

Abstract
Despite metronidazole’s widespread clinical use since the 1960s, the specific enzymes involved in its biotransformation have not been previously identified. Hence, in vitro studies were conducted to identify and characterize the cytochrome P450 enzymes involved in the formation of the major metabolite, 2-hydroxymetronidazole. Formation of 2-hydroxymetronidazole in human liver microsomes was consistent with biphasic, Michaelis-Menten kinetics. Although several cDNA-expressed P450 enzymes catalyzed 2-hydroxymetronidazole formation at a supratherapeutic concentration of metronidazole (2000 μM), at a “therapeutic concentration” of 100 μM only CYPs 2A6, 3A4, 3A5, and 3A7 catalyzed metronidazole 2-hydroxylation at rates substantially greater than control vector, and CYP2A6 catalyzed 2-hydroxymetronidazole formation at rates 6-fold higher than the next most active enzyme. Kinetic studies with these recombinant enzymes revealed that CYP2A6 has a Km = 289 μM which is comparable to the Km for the high-affinity (low-Km) enzyme in human liver microsomes, whereas the Km values for the CYP3A enzymes corresponded with the low-affinity (high-Km) component. The sample-to-sample variation in 2-hydroxymetronidazole formation correlated significantly with CYP2A6 activity (r ≥ 0.970, P < 0.001) at substrate concentrations of 100 and 300 μM. Selective chemical inhibitors of CYP2A6 inhibited metronidazole 2-hydroxylation in a concentration-dependent manner and inhibitory antibodies against CYP2A6 virtually eliminated metronidazole 2-hydroxylation (>99%). Chemical and antibody inhibitors of other P450 enzymes had little or no effect on metronidazole 2-hydroxylation. These results suggest that CYP2A6 is the primary catalyst responsible for the 2-hydroxylation of metronidazole, a reaction that may function as a marker of CYP2A6 activity both in vitro and in vivo.
Introduction
Introduced in 1963 for the treatment of patients with Trichomonas vaginalis, metronidazole is currently approved by the U.S. Food and Drug Administration for the treatment of a variety of anaerobic infections in adults. Although it is not approved for pediatric use, metronidazole is widely used “off-label” to treat serious bacterial and parasitic infections in children. Metronidazole is believed to exert its bactericidal activity through the formation of a toxic, redox intermediate metabolite (nitro reduction) within the bacterium (Edwards, 1980).
Metronidazole undergoes extensive hepatic biotransformation with subsequent renal elimination (Stambaugh et al., 1968; Jensen and Gugler, 1983). The major metabolites found in urine result from the hydroxylation of the 2-methyl group (2-hydroxymetronidazole), oxidation of the 1-ethyl group (1-metronidazole acetic acid), and glucuronide conjugation (on the 1-ethyl group; Fig. 1). A sulfate conjugate, a further oxidation product of 2-hydroxymetronidazole and a glucuronide conjugate of 2-hydroxymetronidazole have also been identified as minor metabolites (Stambaugh et al., 1968; Lamp et al., 1999). Although the parent compound predominates in plasma, along with smaller concentrations of 2-hydroxymetronidazole (Jensen and Gugler, 1983), in urine 2-hydroxymetronidazole is the primary metabolite formed from metronidazole, and accounts for 40–50% of the total metronidazole species present in 24-hour urine samples (Stambaugh et al., 1968). Both metronidazole and 2-hydroxymetronidazole have in vitro activity against most anaerobic bacterial strains (O'Keefe et al., 1982). Of note, the acetic acid metabolite is reported to occur at detectable levels only in patients with renal dysfunction (Lamp et al., 1999).
Fig. 1.

Metabolic scheme for the conversion of metronidazole to its major metabolites.
Despite metronidazole’s clinical use for five decades, the specific enzymes involved in its biotransformation in humans have not been well characterized. Only one previous study has attempted to characterize the human biotransformation of metronidazole in vitro. This study, which was conducted in human liver microsomes (HLMs), did not identify the enzyme(s) responsible for the formation of 2-hydroxymetronidazole at therapeutic substrate concentrations (Loft et al., 1991). Knowledge of the enzyme(s) involved in the biotransformation of metronidazole, and in particular the enzymes responsible for catalyzing the 2-hydroxylation of metronidazole, could be used to predict potential drug-drug and drug-xenobiotic interactions in both adults and children.
Although metronidazole is commonly prescribed to treat anaerobic pathogens in infants and children with complicated intra-abdominal infections, only recently have the pharmacokinetics of metronidazole been characterized in young infants (Suyagh et al., 2011; Cohen-Wolkowiez et al., 2013). The pharmacokinetic data suggest that a developmental component exists that influences metronidazole clearance and the exposure-response relationship. Given the predictable ontogenic profiles for many drug-metabolizing enzymes, identification of the enzymes responsible for the biotransformation of metronidazole is critical for dose optimization in infants and children. Therefore, the current study was undertaken to characterize the human cytochrome P450 enzymes responsible for catalyzing the 2-hydroxylation of metronidazole.
Materials and Methods
Chemicals.
Metronidazole, α-naphthoflavone, clopidogrel, diethyldithiocarbamic acid, (R)-(–)-deprenyl hydrochloride (selegiline), EDTA, (S)-fluoxetine hydrochloride, formic acid, furafylline, glucose-6-phospate, glucose-6-phosphate dehydrogenase, 4-methylpyrazole, 8-methoxypsoralen, mifepristone, β-NADP, quinidine, sulfaphenazole, ticlopidine, tranylcypromine, and troleandomycin (TAO) were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO). Metronidazole-d4, 2-hydroxymetronidazole, paroxetine hydrochloride, montelukast sodium salt, and (S)-(+)-N-3-benzylnirvanol were purchased from Toronto Research Chemicals (Toronto, Ontario, Canada). Ketoconazole was obtained from Research Biochemicals International (Natick, MA). All of the preceding reagents were of analytical grade. Methanol (Optima MS grade) was purchased from Thermo Fisher Scientific (Fair Lawn, NJ). All other reagents were of reagent grade or higher.
Commercial Reagents Biologic.
Adult human liver microsomes prepared from individual donors (1 male, 2 females), a reaction phenotyping kit (Version 7; containing human liver microsomes prepared from 16 individuals: 9 males, 7 females), pooled human liver microsomes (n = 16; mixed genders), inhibitory, mouse monoclonal antibodies to human CYPs 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, and 3A4 and control IgG (from rabbits) were obtained from XenoTech, LLC (Lenexa, KS). Microsomes from baculovirus-infected insect cells (Supersomes) expressing human P450 enzymes (CYPs 1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2J2, 3A4, 3A5, and 3A7) or control vector were purchased from BD Biosciences (Bedford, MA). All recombinant enzymes were coexpressed with human NADPH–cytochrome P450 reductase; some P450s (CYPs 2B6, 2C19, 2E1, 3A4, and 3A7) were also coexpressed with human cytochrome b5. The manufacturers provided protein concentrations, P450 contents, and P450 enzyme activities. Vials of microsomes were stored at –70°C until use. Microsomes were rapidly thawed in room temperature water and placed on ice prior to use.
High-Performance Liquid Chromatography/Mass Spectrometry Analysis of Metronidazole Biotransformation.
Metronidazole and 2-hydroxymetronidazole were resolved on a reversed-phase Agilent Technologies (Palo Alto, CA) Zorbax Eclipse XDB C-18 column (4.6-mm × 75-mm, 3.5-μm particle size) preceded by a Phenomenex C-18 guard column (4-mm × 3-mm i.d., 5-μm particle size) using a Hewlett Packard HP1100 high-performance liquid chromatography (HPLC) system equipped with a HP1100 degasser, binary pump, autosampler, column heater, diode array detector, and mass spectral detector (Hewlett Packard Instruments, Santa Clara, CA). The mobile phase consisted of 0.1% aqueous formic acid (solvent A) and methanol containing 0.1% formic acid (solvent B); analytes were eluted isocratically with an 80:20 mixture of solvent A/solvent B that was delivered at a constant flow of 0.6 ml/minute. The column temperature was maintained at 40°C. Under these conditions, 2-hydroxymetronidazole and metronidazole eluted at ∼2.39 and 2.99 minutes, respectively. The column effluent was monitored by UV detection (313 nm) and by atmospheric pressure electrospray ionization detection with the mass spectrometer operating in a selective positive ion-monitoring mode. Ion detection was optimized for detection of 2-hydroxymetronidazole. The drying gas temperature and flow were maintained at 300°C and 10 l/min, respectively, and the nebulizer pressure was set at 20 pounds per square inch [gauge] (psig). The capillary voltage was set at 3 kV. Under these conditions, 2-hydroxymetronidazole yielded protonated molecular ions (MH+ ions) at a mass charge ratio (m/z) of 188.2; metronidazole and d4-metronidazole (internal standard) were monitored as MH+ ions at m/z 172.2 and 176.2, respectively. Because UV data were used to verify mass spectrometry (MS) data whenever possible, d4-2-hydroxymetronidazole was not used as an internal standard in these experiments. Data were collected and integrated with Hewlett Packard Chemstation software (version B.03.01). 2-Hydroxymetronidazole was quantified by comparison of peak areas with those of analytical standards. The lower limit of quantification for 2-hydroxymetronidazole was 0.03 μM. The analytical method was linear (r2 > 0.999) over a standard concentration range of 0.03–100 μM. Standard curves run on five separate occasions produced intraday CVs that ranged from 0.1 to 9.7%, whereas interday CVs ranged from 1.4 to 12.5%.
In Vitro Incubation Conditions.
In vitro enzyme assays were performed in 96-well microtiter plates. Standard incubation reactions (100 μl) contained human liver microsomes (50 μg of microsomal protein) or insect cell microsomes containing baculovirus-expressed cytochrome P450 enzymes (5 pmol) coexpressed with P450 reductase, potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM), and metronidazole (16 μM–30 mM) dissolved in water at the final concentrations listed. Reactions were initiated by the addition of an NADPH-generating system, consisting of NADP (1 mM), glucose-6-phosphate (5 mM), and glucose-6-phosphate dehydrogenase (1 IU/ml), incubated at 37 ± 0.1°C in a Thermo Forma (Marietta, OH) Benchtop Orbital Shaker, and terminated after 60 minutes by the addition of 100 μl of ice-cold methanol containing the internal standard, metronidazole-d4 (2.5 μM final concentration). Protein was precipitated by centrifugation at 10,000 g for 10 minutes. An aliquot (5–20 μl) of the supernatant was analyzed by HPLC/MS via direct injection.
Preliminary experiments conducted with metronidazole (50, 500, and 5000 μM) and pooled human liver microsomes (0.1, 0.25, and 0.5 mg protein/ml) suggested that the rates of formation for 2-hydroxymetronidazole were proportional to protein concentration (up to 0.5 mg/ml) and incubation time (up to 60 minutes). Biotransformation of metronidazole did not exceed 10% in incubations in vitro and incubations conducted without the NADPH-generating system failed to convert metronidazole to 2-hydroxymetronidazole at detectable levels. Experiments designed to determine kinetic parameters and inhibition experiments with pooled human liver microsomes were performed in duplicate, and all other experiments were performed in triplicate.
Chemical Inhibition Experiments.
Conversion of metronidazole (100 or 300 μM) to 2-hydroxymetronidazole by human liver microsomes was evaluated in the presence or absence (i.e., control) of known P450 isoform-selective inhibitors, using the incubation conditions described previously. To demonstrate concentration dependence with competitive inhibitors, a range of inhibitor concentrations spanning 30-fold or more was used. The lowest inhibitor concentrations chosen were determined using the following equation:
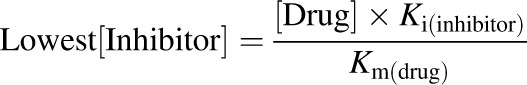 |
(1) |
where [Drug] is the intended final concentration of metronidazole added to the microsomal incubations, Ki is the inhibition constant of the chemical inhibitor for a given enzyme, and Km is the Michaelis constant derived for metronidazole from experiments with pooled human liver microsomes. The Ki values used to calculate the lowest inhibitor concentrations used in this study were based on values reported in the literature (Khojasteh et al., 2011). In theory, this concentration of inhibitor should inhibit ∼50% of the selectively inhibited enzyme’s product formation when [Drug] used in the incubation equals Km. At the other end of the concentration range, the highest concentration of inhibitor used in these studies should virtually abolish product formation (by the inhibited enzyme) under these constraints.
The following competitive chemical inhibitors were examined at the indicated final concentrations: α-naphthoflavone (CYP1A2, 0.15–5 μM), tranylcypromine (CYP2A6, 0.2–6 μM), montelukast (CYP2C8, 0.15–5 μM), sulfaphenazole (CYP2C9, 0.3–10 μM), (S)-(+)-N-3-benzylnirvanol (CYP2C19, 0.3–10 μM), quinidine (CYP2D6, 0.15–5 μM), 3-methylpyrazole (CYP2E1, 2–60 μM), and ketoconazole (CYP3A4/5, 0.03–1 μM). Inhibitors were dissolved in methanol and diluted in the incubation mixtures to a final solvent concentration ≤0.5% (v/v). Control incubations contained an equal volume of methanol.
Incubations containing the mechanism-based inhibitors (at the indicated final concentrations) furafylline (CYP1A2, 1 and 10 μM), selegiline (CYP2A6, 30–1000 μM), 8-methoxypsoralen (CYP2A6, 10 μM), diethyldithiocarbamate (CYP2A6, 50 μM), ticlopidine (CYP2B6, 1 and 3 μM), clopidogrel (CYP2B6, 1 and 3 μM), fluoxetine (CYP2C19, 30 μM), paroxetine (CYP2D6, 1 and 10 μM), troleandomycin (CYP3A4, 100 μM), or mifepristone (CYP3A4, 2 and 100 μM) were preincubated with human liver microsomes and NADPH-generating system for 20 minutes before the reaction was started with substrate. Concentrations of mechanism-based inhibitors were based on literature values (Siu and Tyndale, 2008; Khojasteh et al., 2011) and were dissolved in methanol; inhibitors were appropriately diluted such that the final solvent concentration in the incubation mixtures was ≤0.5% (v/v). Control incubations contained an equal volume of methanol.
Antibody Inhibition Experiments.
Pooled human liver microsomes (50 μg of microsomal protein) were preincubated at room temperature with ascites fluid from mice containing inhibitory monoclonal antibodies against human P450 enzymes or with control IgG, following the manufacturer’s instructions for maximal inhibition of the indicated enzyme’s activity. Following a 15-minute preincubation period, potassium phosphate buffer (50 mM, pH 7.4), MgCl2 (3 mM), EDTA (1 mM), and metronidazole (100 μM) were added to the incubation mixtures. Reactions were initiated by the addition of an aliquot of NADPH-generating system and conducted as described previously.
Data Analysis.
Correlation coefficients (r) between the rates of 2-hydroxymetronidazole formation and the activities of cytochrome P450 enzymes were determined using least-squares regression analysis. Significance was assessed by Pearson’s regression analysis using a two-tailed Student’s t test (Microsoft Excel 2007) with a significance limit set at α = 0.05. Visual inspection of Eadie-Hofstee plots (rate versus rate/[S]) derived from kinetic data with human liver microsomes or with recombinant human P450 enzymes determined whether data were subsequently fit to a one- or two-enzyme Michaelis-Menten model. Kinetic data were subsequently analyzed by nonlinear regression without weighting (GraFit 5; Erithacus Software Ltd, Surrey, UK). For the recombinant enzymes, CYP3A4 and CYP3A5, kinetic data were best fit to a sigmoidal Vmax equation equivalent to the Hill equation:
| (2) |
where S50 is the substrate concentration showing a half-maximal velocity, n is a measure of cooperativity, and Vmax is the maximal velocity.
Results
Biotransformation of Metronidazole by cDNA-Expressed Human P450s.
As an initial screen to determine which P450 enzymes were capable of converting metronidazole to 2-hydroxymetronidazole, a supratherapeutic concentration of metronidazole (2000 μM) was incubated with control vector or cDNA-expressed CYP1A1, 1A2, 1B1, 2A6, 2B6, 2C8, 2C9, 2C18, 2C19, 2D6, 2E1, 2J2, 3A4, 3A5, and 3A7. With the exception of CYPs 1A1, 1B1, 2C18, and 2J2, each of the recombinant enzymes converted metronidazole to 2-hydroxymetronidazole (Fig. 2, upper panel). However, CYP2A6, CYP2E1, and the CYP3A enzymes catalyzed 2-hydroxymetronidazole formation at rates substantially higher than the other recombinant enzymes tested.
Fig. 2.

2-Hydroxymetronidazole formation by human cDNA-expressed cytochrome P450 enzymes. Metronidazole (2 mM, upper panel; 100 μM, lower panel) was incubated with heterologously expressed human P450 enzymes as described under Materials and Methods. Each bar represents the mean ± S.D. of three determinations.
To identify potential candidates for subsequent kinetic analyses, recombinant enzymes were incubated with a therapeutically relevant concentration of metronidazole (100 μM). This concentration was chosen based on a study that examined the pharmacokinetics of different oral dosages of metronidazole in adults (Amon et al., 1978) and corroborated by a recent study which examined its pharmacokinetics in premature neonates (Cohen-Wolkowiez et al., 2012). Following administration of 250, 1000, and 2000 mg in the adult study (Amon et al., 1978), peak plasma concentrations of metronidazole were reported to average 5.1 ± 1.7, 19.6 ± 3.8, and 40.6 ± 9.3 μg/ml (equivalent to: 30 ± 10, 115 ± 22, and 237 ± 54 µM), respectively. At a substrate concentration of 100 μM, only CYPs 2A6, 3A4, 3A5, and 3A7 catalyzed 2-hydroxymetronidazole formation at rates substantially greater than the control vector (Fig. 2, lower panel), with CYP2A6 forming 2-hydroxymetronidazole at rates more than 6-fold greater than the next most active enzyme (CYP3A5). Although CYPs 1A1, 2B6, 2C18, and 2C19 also catalyzed formation of 2-hydroxymetronidazole at this substrate concentration, the rates at which they contributed to 2-hydroxymetronidazole formation were extremely low.
Determination of Kinetic Parameters.
The kinetics of 2-hydroxymetronidazole formation were investigated in pooled adult human liver microsomes, in three individual preparations of adult human liver microsomes [one each with high (H201), moderate (H382), and minimal (H469) CYP2A6 activity], and in microsomes containing recombinant CYP2A6, CYP3A4, CYP3A5, and CYP3A7. Visual inspection of Eadie-Hofstee plots (Fig. 3) derived from kinetic data with three of the human liver microsomal preparations (pool, high, and moderate CYP2A6 activity samples) suggested that the kinetics of 2-hydroxymetronidazole formation might not be consistent with a simple Michaelis-Menten model. For these samples, the relationship between the rate of 2-hydroxymetronidazole formation and substrate concentration was best described by a two-site Michaelis-Menten equation. Kinetic data from the fourth human liver microsomal sample (minimal CYP2A6 activity) could be fit to a single-enzyme Michaelis-Menten model with the kinetic parameters corresponding to the high Km site for the other three preparations. The affinity constant (Km) for the low Km site varied only 1.4-fold and there was a 4.3-fold variation in Vmax (Table 1). The Km values for the high Km site were in the millimolar range and varied 13-fold, whereas the corresponding Vmax values varied 2.0-fold.
Fig. 3.

Effect of substrate concentration on the rate of 2-hydroxymetronidazole formation by human liver microsomes (Eadie-Hofstee plots). Metronidazole (16 μM–30 mM) was incubated with pooled human liver microsomes (0.05 mg microsomal protein) in 100-μl reaction mixtures at 37 ± 0.1°C, and terminated with 100 μl of methanol containing metronidazole-d4 (internal standard) after 60 minutes. Following precipitation of microsomal protein, an aliquot (10 μl) of the supernatant was analyzed by HPLC/MS via direct injection, respectively, as described under Materials and Methods.
TABLE 1.
Apparent kinetic parameters for the formation of 2-hydroxymetronidazole in human liver microsomes
| Low Km Site |
High Km Site |
|||||
|---|---|---|---|---|---|---|
| Sample | Kma | Vmax1a | Vmax1/Km1 | Km2a | Vmax2a | Vmax2/Km2 |
| mM | nmol/mg protein per minute | ml/mg protein per minute | mM | nmol/mg protein per minute | ml/mg protein per minute | |
| H201 | 0.366 ± 0.029 | 0.911 ± 0.044 | 2.48 | 5.7 ± 4.0 | 1.30 ± 0.20 | 0.22 |
| H382 | 0.527 ± 0.056 | 0.235 ± 0.017 | 0.45 | 23.6 ± 3.4 | 1.41 ± 0.10 | 0.06 |
| H469 | — | — | — | 75.1 ± 10.4 | 1.44 ± 0.12 | 0.02 |
| Adult Pool | 0.383 ± 0.040 | 0.213 ± 0.012 | 0.56 | 33.1 ± 6.5 | 0.72 ± 0.10 | 0.02 |
Value ± S.E. of parameter fit.
In general, the formation of 2-hydroxymetronidazole by cDNA-expressed human CYP2A6 and CYP3A7 conformed to a simple, monophasic Michaelis-Menten model. Although there was a slight indication of substrate activation of CYP3A7 at the lowest concentrations of metronidazole studied (as suggested by the hooked portion of its Eadie-Hofstee plot, not shown), the majority of the data points obtained with recombinant CYP3A7 were consistent with Michaelis-Menten kinetics for a single enzyme. The kinetics of 2-hydroxymetronidazole formation by both CYP3A4 and CYP3A5 displayed a sigmoidal appearance (indicative of substrate activation) and data were fit to a derivative of the Hill equation. The apparent Km for CYP2A6 corresponded to microsomal Km values for the low Km site, whereas the Km values obtained for CYP3A4, 3A5, and 3A7 were in the millimolar range and more closely resembled the corresponding Km values for the high Km site (Table 2).
TABLE 2.
Apparent kinetic parameters for the formation of 2-hydroxymetronidazole by cDNA-expressed human P450 enzymes
| 2-Hydroxymetronidazole Formation |
||||
|---|---|---|---|---|
| Sample | Kma or S50a | Vmaxa | Na,b | Clintc |
| mM | nmol/nmol P450 per minute | ml/nmol P450 per minute | ||
| CYP2A6 | 0.289 ± 0.061 | 0.65 ± 0.02 | — | 2.24 |
| CYP3A4 | 5.38 ± 0.42 | 2.49 ± 0.11 | 1.65 ± 0.12 | 0.46 |
| CYP3A5 | 4.45 ± 0.24 | 3.46 ± 0.10 | 1.88 ± 0.11 | 0.77 |
| CYP3A7 | 8.96 ± 1.03 | 1.27 ± 0.06 | — | 0.14 |
Value ± S.E. of parameter fit; bcooperativity coefficient; cintrinsic clearance defined as Vmax/Km or Vmax/S50.
Chemical Inhibition Experiments.
Based on the preceding results, selective chemical inhibitors of P450 enzymes were incubated with metronidazole (100 μM) and pooled human liver microsomes to assess the contribution of CYP2A6 to the formation of 2-hydroxymetronidazole. The effects of various concentrations of selective, competitive P450 inhibitors on the conversion of metronidazole to 2-hydroxymetronidazole are illustrated in Fig. 4 (upper panel). The CYP2A6 inhibitor, tranylcypromine, inhibited the 2-hydroxylation of metronidazole in a concentration-dependent manner, virtually eliminating product formation (>96%) at the highest concentration tested (6 μM). All of the other competitive P450 inhibitors caused little or no inhibition of 2-hydroxymetronidazole formation.
Fig. 4.

Effects of various P450 isoform-selective inhibitors on the formation of 2-hydroxymetronidazole by pooled human liver microsomes. Pooled human liver microsomes were incubated with metronidazole (100 μM) in the presence or absence of various selective, chemical inhibitors, with the major P450 inhibited enclosed in parentheses. Incubations were conducted as described under Materials and Methods. The upper panel shows the effects of selective, competitive P450 inhibitors, whereas the lower panel shows the effects of mechanism-based inhibitors on formation of 2-hydroxymetronidazole. Incubations containing mechanism-based inhibitors were preincubated with human liver microsomes and NADPH-generating system for 20 minutes prior to initiating the reaction with substrate. Each bar represents the mean of duplicate determinations.
The effects of various mechanism-based chemical inhibitors on the formation of 2-hydrozymetronidazole by pooled human liver microsomes were examined at concentrations determined to inhibit >80% of a given P450 isoform’s activity. Diethyldithiocarbamate (DEDC), 8-methoxypsoralen, and selegiline (deprenyl) were the only mechanism-based inhibitors observed to inhibit the conversion of metronidazole to 2-hydroxymetronidazole (Fig. 4, lower panel). 8-Methoxypsoralen and selegiline are CYP2A6 inhibitors, whereas DEDC is capable of inhibiting both CYP2A6 and CYP2E1. However, at the concentration used in this study, DEDC is primarily a CYP2A6 inhibitor (Khojasteh et al., 2011). DEDC inhibited ∼80% of the formation of 2-hydroxymetronidazole, whereas the highest concentrations of 8-methoxypsoralen and selegiline tested inhibited 2-hydroxymetronidazole formation by >95%.
Antibody Inhibition Experiments.
To further assess the contribution of CYP2A6 to metronidazole hydroxylation, the effects of various inhibitory monoclonal antibodies against human P450 enzymes on the conversion of metronidazole (100 μM) to 2-hydroxymetronidazole by pooled human liver microsomes were examined and the results are illustrated in Fig. 5. Under conditions sufficient to cause maximal inhibition of each P450 enzyme’s activity, only anti-CYP2A6 markedly inhibited the formation of 2-hydroxymetronidazole, virtually eliminating formation of the metabolite (>99%).
Fig. 5.

Effects of various inhibitory, monoclonal antibodies against human P450 enzymes on the conversion of metronidazole (100 μM) to 2-hydroxymetronidazole by pooled human liver microsomes. Pooled human liver microsomes (0.05 mg microsomal protein) were preincubated for 15 minutes at room temperature with ascites fluid from mice containing inhibitory monoclonal antibodies against human P450 enzymes or with control IgG followed by incubation with metronidazole (100 μM), as described under Materials and Methods. Volumes of antibodies used followed the manufacturer’s recommendations for maximal P450 inhibition. Each bar represents the mean of duplicate determinations.
Intersubject Variability and Correlation Experiments.
A panel of commercially available human liver microsomes prepared from 16 donors and characterized for P450 isoform activities using marker substrate reactions were examined for their ability to convert metronidazole to 2-hydroxymetronidazole at two substrate concentrations (100 and 300 μM). Based on the results of the preceding inhibition studies, liver microsomes from individuals with high, moderate, and minimal CYP2A6 activity were also examined for their ability to catalyze the 2-hydroxylation of metronidazole at these substrate concentrations. All of the microsomal samples catalyzed the formation of 2-hydroxymetronidazole from metronidazole, with the exception that the microsomal sample with minimal CYP2A6 activity failed to convert 100-μM metronidazole to 2-hydroxymetronidazole. At a substrate concentration of 100 μM, the rate of 2-hydroxymetronidazole formation varied ∼15-fold among samples catalyzing the reaction [range (rates ± S.D.): 20.6 ± 0.4 to 303 ± 20 pmol/mg protein per minute], whereas at the higher substrate concentration, 2-hydroxymetronidazole formation varied ∼100-fold [range (rates ± S.D.): 4.07 ± 0.74 to 410 ± 20 pmol/mg protein per minute] (Fig. 6; 300 μM results were similar to those obtained at 100 μM and thus are not shown). Although the microsomes with minimal CYP2A6 activity were unable to catalyze the formation of 2-hydroxymetronidazole at detectable levels at a substrate concentration of 100 μM, these microsomes were capable of forming small amounts of 2-hydroxymetronidazole at 300 μM metronidazole, which markedly increased the variation observed. If the data for this particular sample are excluded from the analysis, then only a 12-fold variation is observed among the rates of 2-hydroxymetronidazole formation in the microsomal samples at a substrate concentration of 300 μM.
Fig. 6.

Effect of tranylcypromine on the 2-hydroxylation of metronidazole by a panel of human liver microsomes. Human liver microsomes (0.05 mg microsomal protein) were incubated with metronidazole (100 μM) in 100-μl reaction mixtures at 37 ± 0.1°C in the presence or absence of tranylcypromine (6 μM), terminated after 60 minutes with 100 μl of methanol containing metronidazole-d4, and analyzed by HPLC/MS via direct injection, as described under Materials and Methods. Bars representing rates of 2-hydroxymetronidazole formation (inhibited and uninhibited by tranylcypromine) are the mean ± S.D. of three determinations.
The sample-to-sample variation in the rates of 2-hydroxymetronidazole formation correlated significantly with CYP2A6 activity (r ≥ 0.970, P < 0.00001) and with CYP2B6 activity (r ≥ 0.618, P < 0.01) at both substrate concentrations examined in the human liver microsomes from the reaction phenotyping kit (n = 16). Correlations between activities selective for other P450 isoforms and rates of 2-hydroxymetronidazole formation were not statistically significant (Table 3). It should be noted, however, that CYP2A6 activity was significantly correlated with CYP2B6 activity (r ≥ 0.58, P < 0.02) in the panel of microsomes used in these studies, which suggests that the relationship between 2-hydroxymetronidazole formation and CYP2B6 activity may largely be coincidental, particularly since recombinant CYP2B6 failed to catalyze formation of 2-hydroxymetronidazole at a substrate concentration of 100 μM. The observed correlation between CYP2A6 and CYP2B6 activities is not surprising since both enzymes are known to be transcriptionally coregulated by hepatic nuclear factor 4α (HNF-4α), estrogen receptor α (ERα), constitutive androstane receptor (CAR), and the pregnane X receptor (PXR) (Goodwin et al., 2001; Itoh et al., 2006; Higashi et al., 2007; Wortham et al., 2007; Lo et al., 2010).
TABLE 3.
Correlation analysis (r) of the relationship between the rates of metronidazole conversion to 2-hydroxymetronidazole with the sample-to-sample variation in cytochrome P450 activity in human liver microsomes
Values in bold indicate significant correlations between P450 activity and 2-hydroxymetronidazole formation at P < 0.05. Coumarin 7-hydroxlation was significantly correlated (r, P value) with S-mephenytoin N-hydroxylation (0.703, 0.001) and bupropion hydroxylation (0.576, 0.018).
| 2-Hydroxymetronidazole Formation |
||||
|---|---|---|---|---|
| Enzymatic Reaction (Enzyme) | 100-μM Metronidazole a |
300-μM Metronidazole a |
||
| r | P | r | P | |
| 7-Ethoxyresorufin O-deethylation (CYP1A2) | 0.032 | 0.906 | 0.068 | 0.801 |
| Phenacetin O-deethylation (CYP1A2) | 0.107 | 0.692 | 0.128 | 0.635 |
| Coumarin 7-hydroxylation (CYP2A6) | 0.970 | 9.19 × 10−11 | 0.972 | 5.17 × 10−11 |
| S-Mephenytoin N-hydroxylation (CYP2B6) | 0.742 | 0.001 | 0.703 | 0.002 |
| Bupropion hydroxylation (CYP2B6) | 0.648 | 0.006 | 0.618 | 0.010 |
| Paclitaxel 6α-hydroxylation (CYP2C8) | 0.421 | 0.102 | 0.395 | 0.127 |
| Diclofenac 4′-hydroxylation (CYP2C9) | 0.181 | 0.502 | 0.165 | 0.540 |
| S-Mephenytoin 4′-hydroxylation (CYP2C19) | 0.419 | 0.010 | 0.375 | 0.149 |
| Dextromethorphan O-demethylation (CYP2D6) | −0.072 | 0.791 | −0.112 | 0.679 |
| Chlorzoxazone 6-hydroxylation (CYP2E1) | −0.106 | 0.695 | −0.102 | 0.706 |
| Testosterone 6β-hydroxylation (CYP3A4/5) | 0.282 | 0.288 | 0.221 | 0.410 |
| Midazolam 1′-hydroxylation (CYP3A4/5) | 0.298 | 0.260 | 0.245 | 0.359 |
| Lauric acid 12-hydroxylation (CYP4A9/11) | −0.218 | 0.415 | −0.214 | 0.424 |
r, Correlation coefficient.
Concentration of metronidazole present in microsomal incubations.
To further characterize the apparent role of CYP2A6-mediated metronidazole 2-hydroxylation, the panel of human liver microsomes plus the three individual adult microsomal preparations (with high, moderate, and low CYP2A6 activity) were incubated with metronidazole (100 or 300 μM) with or without the CYP2A6 inhibitor, tranylcypromine (6 μM). Tranylcypromine eliminated the formation of 2-hydroxymetronidazole from 100-μM metronidazole in all but the four most active samples, and inhibited the rate of 2-hydroxymetronidazole formation >95% in these micrsosomal samples, as shown in Fig. 6. At a substrate concentration of 300 μM, tranylcypromine inhibited the formation of 2-hydroxymetronidazole by more than 95% in all but three microsomal samples, and in these samples the inhibitor was at least 90% effective in reducing the rate of 2-hydroxymetronidazole formation (results not shown, but the catalytic profile was similar to that shown in Fig. 6 for the reactions conducted at 100-μM metronidazole).
Discussion
Data characterizing the enzymes responsible for metronidazole biotransformation are exceedingly scarce as only one previous in vitro study (Loft et al., 1991) has addressed this issue. The results of this study showed that 2-hydroxymetronidazole formation conformed to biphasic Michaelis-Menten kinetics in human liver microsomes and that the low Km component had an apparent Km ranging from 140 to 320 μM. Chemical inhibition studies conducted by these authors ruled out certain P450 enzymes as the low Km catalyst (namely CYPs 1A2, 2C9, 2C19, 2D6, 2E1, and 3A4), but did not identify the enzyme. Thus, prior to the conduct of the present study, the enzyme(s) responsible for metronidazole biotransformation were unknown.
The present study was undertaken to fill the “information gap” regarding the identity and characterization of the P450 enzyme(s) responsible for metronidazole 2-hydroxylation at substrate concentrations approximating those observed in association with therapeutic doses of the drug. Our results demonstrate that the identity of the low Km enzyme responsible for catalyzing metronidazole 2-hydroxylation is CYP2A6. This assertion is well supported by several lines of evidence: 1) a striking correlation (P < 1 × 10−10) between the rates of 2-hydroxymetronidazole formation and 7-hydroxycoumarin formation in a panel of human liver microsomes, 2) virtual elimination of metronidazole 2-hydroxylation by inhibitory antibodies and selective chemical inhibitors against CYP2A6 but not against other P450 enzymes, 3) an observed Km for recombinant CYP2A6 (290 µM) that is similar to the apparent Km for the high affinity (low Km) enzyme in human liver microsomes (350–500 μM in the present study), and 4) a high rate of 2-hydroxymetronidazole formation was catalyzed by recombinant CYP2A6 at a “therapeutic” concentration of 100 μM, a rate that was ∼6-fold greater than the next most active recombinant enzyme, CYP3A5. Although a number of recombinant P450 enzymes displayed the capacity to catalyze 2-hydroxymetronidazole formation at supratherapeutic concentrations, only CYP2A6 (low Km) and the three CYP3A enzymes (3A4, 3A5, and 3A7, high-Km, low-mM range) catalyzed substantial rates of metabolite formation at metronidazole concentrations approximating those associated with therapeutic drug administrations. Collectively, these data clearly demonstrate the predominant catalytic role of CYP2A6 in the metabolism of metronidazole in humans at “therapeutic” plasma concentrations.
Large interindividual variability exists in the enzymatic activity and protein levels of CYP2A6 in human livers and may vary by as much as 100-fold (Shimada et al., 1994). Part of this variability stems from genetic factors. CYP2A6 is a highly polymorphic enzyme with over 37 variant alleles identified to date (http://www.cypalleles.ki.se/), many of which result in altered activity levels (McDonagh et al., 2012). In a recent study conducted in 139 mono- and dizygotic twins, ∼50% of CYP2A6 activity was attributable to additive genetic influences after taking into account CYP2A6 genotype (Swan et al., 2009). Accordingly, projection of CYP2A6 phenotype from CYP2A6 genotype alone may be difficult and in vivo phenotyping studies will likely be required to obtain an accurate estimate of CYP2A6 activity.
Although CYP2A6 participates in the biotransformation of a number of compounds, including a variety of pharmaceutical agents and several promutagens/procarcinogens (Pelkonen et al., 2000), CYP2A6 plays a major role in the metabolism of only a few chemicals and catalyzes very few reactions specifically. The 7-hydroxylation of coumarin (Miles et al., 1990; Yamano et al., 1990), the 3′-hydroxylation of cotinine (Nakajima et al., 1996) and the 7-hydroxylation of efavirenz (Ogburn et al., 2010) are currently the only reactions recognized as being specifically catalyzed by CYP2A6 at therapeutic concentrations of substrate. The 2-hydroxylation of midazolam can now be added to this list.
In vivo probes for CYP2A6 activity in adults have been limited in the past to coumarin, nicotine, and, to a lesser extent, caffeine (McDonagh et al., 2012). Neither coumarin nor caffeine is an optimal probe. Coumarin is not an optimal probe for pharmacokinetic reasons; it has a short half-life, an extensive and rapid first-pass metabolism and is present in urine and plasma in low concentrations (Pelkonen, 2002). Caffeine’s utility as a probe for CYP2A6 activity requires further scrutiny since CYP1A2 has been shown to contribute significantly to the formation of the metabolite previously attributable to CYP2A6 (1,7-dimethyl uric acid) at therapeutic or dietary concentrations of caffeine (Kimura et al., 2005).
In contrast, nicotine has few issues when determining CYP2A6 phenotype in adults. Nicotine can be administered orally or by transdermal patch, and single dosing is associated with minimal side effects (Benowitz et al., 2009). In addition, the metabolic ratio of 3′-hydroxycotinine to cotinine has been validated as an accurate measure of CYP2A6 activity in vivo (Nakajima et al., 1996), and both of these nicotine metabolites are readily measured in plasma or noninvasively in saliva and urine (Jacob et al., 2011).
To our knowledge, neither coumarin nor nicotine has been administered as an in vivo probe for CYP2A6 activity in children consequent to ethical considerations regarding nontherapeutic drug use in pediatrics (de Wildt et al., 2009). However, suitable medications that are given as a part of routine care or that can be obtained over-the-counter (e.g., dextromethorphan, caffeine) have been used effectively as in vivo phenotyping probes in children.
The evidence presented here suggests that metronidazole 2-hydroxylation certainly can be used to characterize the activity of CYP2A6 in vitro, particularly at substrate concentrations ≤100 μM, and potentially in vivo. The majority of metronidazole and 2-hydroxymetronidazole are excreted via the urine, with 7–12% of the metronidazole dose eliminated unchanged, and 33–44% excreted as 2-hydroxymetronidazole within a 24-hour period (Nilsson-Ehle et al., 1981; Houghton et al., 1982). Hence, it is possible that urine sample collection for phenotyping may be performed noninvasively. The metabolic ratio of 2-hydroxymetronidazole to metronidazole (either in urine or plasma) may serve as a useful tool to support CYP2A6 phenotyping in both adults and children given that the drug is commonly used to treat a variety of infections in infants and children. Its potential use as a probe in children is particularly notable because there are few alternatives to assess CYP2A6 activity in children. In addition, metronidazole as a single agent or a constituent of a multiagent cocktail, could potentially be used as a probe to assess CYP2A6 induction or inhibition in vivo and in vitro. To date, there is only one report of a phenotyping cocktail containing a suitable phenotyping probe for CYP2A6 (nicotine) (Petsalo et al., 2008).
The results of the present study also imply that CYP2A6 genotype could potentially impact the pharmacokinetics and pharmacodynamics of metronidazole. However, the predictive value of specific allelic variants could be limited given that both the parent drug and 2-hydroxymetronidazole are active, have different elimination half-lives, and variable sensitivity profiles with different pathogens (O’Keefe et al., 1982; Lamp et al., 1999). Clearly, further studies will be required to determine the impact of CYP2A6 genotype on metronidazole disposition and effect.
Although progress has been made in recent years to characterize the ontogenic profiles of many drug metabolizing enzymes, including the cytochromes P450, (Kearns et al., 2003; Hines, 2007), little is known regarding the developmental acquisition of CYP2A6 activity. CYP2A6 has been detected by Western blot in both fetal liver and olfactory mucosa; however, protein levels were not quantified (Gu et al., 2000). In another report, where CYP2A6 content was measured immunochemically in liver samples from 4 neonates, 6 infants, and 10 pediatric donors, it was found that CYP2A6 was expressed in all livers, but that there was no difference in the protein expression pattern between the pediatric-aged donors and the combined group of neonates and infants (Tateishi et al., 1997).
In that 2-hydroxymetronidazole is the predominant metabolite formed from metronidazole, it follows that CYP2A6 activity could serve as a major determinant of metronidazole plasma clearance. Consequently, characterizing the pharmacokinetics of the drug in neonates, infants, and children could provide a phenotypic depiction of the impact of ontogeny on CYP2A6 activity. This assertion is supported by limited pharmacokinetic data in the literature.
An early study that compared metronidazole pharmacokinetics between children and adults failed to yield appreciable age-related differences (Amon et al., 1983). In contrast, pharmacokinetic parameters in young infants appear to be markedly different from those in adults. For example, the apparent elimination half-life of metronidazole is increased 2- to 3-fold in young infants and decreases with increasing postnatal age (Jager-Roman et al., 1982; Hall et al., 1983; Upadhyaya et al., 1988). Another previous report demonstrated that in seven infants less than 8 weeks of age, the mean metronidazole elimination half-life was 18.4 hours compared with a corresponding value of 7 hours in four infants over 8 weeks of age (Rubenson and Rosetzsky, 1986). Finally two recent studies in preterm infants demonstrated significant increases in metronidazole clearance over the first weeks and months of life (Suyagh et al., 2011; Cohen-Wolkowiez et al., 2013). Despite these apparent developmental differences in metronidazole elimination, it is not yet known at what age the clearance or elimination half-life of metronidazole becomes comparable between children and adults.
In summary, the in vitro studies presented here suggest that CYP2A6 is the predominant catalyst of metronidazole 2-hydroxylation in humans at therapeutic concentrations. Furthermore, at a substrate concentration of 100 μM, this reaction appears to function as a biomarker of CYP2A6 activity, with ≥96% of 2-hydroxymetronidazole formation attributed to CYP2A6. The current in vitro studies coupled with recent pharmacokinetic data in preterm and term neonates (Suyagh et al., 2011; Cohen-Wolkowiez et al., 2013) suggest that metronidazole may be a sensitive probe for CYP2A6 activity that may be sufficient to characterize the impact of ontogeny on the activity of this enzyme.
Abbreviations
- DEDC
diethyldithiocarbamate
- HPLC
high-performance liquid chromatography
- MS
mass spectrometry
- P450
cytochrome P450
Authorship Contributions
Participated in research design: Pearce.
Conducted experiments: Pearce.
Performed data analysis: Pearce.
Wrote or contributed to the writing of the manuscript: Pearce, Cohen-Wolkowiez, Sampson, Kearns.
Footnotes
This work was supported by the National Institutes of Health National Institute of General Medical Sciences [Training Grant T32GM086330] (to M.R.S.).
References
- Amon I, Amon K, Hüller H. (1978) Pharmacokinetics and therapeutic efficacy of metronidazole at different dosages. Int J Clin Pharmacol Biopharm 16:384–386 [PubMed] [Google Scholar]
- Amon I, Amon K, Scharp H, Franke G, Nagel F. (1983) Disposition kinetics of metronidazole in children. Eur J Clin Pharmacol 24:113–119 [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Hukkanen J and Jacob P, 3rd (2009) Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol 192:29–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Wolkowiez M, Ouellet D, Smith PB, James LP, Ross A, Sullivan JE, Walsh MC, Zadell A, Newman N, White NR, et al. (2012) Population pharmacokinetics of metronidazole evaluated using scavenged samples from preterm infants. Antimicrob Agents Chemother 56:1828–1837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen-Wolkowiez M, Sampson M, Bloom BT, Arrieta A, Wynn JL, Martz K, Harper B, Kearns GL, Capparelli EV, Siegel D, Benjamin DK, Jr. and Smith PB (2013) Determining population and developmental pharmacokinetics of metronidazole using plasma and dried blood spot samples from premature infants. Pediatr Infect Dis J [EPub ahead of print]. [DOI] [PMC free article] [PubMed]
- de Wildt SN, Ito S, Koren G. (2009) Challenges for drug studies in children: CYP3A phenotyping as example. Drug Discov Today 14:6–15 [DOI] [PubMed] [Google Scholar]
- Edwards DI. (1980) Mechanisms of selective toxicity of metronidazole and other nitroimidazole drugs. Br J Vener Dis 56:285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin B, Moore LB, Stoltz CM, McKee DD, Kliewer SA. (2001) Regulation of the human CYP2B6 gene by the nuclear pregnane X receptor. Mol Pharmacol 60:427–431 [PubMed] [Google Scholar]
- Gu J, Su T, Chen Y, Zhang QY, Ding X. (2000) Expression of biotransformation enzymes in human fetal olfactory mucosa: potential roles in developmental toxicity. Toxicol Appl Pharmacol 165:158–162 [DOI] [PubMed] [Google Scholar]
- Hall P, Kaye CM, McIntosh N, Steele J. (1983) Intravenous metronidazole in the newborn. Arch Dis Child 58:529–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi E, Fukami T, Itoh M, Kyo S, Inoue M, Yokoi T, Nakajima M. (2007) Human CYP2A6 is induced by estrogen via estrogen receptor. Drug Metab Dispos 35:1935–1941 [DOI] [PubMed] [Google Scholar]
- Hines RN. (2007) Ontogeny of human hepatic cytochromes P450. J Biochem Mol Toxicol 21:169–175 [DOI] [PubMed] [Google Scholar]
- Houghton GW, Hundt HK, Muller FO, Templeton R. (1982) A comparison of the pharmacokinetics of metronidazole in man after oral administration of single doses of benzoylmetronidazole and metronidazole. Br J Clin Pharmacol 14:201–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Nakajima M, Higashi E, Yoshida R, Nagata K, Yamazoe Y, Yokoi T. (2006) Induction of human CYP2A6 is mediated by the pregnane X receptor with peroxisome proliferator-activated receptor-gamma coactivator 1alpha. J Pharmacol Exp Ther 319:693–702 [DOI] [PubMed] [Google Scholar]
- Jacob P, 3rd, Yu L, Duan M, Ramos L, Yturralde O, Benowitz NL. (2011) Determination of the nicotine metabolites cotinine and trans-3′-hydroxycotinine in biologic fluids of smokers and non-smokers using liquid chromatography-tandem mass spectrometry: biomarkers for tobacco smoke exposure and for phenotyping cytochrome P450 2A6 activity. J Chromatogr B Analyt Technol Biomed Life Sci 879:267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager-Roman E, Doyle PE, Baird-Lambert J, Cvejic M, Buchanan N. (1982) Pharmacokinetics and tissue distribution of metronidazole in the new born infant. J Pediatr 100:651–654 [DOI] [PubMed] [Google Scholar]
- Jensen JC, Gugler R. (1983) Single- and multiple-dose metronidazole kinetics. Clin Pharmacol Ther 34:481–487 [DOI] [PubMed] [Google Scholar]
- Kearns GL, Abdel-Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. (2003) Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med 349:1157–1167 [DOI] [PubMed] [Google Scholar]
- Khojasteh SC, Prabhu S, Kenny JR, Halladay JS, Lu AY. (2011) Chemical inhibitors of cytochrome P450 isoforms in human liver microsomes: a re-evaluation of P450 isoform selectivity. Eur J Drug Metab Pharmacokinet 36:1–16 [DOI] [PubMed] [Google Scholar]
- Kimura M, Yamazaki H, Fujieda M, Kiyotani K, Honda G, Saruwatari J, Nakagawa K, Ishizaki T, Kamataki T. (2005) Cyp2a6 is a principal enzyme involved in hydroxylation of 1,7-dimethylxanthine, a main caffeine metabolite, in humans. Drug Metab Dispos 33:1361–1366 [DOI] [PubMed] [Google Scholar]
- Lamp KC, Freeman CD, Klutman NE, Lacy MK. (1999) Pharmacokinetics and pharmacodynamics of the nitroimidazole antimicrobials. Clin Pharmacokinet 36:353–373 [DOI] [PubMed] [Google Scholar]
- Lo R, Burgoon L, Macpherson L, Ahmed S, Matthews J. (2010) Estrogen receptor-dependent regulation of CYP2B6 in human breast cancer cells. Biochim Biophys Acta 1799:469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loft S, Otton SV, Lennard MS, Tucker GT, Poulsen HE. (1991) Characterization of metronidazole metabolism by human liver microsomes. Biochem Pharmacol 41:1127–1134 [DOI] [PubMed] [Google Scholar]
- McDonagh EM, Wassenaar C, David SP, Tyndale RF, Altman RB, Whirl-Carrillo M, Klein TE. (2012) PharmGKB summary: very important pharmacogene information for cytochrome P-450, family 2, subfamily A, polypeptide 6. Pharmacogenet Genomics 22:695–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles JS, McLaren AW, Forrester LM, Glancey MJ, Lang MA, Wolf CR. (1990) Identification of the human liver cytochrome P-450 responsible for coumarin 7-hydroxylase activity. Biochem J 267:365–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima M, Yamamoto T, Nunoya K, Yokoi T, Nagashima K, Inoue K, Funae Y, Shimada N, Kamataki T, Kuroiwa Y. (1996) Characterization of CYP2A6 involved in 3′-hydroxylation of cotinine in human liver microsomes. J Pharmacol Exp Ther 277:1010–1015 [PubMed] [Google Scholar]
- Nilsson-Ehle I, Ursing B, Nilsson-Ehle P. (1981) Liquid chromatographic assay for metronidazole and tinidazole: pharmacokinetic and metabolic studies in human subjects. Antimicrob Agents Chemother 19:754–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Keefe JP, Troc KA, Thompson KD. (1982) Activity of metronidazole and its hydroxy and acid metabolites against clinical isolates of anaerobic bacteria. Antimicrob Agents Chemother 22:426–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogburn ET, Jones DR, Masters AR, Xu C, Guo Y, Desta Z. (2010) Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab Dispos 38:1218–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelkonen O. (2002) Human CYPs: in vivo and clinical aspects. Drug Metab Rev 34:37–46 [DOI] [PubMed] [Google Scholar]
- Pelkonen O, Rautio A, Raunio H, Pasanen M. (2000) CYP2A6: a human coumarin 7-hydroxylase. Toxicology 144:139–147 [DOI] [PubMed] [Google Scholar]
- Petsalo A, Turpeinen M, Pelkonen O, Tolonen A. (2008) Analysis of nine drugs and their cytochrome P450-specific probe metabolites from urine by liquid chromatography-tandem mass spectrometry utilizing sub 2 microm particle size column. J Chromatogr A 1215:107–115 [DOI] [PubMed] [Google Scholar]
- Rubenson A, Rosetzsky A. (1986) Single dose prophylaxis with metronidazole in infants during abdominal surgery: a pharmacokinetic study. Eur J Clin Pharmacol 29:625–628 [DOI] [PubMed] [Google Scholar]
- Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. (1994) Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther 270:414–423 [PubMed] [Google Scholar]
- Siu EC, Tyndale RF. (2008) Selegiline is a mechanism-based inactivator of CYP2A6 inhibiting nicotine metabolism in humans and mice. J Pharmacol Exp Ther 324:992–999 [DOI] [PubMed] [Google Scholar]
- Stambaugh JE, Feo LG, Manthei RW. (1968) The isolation and identification of the urinary oxidative metabolites of metronidazole in man. J Pharmacol Exp Ther 161:373–381 [PubMed] [Google Scholar]
- Suyagh M, Collier PS, Millership JS, Iheagwaram G, Millar M, Halliday HL, McElnay JC. (2011) Metronidazole population pharmacokinetics in preterm neonates using dried blood-spot sampling. Pediatrics 127:e367–e374 [DOI] [PubMed] [Google Scholar]
- Swan GE, Lessov-Schlaggar CN, Bergen AW, He Y, Tyndale RF, Benowitz NL. (2009) Genetic and environmental influences on the ratio of 3′hydroxycotinine to cotinine in plasma and urine. Pharmacogenet Genomics 19:388–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tateishi T, Nakura H, Asoh M, Watanabe M, Tanaka M, Kumai T, Takashima S, Imaoka S, Funae Y, Yabusaki Y, et al. (1997) A comparison of hepatic cytochrome P450 protein expression between infancy and postinfancy. Life Sci 61:2567–2574 [DOI] [PubMed] [Google Scholar]
- Upadhyaya P, Bhatnagar V, Basu N. (1988) Pharmacokinetics of intravenous metronidazole in neonates. J Pediatr Surg 23:263–265 [DOI] [PubMed] [Google Scholar]
- Wortham M, Czerwinski M, He L, Parkinson A, Wan YJ. (2007) Expression of constitutive androstane receptor, hepatic nuclear factor 4 alpha, and P450 oxidoreductase genes determines interindividual variability in basal expression and activity of a broad scope of xenobiotic metabolism genes in the human liver. Drug Metab Dispos 35:1700–1710 [DOI] [PubMed] [Google Scholar]
- Yamano S, Tatsuno J, Gonzalez FJ. (1990) The CYP2A3 gene product catalyzes coumarin 7-hydroxylation in human liver microsomes. Biochemistry 29:1322–1329 [DOI] [PubMed] [Google Scholar]