

Abstract
The multidrug and toxin extruders 1- and 2-K (MATE1 and MATE2-K) are expressed in the luminal membrane of renal proximal tubule cells and provide the active step in the secretion of molecules that carry a net positive charge at physiologic pH, so-called organic cations. The present study tested whether structurally distinct MATE substrates can display different quantitative profiles of inhibition when interacting with structurally distinct ligands. The tested ligands were three structurally similar cationic ionic liquids (ILs, salts in the liquid state: N-butylpyridinium, NBuPy; 1-methyl-3-butylimidazolium, Bmim; and N-butyl-N-methylpyrrolidinium, BmPy). Uptake was measured using Chinese hamster ovary cells that stably expressed MATE1 or MATE2-K. By trans-stimulation, all three ILs were transported by both MATE transporters. The three ILs also inhibited uptake of three structurally distinct MATE substrates: 1-methyl-4-phenylpyridinium (MPP), triethylmethylammonium (TEMA), and N,N,N-trimethyl-2-[methyl(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]ethanaminium (NBD-MTMA). MATE1 displayed a higher affinity for the pyridinium-based NBuPy (IC50 values, 2–4 µM) than for either the pyrrolidinium- (BmPy; 20–70 µM) or imidazolium-based ILs (Bmim; 15–60 µM). Inhibition of MPP, TEMA, and NBD-MTMA transport by NBuPy was competitive, with comparable Ki values against all substrates. Bmim also competitively blocked the three substrates but with Ki values that differed significantly (20 µM against MPP and 30 µM against NBD-MTMA versus 60 µM against TEMA). Together, these data indicate that renal secretion of ILs by the human kidney involves MATE transporters and suggest that the mechanism of transport inhibition is ligand-dependent, supporting the hypothesis that the binding of substrates to MATE transporters involves interaction with a binding surface with multiple binding sites.
Introduction
The kidney is the primary route for elimination from the body of a structurally diverse array of organic compounds, many of which are exogenous (i.e., xenobiotic) in origin. These include many plant-derived compounds found in typical diets, clinically relevant synthetic pharmaceuticals, and environmental toxins. The renal proximal tubule (RPT) is the principal site of active secretion of organic compounds that carry a net positive charge at physiologic pH, so-called organic cations (OCs) (Hagenbuch, 2010), by a process that involves two distinct steps (Pelis and Wright, 2011). The first step involves entry of substrate from the blood into RPT cells across the basolateral (peritubular) membrane and involves electrogenic uniport mediated by one or more members of the SLC22A family of solute carriers; in humans, this is the organic cation transporter OCT2 (Motohashi et al., 2002), whereas in rodents, both Oct1 and Oct2 are involved Karbach et al., 2000; Jonker and Schinkel, 2004). The second step in renal OC secretion, exit from RPT cells into the tubular filtrate across the apical (luminal) membrane, involves one or more members of the solute carrier SLC47A [multidrug and toxin extrusion (MATE)] family of electroneutral OC/H+ exchangers (Terada and Inui, 2008); in humans, this includes MATE1 and MATE2/2-K (Otsuka et al., 2005; Komatsu et al., 2011); in rodents, it is restricted to Mate1 (Lickteig et al., 2008). It is this second luminal step that is the active and rate-limiting element of OC secretion in renal tubules (Ross and Holohan, 1983; Schäli et al., 1983).
Despite the central role in renal OC secretion played by MATEs, comparatively little is known about the mechanistic basis of ligand interaction with these transport proteins. We recently used the profiles of inhibition of 1-methyl-4-phenylpyridinium (MPP) transport displayed by a set of structurally distinct ligands to develop a three-dimensional pharmacophore of inhibitory ligand interaction with MATE1 (Astorga et al., 2012). The model revealed hydrophobic regions, hydrogen bond donor and acceptor sites, and an ionizable (cationic) feature as key determinants for binding of inhibitory ligands to MATE1. That study also developed a pharmacophore based on results reported by Kido et al. (2011) for ligand inhibition of MATE1-mediated transport of a structurally distinct substrate, 4-(4-(dimethylamino)styryl)-N-methylpyridinium. It is noteworthy that this model had features arranged in a spatial configuration that differed substantially from that of the model based on inhibition of MPP transport. Although the basis of this discrepancy could reflect differences in methods used in these studies (with respect to measurement of transport), we suggest another, heretofore unacknowledged, complicating factor associated with efforts to develop predictive models of inhibitory ligand interaction with MATEs, namely, the influence of substrate on inhibitor interaction.
The present study tests the hypothesis that structurally distinct substrates of MATE1 and MATE2-K (Fig. 1) can display different profiles of inhibition when interacting with structurally distinct inhibitory ligands. In our choice of test compounds, we took the opportunity to assess the basis of interaction with MATE transporters of a novel class of compounds within the larger group of organic cations: the so-called ionic liquids, i.e., salts in the liquid state (ILs). These compounds are of increasing interest for their utility for a variety of industrial applications. Although these characteristics make the ILs very appealing from an industrial perspective, their growing use comes with an increased risk of human exposure. Three ILs, N-butylpyridinium (NBuPy), 1-butyl-1-methylpyrrolidinium (BmPy), and 1-butyl-3-methylimidazolium (Bmim) (Fig. 1), have been nominated by the National Toxicology Program as models for IL toxicological testing because they are representative of the most common cationic classes of ILs.
Fig. 1.

Structures of the three test substrates (MPP, TEMA, and NBD-MTMA) and three test inhibitors (NBuPy, Bmim, and BmBy) used in this study and the table of Tanimoto similarity coefficients reveal their comparative structural dissimilarity.
Previous studies showed that the three aforementioned model ILs are, in fact, actively secreted in urine of both mice and rats (Cheng et al., 2009; Knudsen et al., 2009), and they both inhibit and serve as substrate for the human and rat orthologs of OCT2 (Cheng et al., 2009, 2011; Knudsen et al., 2009). Although it might reasonably be inferred that secreted ILs are substrates for MATE transporters, the nature of the interaction of ILs with the MATEs has not been fully examined. Here we show that the three model ILs are effective substrates as well as inhibitors of MATE1 and MATE2-K. In addition, we show that their quantitative inhibitory effect on MATE1 activity depends on the transported substrate, which is not consistent with simple competition for a common binding site and, instead, suggests that ligand interaction with MATE1 can involve binding to distinct sites within a larger binding surface.
Materials and Methods
Chemicals.
[3H]1-Methyl-4-phenylpyridinium ([3H]MPP; specific activity (S.A.) 80 Ci/mmol), [3H]triethylmethylammonium ([3H]TEMA; S.A. 8.5 Ci/mmol), and [3H]N,N,N-trimethyl-2-[methyl(7-nitrobenzo[c][1,2,5]oxadiazol-4-yl)amino]ethanaminium ([3H]NBD-MTMA; S.A. 80 Ci/mmol) were synthesized by the Department of Chemistry and Biochemistry, University of Arizona (Tucson, AZ). [14C]1-Ethyl-3-butylimidazolium ([14C]Bmim); S.A. 27.5 mCi/mmol) was obtained from RTI International (Research Triangle Park, NC). TEMA, MPP, Hams’ F-12 Kaighn’s modified medium, and Dulbecco’s modified Eagle’s medium were obtained from Sigma-Aldrich Co. (St. Louis, MO). [3H]NBD-MTMA was synthesized by the Synthesis Core of the Southwest Environmental Health Sciences Center, Department of Chemistry of the University of Arizona (Tucson, AZ) (Belzer et al., 2013); analysis by paper chromatography showed that the compound was >90% pure. The chloride salts of NBuPy, Bmim, and BmPy were obtained from Merck KGaA (Darmstadt, Germany). Other reagents were of analytical grade and commercially obtained from routine sources.
Cell Culture and Stable Expression of MATE1.
Chinese hamster ovary (CHO) cells containing a single integrated Flp Recombination Target site were obtained from Invitrogen Corporation (Carlsbad, CA) and used for stable expression of MATE1. The full-length human MATE1 sequence used in this study was generously provided by Dr. Kathleen Giacomini (University of California, San Francisco, CA) (Chen et al., 2007) (GenBank accession number NP_060712.2). The full-length human MATE2-K sequence used in this study was generously provided by Dr. Ken-ichi Inui (Kyoto University) (Masuda, et al., 2006) (GenBank accession number NM_001099646.1). Stable cells expressing MATE1 and MATE2-K were prepared using methods previously described (Astorga et al., 2012) and were maintained under selection pressure with hygromycin B (100 μg/ml; Invitrogen) under 5% CO2-95% air in a humidified incubator (Nuaire, Plymouth, MN) at 37°C. Subculture of the cells was performed every 3 to 4 days.
Uptake Experiments.
CHO cells expressing MATE1 were plated in 24-well cell culture plates (Greiner, Monroe, NC) at 6.0 × 105 cells per well, a density sufficient for the cells to reach confluence within 24 hours (or within 48 hours if seeded at 3.0 × 105 cells per well), at which time they were used in transport experiments. Before each experiment, cells were rinsed twice with 500 μl of Waymouth’s buffer (135 mM NaCl, 13 mM HEPES-NaOH, 28 mM d-glucose, 5 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, and 0.8 mM MgSO4), pH 8.4, at room temperature. For time-course experiments, cells were incubated in 200 μl of Waymouth’s buffer containing radiolabeled substrate (∼15 nM [3H]MPP, ∼15 nM [3H]NBD-MTMA, or ∼150 nM [3H]TEMA) for 2–10 minutes. To stop the transport process, each well was aspirated and rinsed three times with 1 ml of ice-cold Waymouth’s buffer. The cells were then solubilized in 200 µl of 0.5 N NaOH with 1% SDS and gently shaken for 30 minutes. For each sample, 100 µl of 1 N HCl was added to neutralize the cell lysate, and then aliquots of 250 µl were placed in liquid scintillation vials later filled with 3 ml of scintillation cocktail (MP Biomedicals, Santa Ana, CA). Accumulated radioactivity was determined by liquid scintillation spectrometry (Beckman LS6000IC; Beckman-Coulter, Santa Ana, CA). Individual transport observations were typically performed in duplicate for each experiment, and observations were usually confirmed at least three times in separate experiments using cells of a different passage. Over the course of the study, cells at passages 10–35 were used; there was no systematic variation in the kinetics of MATE-mediated transport.
Efflux Experiments.
MATE-expressing CHO cells were plated in 35-mm cell culture dishes (BD Biosciences, San Jose, CA) at 2.4 × 106 cells per plate, sufficient to reach confluence within 24 hours (or within 48 hours if plated at 1.2 × 106 cells), at which time they were used. Before the experiments, cells were rinsed twice with 2.0 ml of Waymouth’s buffer, pH 7.4, and were equilibrated in 2.0 ml of Waymouth’s buffer, pH 8.5, at room temperature, for 15 minutes. The cells were then incubated in Waymouth’s buffer (pH 8.5) containing 25 nM [3H]MPP for 20 minutes. To initiate efflux of the labeled substrate, 500 μl of Waymouth’s buffer (pH 8.5) containing a test compound was added (time zero) and efflux was monitored by collecting this volume and immediately replacing it every 15 seconds. Scintillation cocktail was added to the effluent samples, and radioactivity was determined using a liquid scintillation counter. After 3 minutes, efflux was stopped and the remaining intracellular [3H]MPP was determined by lysing the cells in 600 µl of 0.5 N NaOH with 1% SDS. After this mixture was gently shaken for 30 minutes, 300 µl of 1.0 N HCl was used to neutralize the resulting lysate. Aliquots of 800 µl were placed in liquid scintillation vials and filled with 5 ml of scintillation cocktail; radioactivity was determined by liquid scintillation spectrometry.
Data Analysis.
As shown in eq. 1, the kinetics of transport was assessed using the substrate displacement method of Malo and Berteloot (1991):
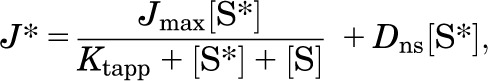 |
(1) |
where J* is the rate of transport of the radiolabeled substrate (for example, [3H]MPP) from a concentration of the labeled substrate equal to [S*], Jmax is the maximal rate of mediated substrate transport, Ktapp is the apparent Michaelis constant of the transported substrate, [S] is the concentration of unlabeled substrate, and Dns is a rate constant that describes the nonsaturable component of labeled substrate accumulation (reflecting the combined influences of diffusion, nonspecific binding, and incomplete rinsing of [3H]MPP from the cell culture well). The kinetics of ligand inhibition of MATE-mediated transport was adequately described by the relationship shown in eq. 2:
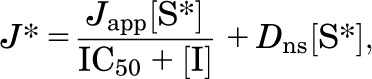 |
(2) |
where Japp is a constant that includes both the Jmax for substrate uptake modified by the other rate constants for the transported and inhibitory ligands (i.e., Ktapp and Ki), [I] is the concentration of the test agent (e.g., NBuPy), and IC50 is concentration of inhibitor that blocked 50% of mediated substrate transport. Results are presented as means ± S.E. Statistical analyses were performed using either analysis of variance or, when appropriate, a two-tailed unpaired Student’s t test, and observed differences were considered significant when P < 0.05 (Prism 5.03; GraphPad Software Inc., San Diego, CA).
Results
Characterization of MATE1 Transport Activity.
Before assessing the kinetic basis of IL interaction with MATE1, we established the transport characteristics of the probe substrates, MPP, NBD-MTMA, and TEMA. The functional expression of MATE1 was assessed by measuring the uptake of [3H]MPP in CHO-MATE1 (Fig. 2). To minimize the inhibitory effect of extracellular H+ on MATE-mediated OC transport (Tsuda et al., 2007; Dangprapai and Wright, 2011), transport was measured at an extracellular pH of 8.4. [3H]MPP transport was 20-fold greater in CHO-MATE1 compared with that in wild-type CHO cells after 10 minutes of uptake (Fig. 2A). Uptake in MATE1 cell line was nearly linear for 5 minutes (Fig. 2B); therefore, 5-minute uptakes were used to provide estimates of the initial rate of transport in subsequent studies of the kinetics of MATE-mediated transport.
Fig. 2.

(A) Transport of [3H]MPP mediated by CHO-wild type (WT) cells and CHO-MATE1. Uptakes (10 minutes; expressed relative to uptake in CHO WT cells) of [3H]MPP (∼15 nM) were measured at pH 8.4, in the presence and absence of 1 mM unlabeled MPP. The height of each bar is the mean (+S.E.) of uptake measured in three wells of a single representative experiment. (B) Time course of [3H]MPP (∼15 nM) uptake (pH 8.4) into CHO cells that stably expressed MATE1. Each point is the mean of triplicate measures of uptake determined in a single representative experiment, measured in the presence or absence of 1 mM unlabeled MPP (as indicated).
To determine the kinetics of probe substrate transport by MATE1, the uptake of [3H]substrate (15 nM) was measured in the presence of increasing concentrations of unlabeled substrate (Fig. 3). In seven separate experiments, the Ktapp was 5.8 ± 0.8 µM, and the Jmax was 1.8 ± 0.3 pmol cm−2 min−1 (Table 1).
Fig. 3.

Kinetics of MPP, TEMA, and NBD-MTMA uptake in CHO cells that stably expressed MATE1. Five-minute uptakes (pH 8.4) of 15 nM of [3H]MPP, 150 nM [3H]TEMA, or 14 nM [3H]NBD-MTMA were measured in the presence of increasing concentrations of unlabeled MPP, TEMA, or NBD-MTMA; each point represents the mean (±S.E.) of several separate experiments (n = 4, 5, or 7 for MPP, TEMA, or NBD-MTMA, respectively) selected for this summary presentation because they used a common set of substrate concentrations. Uptakes were normalized to the level of [3H]MPP, [3H]TEMA, or [3H]NBD-MTMA transport measured in the absence of unlabeled MPP, TEMA, or NBD-MTMA (% control).
TABLE 1.
Kinetics of MATE1-mediated transport of MPP, TEMA, NBD-MTMA, and the ionic liquid, Bmim
| Substrate |
MATE1 |
||
|---|---|---|---|
|
Jmax |
Ktapp |
TE |
|
| pmol cm−2 min−1 | μM | 10−6 cm s−1 | |
| [3H]MPP (n = 7) | 1.8 ± 0.3 | 5.8 ± 0.8 | 5.5 ± 0.8 |
| [3H]TEMA (n = 8) | 3.1 ± 0.5 | 80.2 ± 8.4 | 0.6 ± 0.01 |
| [3H]NBD-MTMA (n = 7) | 3.8 ± 1.2 | 19.8 ± 3.5 | 2.7 ± 0.5 |
| [3H]Bmim (n = 4) | 7.0 ± 1.6 | 33.9 ± 14.2 | 6.0 ± 2.6 |
MPP is a comparatively amphiphilic, planar, heterocyclic ring compound. Given the characteristic multiselectivity of MATEs (Tanihara et al., 2007) and the potential of xenobiotic transporters to display kinetically complex interactions with substrates and inhibitory ligands (e.g., Gorboulev et al., 2005; Harper and Wright, 2013), we elected to establish the kinetics of MATE1-mediated transport of two structurally dissimilar OCs, namely, the tetra-alkylammonium compound TEMA and the fluorescent substrate NBD-MTMA (Fig. 1). The 5-minute uptake of [3H]TEMA was measured against increasing concentrations of unlabeled TEMA (Fig. 3), and the resulting decrease in the uptake of the radiolabeled TEMA (150 nM) revealed a Ktapp of 80.2 ± 8.4 µM and Jmax of 3.1 ± 0.5 pmol cm−2 min−1 (n = 8; Table 1). The uptake of [3H]NBD-MTMA (15 nM) was measured against increasing concentrations of unlabeled NBD-MTMA (Fig. 3), revealing a Ktapp of 19.8 ± 3.5 µM and Jmax of 3.8 ± 1.2 pmol cm−2 min−1 (n = 7; Table 1).
Transport efficiency, which is defined as the ratio of Jmax/Kt, is a useful measure of the relative impact that OC transporters have on mediating the transmembrane flux from the comparatively low concentrations (generally < < than the Kt for the process) of substrate to which they are typically exposed (Schomig, et al., 2006). The typical units for the transport efficiency ratio, that is, μl min−1 mg protein−1 (Schomig et al., 2006), reflect those typically used to express Jmax (mol min−1 mg−1) and Kt (mol l−1). Our transport rates are presented in conventional units of flux, that is, flow/[(unit area)(unit time)], with the units of transport efficiency reduced to centimeters per second, which are the classic units of permeability. In other words, transport efficiency can be viewed as a measure of the contribution of the transporter in question to the carrier-mediated permeability of a membrane to the substrate in question. To facilitate comparison with transport efficiency values reported in the literature, the interested reader can convert our values to microliters min−1 mg protein−1 using the conversion factor 0.050 mg/cm2 of protein. MATE1 transported MPP with greater efficiency than that noted for TEMA or NBD-MTMA. Although MATE1 had a 2-fold greater Jmax for TEMA or NBD-MTMA (suggestive of a higher turnover number for translocation of this substrate), Ktapp values for MPP were 15 lower compared with that for TEMA and 4 times lower compared with NBD-MTMA. The result was a transport efficiency of 5.5 × 10 −6 cm s−1 for MPP versus 0.6 × 10 −6 cm s−1 for TEMA and 2.7 × 10−6 cm s−1 for NBD-MTMA (Table 1).
Inhibitory Interactions of ILs with MATE1 and MATE2-K
The inhibition of [3H]MPP by NBuPy, Bmim, and BmPy generated (respectively) IC50 values (in microliters) of 2.9 ± 0.4, 15.9 ± 1.5, and 18.8 ± 1.9 µM (Fig. 4A; Table 2). The three ILs generated similar profiles of inhibition of MATE2-K-mediated MPP transport as well (Table 2).
Fig. 4.

Inhibition by NBuPy, Bmim, or BmPy of (A) MPP, (B) TEMA, or (C) NBD-MTMA uptake in MATE1 expressing CHO cells. Five-minute uptakes (pH 8.4) of 15 nM [3H]MPP, 160 nM [3H]TEMA, or 13 nM [3H]NBD-MTMA were measured in the presence of increasing concentrations of each inhibitor. Each point is the mean (±S.E.) of results determined in two or three separate experiments. The IC50 values listed in the individual panels represent the mean values of individual experiments (see Table 2).
TABLE 2.
IC50/Ki values for ionic liquid inhibition of MATE-mediated transport of MPP, TEMA, and NBD-MTMA
| [3H]MPP |
[3H]TEMA |
[3H]NBD-MTMA |
||||
|---|---|---|---|---|---|---|
| IC50 | Ki | IC50 | Ki | IC50 | Ki | |
| μM | ||||||
| MATE1 | ||||||
| NBuPy | 2.9 ± 0.4 (n = 4) | 3.6 ± 1.3 (n = 3) | 3.8 ± 1.0 (n = 2) | 17.7 ± 8.4 (n = 3) | 1.7 ± 0.2 (n = 3) | 0.8 ± 0.4 (n = 3) |
| Bmim | 15.9 ± 1.5 (n = 3) | 24.3 ± 6.2 (n = 3) | 63.0 ± 0.5 (n = 2) | 63.4 ± 18.7 (n = 2) | 34.2 ± 3.6 (n = 3) | 28.4 ± 5 (n = 3) |
| BmPy | 18.8 ± 1.9 (n = 3) | — | 71.6 ± 17.0 (n = 3) | — | 60.0 ± 8.4 (n = 3) | — |
| MATE2-K | ||||||
| NBuPy | 1.6 ± 0.2 (n = 2) | — | 5.0 ± 2.8 (n = 2) | — | — | — |
| Bmim | 15.7 ± 0.7 (n = 3) | — | 33.5 ± 1.7 (n = 2) | — | — | — |
| BmPy | 19.0 ± 6.5 (n = 3) | — | 50.4 ± 12.6 (n = 3) | — | — | — |
—, not determined.
A parallel set of IC50 values was generated against transport of [3H]TEMA and [3H]NBD-MTMA to assess the potential role of substrate structure on the inhibitory interaction of the ILs with MATE1. The uptake of approximately 160 nM [3H]TEMA and 13 nM [3H]NBD-MTMA, concentrations well below the Ktapp for transport of each substrate, was measured in the presence of increasing concentrations of NBuPy, Bmim, or BmPy. Figure 4, B and C shows that, as seen for MPP, all the ILs inhibited the uptake of TEMA and NBD-MTMA. The IC50 values for NBuPy’s inhibition of TEMA (3.8 μM) and NBD-MTMA (1.7 μM) transport were not different from the IC50 value noted earlier for inhibition of MPP (2.9 µM) (P > 0.05; Table 2), which was expected if NBuPy competes with MPP, TEMA, and NBD-MTMA for a common binding site (or a set of mutually exclusive or overlapping binding sites). In contrast, the IC50 values for inhibition of TEMA and NBD-MTMA observed for Bmim and BmPy were both substantially higher than those for NBuPy (indicating a lower affinity of MATE1 for these two ILs, a profile shared by MATE2-K as well; Table 2) and, more intriguingly, significantly different (P < 0.05) from the IC50 values for inhibition by these compounds of MATE-mediated MPP transport noted already. For Bmim, whereas the IC50 value for MATE1-mediated MPP transport (in μM) was 15.9 ± 1.5, the values were 34.2 ± 3.6 and 63.0 ± 0.5 for NBD-MTMA and TEMA, respectively (P < 0.05); for BmPy, the IC50 value for inhibition of MPP transport was 18.8 ± 1.9, compared with 60.0 ± 8.4 and 71.6 ± 17 (P < 0.05; Table 2). Thus, although the data indicate that the test ILs were effective inhibitors of MATE1 (and MATE2-K), the mechanism(s) of that interaction is(are) unclear. As noted earlier, if ILs share a common binding site with MPP, TEMA, and NBD-MTMA, the IC50 (if representative of a competitive Ki) for inhibition of MPP transport generated for an IL should be the same as its value for inhibition of TEMA and NBD-MTMA transport (Christensen, 1975; Segel, 1975). Whereas this was the case for NBuPy (Table 2), the IC50 values for Bmim and BmPy inhibition of MPP uptake were consistently lower (2- to 4-fold) than those noted for TEMA and NBD-MTMA uptake (Table 2).
The similarity of IC50 and Ki values for NBuPy inhibition of MATE1-mediated transport of MPP, TEMA, and NBD-MTMA was consistent with a competitive interaction of these ligands. To assess more rigorously the mechanism of this interaction, we measured the kinetics of transport of MPP in the presence and absence of 30 µM NBuPy in CHO cells expressing MATE1 (Fig. 5). For MATE1, the presence of NBuPy caused a significant increase in the Ktapp, from 6.3 ± 0.7 µM to 61.5 ± 15.9 µM (P < 0.05), without significantly changing the Jmax (2.3 ± 0.45 versus 1.8 ± 0.16 pmol cm−2 min−1; P > 0.05) (Table 3). Because these data were consistent with competitive inhibition for a common binding site, we calculated the Ki for NBuPy inhibition of MPP transport using the following relationship: Ktapp(inhib) = Ktapp[1+([I]/Kiapp)], where Ktapp(inhib) is the Michaelis constant for the transport of the test substrate determined in the presence of an inhibitor at concentration [I], Ktapp is the apparent Michaelis constant for the test substrate measured in the absence of the inhibitor, and Kiapp is the apparent Michaelis constant of the competitive inhibitor. The calculated Kiapp value for inhibition of MATE1-mediated MPP transport by NBuPy was 3.6 ± 1.3 µM (Table 2), which was not different (P > 0.05) from the measured IC50 value for NBuPy inhibition of MPP transport (Table 2).
Fig. 5.

Eadie-Hostee plot showing the effect of extracellular NBuPy (30 μM) on the kinetics of MPP transport in CHO cells that stably expressed MATE1. Five-minute uptakes (pH 8.4) of 16 nM [3H]MPP were measured in a transport buffer containing increasing concentrations of the unlabeled substrate, plus or minus 30 µM unlabeled NBuPy. MATE-mediated substrate uptake was corrected for the nonsaturable component of total uptake. Each point is the mean (±S.E.) of results obtained in two separate experiments.
TABLE 3.
Kinetic basis of the inhibition by NBuPy and Bmim of MATE1-mediated transport of MPP, TEMA, and NBD-MTMA
| Substrate | MATE1 |
|||
|---|---|---|---|---|
| Jmax(control)a | Jmax(inhib)b | Ktapp(control)a | Ktapp(inhib)b | |
| pmol cm−2 min−1 | pmol cm−2 min−1 | μM | μM | |
| NBuPy (30 µM) | ||||
| [3H]MPP (n = 3) | 2.3 ± 0.45 | 1.8 ± 0.16 | 6.3 ± 0.7 | 61.5 ± 15.9 |
| P > 0.05 | P < 0.05 | |||
| [3H]TEMA (n = 3) | 3.4 ± 0.63 | 4.8 ± 1.17 | 91.5 ± 9.1 | 333 ± 140 |
| P > 0.05 | P = 0.16 | |||
| NBuPy (2 µM) | ||||
| [3H]NBD-MTMA (n = 7) | 3.8 ± 1.2 | 2.9 ± 0.7 | 20.0 ± 3.5 | 108.9 ± 33.8 |
| P > 0.05 | P < 0.05 | |||
| Bmim (120 µM) | ||||
| [3H]MPP (n = 3) | 6.2 ± 1 | 7.6 ± 2.9 | 13.3 ± 1.4 | 71.3 ± 11.7 |
| P > 0.05 | P < 0.05 | |||
| [3H]TEMA (n = 2) | 6.5 ± 1.1 | 8.1 ± 2.2 | 124.4 ± 26.6 | 366.1 ± 5.9 |
| P > 0.05 | P < 0.05 | |||
| Bmim (30 µM) | ||||
| [3H] NBD-MTMA (n = 3) | 2 ± 0.8 | 1.9 ± 0.8 | 15.6 ± 2.7 | 33.9 ± 5.4 |
| P > 0.05 | P < 0.05 | |||
Values marked control were measured in the absence of inhibitor.
“inhib” values were measured in the presence of indicated concentrations of either NBuPy or Bmim. Neither inhibitor influenced Jmax values, whereas Ktapp values were significantly increased over the control values.
Similar comparisons were performed for inhibition by NBuPy of MATE1-mediated transport of [3H]NBD-MTMA and [3H]TEMA (Table 3). For NBD-MTMA, the presence of 2 µM NBuPy had no effect (P > 0.05) on the Jmax (3.8 ± 1.2 pmol cm−2 min−1 versus 2.9 ± 0.7 pmol cm−2 min−1) but increased (P < 0.05) the Ktapp from 20.0 ± 3.5 μM to 108.9 ± 33.8 μM. The calculated Kiapp values for NBuPy inhibition of NBD-MTMA was 0.8 ± 0.4 µM, which is not different from its IC50 for inhibition of MPP transport (Table 2). For TEMA, the presence of 30 µM NBuPy had no effect on Jmax (3.4 ± 0.63 pmol cm−2 min−1, versus 4.8 ± 1.17 pmol cm−2 min−1), whereas Ktapp went from 91.5 ± 9.1 μM to 333 ± 140 μM. The calculated Kiapp values for NBuPy inhibition of TEMA was 17.7 ± 8.4 µM, which is not different from its IC50 for inhibition of MPP transport (Table 2).
As noted previously, the disparity in IC50 values for Bmim’s inhibition of the MATE1-mediated transport of three probe substrates (Table 2) suggested that these ligands do not share a common binding site. The kinetic basis of the inhibitory interaction between Bmim and transport of MPP, TEMA, and NBD-MTMA was determined by assessing the effect of Bmim on the kinetics of transport of MPP and TEMA (120 µM Bmim) and NBD-MTMA (30 µM Bmim). In each case, the inhibitory profiles were consistent with competition between Bmim and probe substrate, that is, significant increases in apparent Kt values with no effect on Jmax (Table 3). However, consistent with the disparity in Bmim IC50 values for inhibition of MATE1-mediated MPP and TEMA transport noted earlier, and in contrast to the results with NBuPy noted previously, the calculated Ki values for Bmim inhibition of the three probe substrates were not the same (P < 0.05) (Table 2); the Bmim Ki for inhibition of MPP transport was 24.3 µM, significantly less than the Ki of 63.4 µM for inhibition of TEMA transport. Similarly, the IC50 for Bmim’s inhibition of NBD-MTMA transport, 28.4 µM, differed significantly from that for inhibition of TEMA. These observations, summarized in Fig. 6, necessitate ligand interactions with the MATEs that are more complex than those limited to the classic model of competitive inhibition.
Fig. 6.

Comparison of inhibitor constants (IC50 or Ki) generated for NBuPy and Bmim against MATE1-mediated transport of MPP, TEMA, and NBD-MTMA. Data were taken from Tables 1, 2, and 3. *Differences that were significant at the level of P < 0.05.
MATE-Mediated Transport of ILs
Inhibition of transport is not evidence that the inhibitor is itself transported. Figure 7 shows that unlabeled Bmim inhibited the MATE1-mediated uptake of [14C]Bmim in a concentration-dependent manner, revealing Ktapp and Jmax values of 33.9 ± 14.2 µM and 7.0 ± 1.6 pmol cm−2 min−1. The resulting transport efficiency was 6 × 10−6 cm s−1, which was intermediate to the transport efficiency values for MATE-mediated MPP, TEMA, and NBD-MTMA transport efficiency noted earlier. Interestingly, the Kt for MATE1-mediated Bmim transport (34 µM) was intermediate to the constants it generated for inhibition of MPP (∼20 µM) and TEMA (∼60 µM) but identical to the constant generated against NBD-MTMA (34 µM) (Fig. 6).
Fig. 7.

Kinetics of Bmim transport mediated by MATE1. Five-minute uptakes (pH 8.4) of 23 µM [14C]Bmim were measured in the presence of increasing concentrations of unlabeled Bmim. Each point is the mean (±S.E.) of results obtained in three separate experiments.
To address whether NBuPy and BmPy, for which radiolabeled forms were not available, were substrates as well as inhibitors of MATE1 and MATE2-K, we determined their effectiveness as trans-stimulators of efflux of [3H]MPP preloaded into MATE1- and MATE2-K- expressing cells. After a 20-minute incubation of the MATE-expressing cells in buffer containing ∼25 nM [3H]MPP, the cells were briefly rinsed and then exposed to buffers containing one of the ILs at a concentration ∼20 times its IC50 or Kiapp value, that is, presumably a near-saturating concentration. Figure 8 shows that all three ILs successfully stimulated the rate of [3H]MPP efflux compared with that occurring under the control condition (no external OC substrate). Preliminary experiments showed that the presence of a 5 mM concentration of the organic anion p-aminohippurate had no effect on the rate of [3H]MPP efflux (data not shown), lending credence to the conclusion that the trans-stimulation of efflux reflected mediated exchange of intracellular MPP for extracellular IL. These results indicate that the ILs are not only effective inhibitors of, but also transported substrates for, MATE1 and MATE2-K. NBuPy, Bmim, and BmPy stimulated the rate of efflux at virtually the same rate, suggesting that the turnover numbers for each transporter-IL complex are similar.
Fig. 8.

Trans-stimulation, produced by inwardly directed gradients of NBuPy, Bmim, or BmPy, of [3H]MPP efflux from CHO cells stably expressing MATE1 or MATE2-K. Cells were preloaded with labeled substrate by a 20-minute incubation in Waymouth’s buffer (WB) containing 100 nM [3H]MPP (pH 8.5). Efflux buffers (pH 8.5) contained NBuPy (100 μM), Bmim (300 μM), or BmPy (300 μM) (see text for details). Each point is the mean of results obtained in two to four separate experiments (error bars omitted for clarity). Cell content (of [3H]MPP) is expressed as percentage on initial (time zero) content.
Discussion
The three cationic ILs selected by the National Toxicology Program as models for toxicological testing are substrates for renal secretion in rats and mice (Sipes et al., 2008; Cheng et al., 2009; Knudsen et al., 2009), and two of them (BmPy and NBuPy) are transported by the basolateral entry step in OC secretion in human renal proximal tubule, OCT2 (Cheng et al., 2009; Knudsen, et al., 2009). If the basolateral entry step in the secretion of ILs involves the passive, electrogenic uniporter, OCT2, the active step in renal secretion of ILs must reside in the luminal membrane. Here we showed that all three of the model ILs are transported substrates, as well as inhibitors, of the secondary active luminal OC transporters, MATE1 and MATE2-K. That ILs are effective inhibitors of the MATEs was expected; they are positively charged, comparatively small [mol. wt. 172–178 (as the chloride salts)], and moderately hydrophilic (logP values of −2.78 to −2.24, as predicted by the algorithm ALOGP, http://www.vcclab.org/lab/alogps), all of which are characteristic of the so-called type I OCs (Meijer et al., 1990), which have generally proven to be effective inhibitors of the multispecific OC+/H+ exchange activity of intact renal tubules (David et al., 1995) and isolated renal brush-border membrane vesicles (Wright et al., 1995; Wright and Wunz, 1999).1 However, the inhibition of MATE1 activity by ILs suggested a kinetically complex mechanism of substrate-inhibitor interaction with the transport protein. The inhibition of each of the probe substrates did appear to be classically competitive (i.e., the presence of inhibitor increased apparent Kt without influencing Jmax; but whereas NBuPy was equally effective as an inhibitor of MPP, TEMA, or NBD-MTMA transport (i.e., had equal IC50 values for inhibition of all three substrates), as expected, if these ligands compete for a common (or overlapping) binding site(s), Bmim and BmPy were significantly more effective inhibitors of MPP transport than of either TEMA or NBD-MTMA transport. This substrate-dependent inhibitor interaction is inconsistent with a purely competitive model of substrate-inhibitor interaction (Segel, 1975). Instead, this behavior was reminiscent of corticosterone’s inhibition of transport mediated by selected site-directed mutants of rOct1, which displayed IC50 values for MPP versus TEA that differed by 5-fold (Gorboulev et al., 2005). Similarly, Zolk et al. (2009) reported that a set of structurally diverse cationic drugs were markedly more effective inhibitors of OCT2-mediated metformin transport than of MPP transport. These observations with OCTs, and those presented here for the interaction of selected ILs with the MATEs, support the view that ligand interactions with multidrug transporters can include close-order (competitive) interactions and distant (allosteric) interactions (Gorboulev et al., 2005). We suggest that rather than having a single unique binding site, the MATEs have a binding surface that permits spatially distinct interactions with structurally distinct ligands. Similar ideas have been invoked to explain kinetically complex substrate-inhibitor interactions in other efflux transporters (e.g., the prokaryotic multidrug transporter, BmrR) (Vazquez-Laslop et al., 2000) and with the organic cation transporters (OCTs) (Gorboulev et al., 2005), providing a potential mechanistic basis for the polyselectivity of xenobiotic transporters. With respect to these latter ideas, it is interesting to note that the central mass of NBuPy (and MPP) is a six-membered ring (i.e., pyridinium), whereas Bmim and BmPy include smaller, five-membered rings (imidazolium and pyrrolidinium, respectively). This common structural difference could account for Bmim and BmPy interactions at sites spatially (and kinetically) distinct from those accessed by the larger NBuPy.
The three model ILs were not only effective inhibitors of MATE1-mediated transport; they also proved to be substrates for these processes. Transport of Bmim was assessed directly using the radiolabeled substrate (Fig. 7). Interestingly, the Kt for MATE1-mediated Bmim transport (34 µM) was identical to the constant generated against NBD-MTMA inhibition but intermediate to the constants it generated for inhibition of MPP (∼20 µM) and TEMA (∼60 µM) (Fig. 6). The Jmax for MATE1-mediated Bmim transport was severalfold higher than that for MPP and generally comparable to those for TEMA and NBD-MTMA (Table 1). We also used a trans-stimulation assay to provide an indirect measure of Bmim transport, showing that an inwardly directed chemical gradient of Bmim was sufficient to stimulate the efflux of preloaded [3H]MPP from CHO cells that stably expressed MATE1 and MATE2-K (Fig. 8). Although trans-stimulation of the rate of transport of compound A by an oppositely oriented gradient of compound B is not proof that A and B are both transported by a common process, it is the simplest explanation for observations like those shown in Fig. 8 (Stein, 1986). The extracellular concentration of Bmim used in these experiments (300 μM) was ∼20 times the Kt for Bmim transport and was selected to ensure that the transporters were >90% saturated with the substrate. Consequently, the stimulation of MPP efflux produced by this condition presumably reflected transporter turnover at or near the observed Jmax of Bmim uptake. Thus, the observation that extracellular concentrations of NBuPy and BmPy equal to ∼20 times their respective IC50 values also trans-stimulated MPP efflux mediated by MATE1 and MATE2-K at rates comparable to those driven by Bmim supports the conclusions that 1) NBuPy and BmPy are transported substrates of both MATEs and 2) the maximal rates of transport of both are similar to that for Bmim.
The existence of a common pathway(s) for the secretion of many OCs (e.g., cimetidine, pindolol, metformin) in the kidney and liver sets the stage for unwanted drug-drug interactions (Endres et al., 2006; Giacomini et al., 2010). Environmental chemicals, like the model ILs, can also exert unwanted interactions at the level of renal secretion. Cheng et al. (2011) recently showed that infusion of NBuPy reduced the plasma clearance of metformin by 65% in rats (with an associated increase in the plasma metformin concentration). NBuPy can block both the OCT2-mediated entry of metformin into RPT cells and the MATE1-mediated exit of metformin from these cells (rat kidney expresses MATE1 but not MATE2-K; (Ohta et al., 2006; Klaassen and Aleksunes, 2010)), but the observation that the NBuPy-induced reduction in plasma clearance of metformin was accompanied by an increase in metformin content (3.7-fold) (Cheng et al., 2011) in renal tissue suggests that NBuPy exerted its principal inhibitory effect on the MATE1-mediated exit step. It should be noted that the doses of NBuPy required to inhibit metformin clearance were quite high and most likely resulted in blood levels that would not be achieved in humans exposed orally or dermally to environmental or occupational levels of NBuPy-Cl or other ILs (Cheng et al., 2011). Nevertheless, the results presented here indicate that ILs are potentially capable of interfering with MATE-mediated OC transport and, consequently, of influencing the distribution and pharmacokinetics of cationic compounds that rely on renal (or hepatic) secretory pathways.
In conclusion, we showed that the National Toxicology Program’s three model cationic ILs (NBuPy, Bmim, and BmPy) are potent inhibitors, as well as transported substrates, of MATE1 and MATE2-K. These interactions support the hypothesis that MATE transporters serve as the active step in secretion of these compounds across the RPT. In addition, the substrate dependency of the inhibitory profiles generated by these ILs against MATE1-mediated transport of structurally distinct substrates supports the view ligands can interact with this multidrug-binding protein at multiple sites within a larger binding surface.
Abbreviations
- Bmim
1-butyl-3-methylimidazolium chloride
- BmPy
N-butyl-N-methylpyrrolidinium chloride
- CHO
Chinese hamster ovary
- IL
ionic liquid
- MATE
multidrug and toxin extrusion transporter
- MPP
1-methyl-4-phenylpyridinium
- NBuPy
N-butylpyridinium chloride
- OC
organic cation
- OCT
organic cation transporter
- RPT
renal proximal tubule
- S.A.
specific activity
- SLC
solute carrier
- TEMA
triethylmethylammonium
Authorship Contributions
Participated in research design: Martínez-Guerrero, Wright.
Conducted experiments: Martínez-Guerrero.
Performed data analysis: Martínez-Guerrero.
Wrote or contributed to the writing of the manuscript: Martínez-Guerrero, Wright.
Footnotes
This work was supported in part by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant 1R01DK080801] and National Institutes of Health National Institute of Environmental Health Sciences [Grant 5P30ES006694]. The authors gratefully acknowledge the support for Lucy J. Martínez-Guerrero provided by the Fulbright International Educational Exchange Program.
Given the structural diversity of organic cations, it is useful to refer to the type I and type II classifications for different structural classes of organic cations developed to describe OC secretion in the liver (Meijer et al., 1990). Whereas type II OCs are generally bulky (typically >500 Da) and frequently polyvalent (e.g., d-tubocurarine, vercuronium), and hexafluorenium, type I OCs are generally small (typically <400 Da) monovalent compounds that include the prototypic substrates of renal organic cation transporters (i.e., the OCTs and MATEs), MPP and TEA. Importantly, most cationic drugs from a wide array of clinical classes, including antihistamines, skeletal muscle relaxants, antiarrhythmics, and β-adrenoceptor blocking agents, are adequately described as being type I OCs.
References
- Astorga B, Ekins S, Morales M, Wright SH. (2012) Molecular determinants of ligand selectivity for the human multidrug and toxin extruder proteins MATE1 and MATE2-K. J Pharmacol Exp Ther 341:743–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzer M, Morales M, Jagadish B, Mash MA, Wright SH. (2013) Substrate-dependent ligand inhibition of the human organic cation transporter OCT2. J Pharmacol Exp Ther 346:300–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang S, Sorani M, Giacomini KM. (2007) Transport of paraquat by human organic cation transporters and multidrug and toxic compound extrusion family. J Pharmacol Exp Ther 322:695–700 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Martinez-Guerrero LJ, Wright SH, Kuester RK, Hooth MJ, Sipes IG. (2011) Characterization of the inhibitory effects of N-butylpyridinium chloride and structurally related ionic liquids on organic cation transporters 1/2 and human toxic extrusion transporters 1/2-k in vitro and in vivo. Drug Metab Dispos 39:1755–1761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Wright SH, Hooth MJ, Sipes IG. (2009) Characterization of the disposition and toxicokinetics of N-butylpyridinium chloride in male F-344 rats and female B6C3F1 mice and its transport by organic cation transporter 2. Drug Metab Dispos 37:909–916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen HN. (1975) Biological Transport, W. A. Benjamin, Inc., London [Google Scholar]
- Dangprapai Y, Wright SH. (2011) Interaction of H+ with the extracellular and intracellular aspects of hMATE1. Am J Physiol Renal Physiol 301:F520–F528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David C, Rumrich G, Ullrich KJ. (1995) Luminal transport system for H+/organic cations in the rat proximal tubule. Kinetics, dependence on pH; specificity as compared with the contraluminal organic cation-transport system. Pflugers Arch 430:477–492 [DOI] [PubMed] [Google Scholar]
- Endres CJ, Hsiao P, Chung FS, Unadkat JD. (2006) The role of transporters in drug interactions. Eur J Pharm Sci 27:501–517 [DOI] [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorboulev V, Shatskaya N, Volk C, Koepsell H. (2005) Subtype-specific affinity for corticosterone of rat organic cation transporters rOCT1 and rOCT2 depends on three amino acids within the substrate binding region. Mol Pharmacol 67:1612–1619 [DOI] [PubMed] [Google Scholar]
- Hagenbuch B. (2010) Drug uptake systems in liver and kidney: a historic perspective. Clin Pharmacol Ther 87:39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JN, Wright SH. (2013) Multiple mechanisms of ligand interaction with the human organic cation transporter, OCT2. Am J Physiol Renal Physiol 304:F56–F67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonker JW, Schinkel AH. (2004) Pharmacological and physiological functions of the polyspecific organic cation transporters: OCT1, 2, and 3 (SLC22A1-3). J Pharmacol Exp Ther 308:2–9 [DOI] [PubMed] [Google Scholar]
- Karbach U, Kricke J, Meyer-Wentrup F, Gorboulev V, Volk C, Loffing-Cueni D, Kaissling B, Bachmann S, Koepsell H. (2000) Localization of organic cation transporters OCT1 and OCT2 in rat kidney. Am J Physiol Renal Physiol 279:F679–F687 [DOI] [PubMed] [Google Scholar]
- Kido Y, Matsson P, Giacomini KM. (2011) Profiling of a prescription drug library for potential renal drug-drug interactions mediated by the organic cation transporter 2. J Med Chem 54:4548–4558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaassen CD, Aleksunes LM. (2010) Xenobiotic, bile acid, and cholesterol transporters: function and regulation. Pharmacol Rev 62:1–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen GA, Cheng Y, Kuester RK, Hooth MJ, Sipes IG. (2009) Effects of dose and route on the disposition and kinetics of 1-butyl-1-methylpyrrolidinium chloride in male F-344 rats. Drug Metab Dispos 37:2171–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu T, Hiasa M, Miyaji T, Kanamoto T, Matsumoto T, Otsuka M, Moriyama Y, Omote H. (2011) Characterization of the human MATE2 proton-coupled polyspecific organic cation exporter. Int J Biochem Cell Biol 43:913–918 [DOI] [PubMed] [Google Scholar]
- Lickteig AJ, Cheng X, Augustine LM, Klaassen CD, Cherrington NJ. (2008) Tissue distribution, ontogeny and induction of the transporters Multidrug and toxin extrusion (MATE) 1 and MATE2 mRNA expression levels in mice. Life Sci 83:59–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malo C, Berteloot A. (1991) Analysis of kinetic data in transport studies: new insights from kinetic studies of Na(+)-D-glucose cotransport in human intestinal brush-border membrane vesicles using a fast sampling, rapid filtration apparatus. J Membr Biol 122:127–141 [DOI] [PubMed] [Google Scholar]
- Masuda S, Terada T, Yonezawa A, Tanihara Y, Kishimoto K, Katsura T, Ogawa O, Inui KI. (2006) Identification and functional characterization of a new human kidney-specific H+/organic cation antiporter, kidney-specific multidrug and toxin extrusion 2. J Am Soc Nephrol 17:2127–2135 [DOI] [PubMed] [Google Scholar]
- Meijer DKF, Mol WEM, Müller M, Kurz G. (1990) Carrier-mediated transport in the hepatic distribution and elimination of drugs, with special reference to the category of organic cations. J Pharmacokinet Biopharm 18:35–70 [DOI] [PubMed] [Google Scholar]
- Motohashi H, Sakurai Y, Saito H, Masuda S, Urakami Y, Goto M, Fukatsu A, Ogawa O, and Inui K (2002) Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J Am Soc Nephrol 13:866-874. [DOI] [PubMed]
- Ohta KY, Inoue K, Hayashi Y, Yuasa H. (2006) Molecular identification and functional characterization of rat multidrug and toxin extrusion type transporter 1 as an organic cation/H+ antiporter in the kidney. Drug Metab Dispos 34:1868–1874 [DOI] [PubMed] [Google Scholar]
- Otsuka M, Matsumoto T, Morimoto R, Arioka S, Omote H, Moriyama Y. (2005) A human transporter protein that mediates the final excretion step for toxic organic cations. Proc Natl Acad Sci USA 102:17923–17928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelis RM, Wright SH. (2011) Renal transport of organic anions and cations. Compr Physiol 1:1795–1835 [DOI] [PubMed] [Google Scholar]
- Ross CR, Holohan PD. (1983) Transport of organic anions and cations in isolated renal plasma membranes. Annu Rev Pharmacol Toxicol 23:65–85 [DOI] [PubMed] [Google Scholar]
- Schäli C, Schild L, Overney J, Roch-Ramel F. (1983) Secretion of tetraethylammonium by proximal tubules of rabbit kidneys. Am J Physiol 245:F238–F246 [DOI] [PubMed] [Google Scholar]
- Schomig E, Lazar A and Grundemann D (2006) Extraneuronal monoamine transporter and organic cation transporters 1 and 2: a review of transport efficiency. Handb Exp Pharmacol 175:151–180. [DOI] [PubMed]
- Segel IH. (1975) Enzyme Kinetics, pp 100–111, John Wiley & Sons, New York [Google Scholar]
- Sipes IG, Knudsen GA, Kuester RK. (2008) The effects of dose and route on the toxicokinetics and disposition of 1-butyl-3-methylimidazolium chloride in male F-344 rats and female B6C3F1 mice. Drug Metab Dispos 36:284–293 [DOI] [PubMed] [Google Scholar]
- Stein WD. (1986) Transport and Diffusion across Cell Membranes, Academic Press, New York [Google Scholar]
- Tanihara Y, Masuda S, Sato T, Katsura T, Ogawa O, Inui KI. (2007) Substrate specificity of MATE1 and MATE2-K, human multidrug and toxin extrusions/H(+)-organic cation antiporters. Biochem Pharmacol 74:359–371 [DOI] [PubMed] [Google Scholar]
- Terada T, Inui K. (2008) Physiological and pharmacokinetic roles of H+/organic cation antiporters (MATE/SLC47A). Biochem Pharmacol 75:1689–1696 [DOI] [PubMed] [Google Scholar]
- Tsuda M, Terada T, Asaka J, Ueba M, Katsura T, Inui K. (2007) Oppositely directed H+ gradient functions as a driving force of rat H+/organic cation antiporter MATE1. Am J Physiol Renal Physiol 292:F593–F598 [DOI] [PubMed] [Google Scholar]
- Vazquez-Laslop N, Zheleznova EE, Markham PN, Brennan RG, Neyfakh AA. (2000) Recognition of multiple drugs by a single protein: a trivial solution of an old paradox. Biochem Soc Trans 28:517–520 [PubMed] [Google Scholar]
- Wright SH, Wunz TM. (1999) Influence of substrate structure on substrate binding to the renal organic cation/H+ exchanger. Pflugers Arch 437:603–610 [DOI] [PubMed] [Google Scholar]
- Wright SH, Wunz TM, Wunz TP. (1995) Structure and interaction of inhibitors with the TEA/H+ exchanger of rabbit renal brush border membranes. Pflugers Arch 429:313–324 [DOI] [PubMed] [Google Scholar]
- Zolk O, Solbach TF, König J, Fromm MF. (2009) Structural determinants of inhibitor interaction with the human organic cation transporter OCT2 (SLC22A2). Naunyn Schmiedebergs Arch Pharmacol 379:337–348 [DOI] [PubMed] [Google Scholar]