ramamoorthy A. J Chem Phys. 2008;128:052308. doi: 10.1063/1.2826323. [DOI] [PubMed] [Google Scholar]; d) Traaseth NJ, Gopinath T, Veglia G. J Phys Chem B. 2010;114:13872–13880. doi: 10.1021/jp105718r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banigan JR, Gayen A, Traaseth NJ. J Biomol NMR. 2013;55:391–399. doi: 10.1007/s10858-013-9724-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodsell DS, Olson AJ. Ann Rev of Biophys and Biomolec Structure. 2000;29:105–153. doi: 10.1146/annurev.biophys.29.1.105. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.