

Abstract
Activation of the transcription factor aryl hydrocarbon receptor by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) prevents the formation of the epicardium and leads to severe heart malformations in developing zebrafish (Danio rerio). The downstream genes that cause heart malformation are not known. Because TCDD causes craniofacial malformations in zebrafish by downregulating the sox9b gene, we hypothesized that cardiotoxicity might also result from sox9b downregulation. We found that sox9b is expressed in the developing zebrafish heart ventricle and that TCDD exposure markedly reduces this expression. Furthermore, we found that manipulation of sox9b expression could phenocopy many but not all of the effects of TCDD at the heart. Loss of sox9b prevented the formation of epicardium progenitors comprising the proepicardium on the pericardial wall, and prevented the formation and migration of the epicardial layer around the heart. Zebrafish lacking sox9b showed pericardial edema, an elongated heart, and reduced blood circulation. Fish lacking sox9b failed to form valve cushions and leaflets. Sox9b is one of two mammalian Sox9 homologs, sox9b and sox9a. Knock down of sox9a expression did not cause cardiac malformations, or defects in epicardium development. We conclude that the decrease in sox9b expression in the heart caused by TCDD plays a role in many of the observed signs of cardiotoxicity. We find that while sox9b is expressed in myocardial cells, it is not normally expressed in the affected epicardial cells or progenitors. We therefore speculate that sox9b is involved in signals between the cardiomyocytes and the nascent epicardial cells.
Introduction
The zebrafish (Danio rerio) has been used as a model for studying the toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) (Henry et al., 1997). By using zebrafish, it has been possible to determine that TCDD exposure during development causes heart failure and circulation collapse (Belair et al., 2001; Antkiewicz et al., 2005; Heideman et al., 2005). Interestingly, it is only during heart development that fish are sensitive to TCDD cardiotoxic effects: TCDD does not appear to harm the juvenile or adult heart (Lanham et al., 2012; Hofsteen et al., 2013).
In zebrafish, TCDD-induced heart malformation is associated with the loss of epicardium and the proepicardium (PE) (Plavicki et al., 2013). During the period before 48 hours postfertilization (hpf), TCDD exposure has no discernible effect on development of the zebrafish heart. However, after 48 hpf, TCDD-exposed hearts begin to deteriorate and unloop. The manifestation of cardiotoxicity corresponds to the timing of epicardium formation (Plavicki et al., 2013). The epicardium and epicardium-derived progenitor cells are thought to play a critical role in cardiomyocyte proliferation, valve development, heart looping, generation of fibroblasts, cardiac morphogenesis, development of the coronary vasculature, and adult cardiac regeneration (Lepilina et al., 2006; Lie-Venema et al., 2007; Olivey and Svensson, 2010; Svensson, 2010). TCDD-induced epicardium failure accounts for most if not all of the observed cardiotoxicity.
TCDD-induced toxicity in zebrafish is mediated through the aryl hydrocarbon receptor (AHR) (Prasch et al., 2003). AHR is a ligand-activated transcription factor belonging to the basic helix-loop-helix per-ARNT-Sim (PAS) family of DNA-binding proteins (Schmidt and Bradfield, 1996). TCDD-activation of AHR leads to altered gene expression. While identification of DNA sequence motifs recognized by AHR allows us to better understand the activation of genes encoding cytochrome P-450s and other AHR-battery genes (Chang and Puga, 1998), it has remained difficult to link AHR regulation of a specific target gene with toxic responses. A recent study showed that TCDD-induced jaw malformation in developing zebrafish is caused by downregulation of sox9b (Xiong et al., 2008).
Sox9b is a critical chondrogenic transcription factor, derived from an ancestral genome duplication in teleost fish that produced two sox9 homologs in zebrafish: sox9a and sox9b (Yan et al., 2005). In humans, SOX9 mutations cause campomelic dysplasia, producing defects in long bones, jaw, palate, axial skeleton, heart development, and reproductive systems. It is particularly interesting that campomelic dysplasia patients suffer from a defect in heart development known as the Tetralogy of Fallot (Houston et al., 1983). The discovery that TCDD downregulates sox9b and the similarity between known developmental effects of sox9b mutation and TCDD developmental toxicity led us to the question: Might TCDD-induced heart malformation in zebrafish larvae be caused by downregulation of sox9b?
Here we report that sox9b is expressed in the developing zebrafish heart, and that this expression is reduced by TCDD. We also show that loss of sox9b causes cardiac malformation, pericardial edema, and decreased circulation. Furthermore, sox9b is required for PE, epicardium, and valve formation.
Materials and Methods
Zebrafish Husbandry.
Lines used were: AB wild-type, sox9bb971 (Yan et al., 2005), pard3:EGFP [ET(Krt4:EGFP)spe27] (Poon et al., 2010), tcf21:DsRed [Tg(tcf21:DsRed2)pd32] (Kikuchi et al., 2011), sox9b:EGFP [Tg(−2450/0sox9b:EGFP)] (J. Plvicki, F. Burns, T. Baker, P. Hofsteen, K. Xiong, R. Peterson, and W. Heideman, manuscript in preparation), Tg(cmlc2:GFP) and Tg(flk:GFP) (Cross et al., 2003). All embryos were housed in water buffered with Instant Ocean salts (60 mg/l; Aquarium Systems, Mentor, OH) at 27°C with a 14-hour/10-hour light/dark cycle. All procedures involving zebrafish were approved by the Animal Care and Use Committee of the University of Wisconsin-Madison, and adhered to the National Institutes of Health’s Guide for the Care and Use of Laboratory Animals.
TCDD Exposure.
Embryos were collected at 2–4 hpf and exposed to TCDD (1 ng/ml, 99% purity; Chemsyn, Lenexa, KS) or dimethyl sulfoxide (DMSO) vehicle (0.1%) with gentle rocking for 1 hour in glass scintillation vials (10 embryos/ml) as described (Carney et al., 2006a). After the exposure, embryos were rinsed with water and returned to culture vessels.
Heart Extraction and Quantitative Polymerase Chain Reaction.
Hearts were extracted from 72-hpf cmlc2:GFP embryos using shearing as previously described (Burns and MacRae, 2006; Carney et al., 2006a). Three independent replicate experiments were conducted, with each replicate using total RNA from 200 hearts for both TCDD and controls. The RNA was extracted using a QIAGEN RNeasy Minikit following the manufacturer’s protocol, and cDNA was synthesized using oligo(dT) primers and a Superscript II RT cDNA synthesis kit (Invitrogen, Carlsbad, CA).
The quantitative reverse-transcription polymerase chain reaction was performed as described previously using a Light Cycler (Roche Applied Science, Indianapolis, IN) and SYBR green (Carney et al., 2006a). Standard curves were made using serial dilutions of sox9b and β-actin plasmid DNA. Primers were: β-actin: forward, 5′-aag cag gag tac gat gag tc-3′; reverse, 5′-tgg agt cct cag atg cat tg-3′ and sox9b: forward primer, 5′-tga cga gtt gtt ctc cag ag-3′; reverse primer, 5′-agg cca cac gtc tat aac cc-3′. Sox9b mRNA levels were normalized to β-actin to generate a relative expression ratio. Statistical analysis was performed using Minitab 12. Significance was determined using one-way analysis of variance followed by Fisher’s least significant difference test (P < 0.05).
Pericardial Edema and Heart Length.
Larvae were mounted in 3% methylcellulose and imaged laterally at 8.5× on a Leica MZ16 stereomicroscope using a 1.5× lens. Pericardial area and heart length were measured using NIH Image J 1.44 software (http://rsb.info.nih.gov/nih-image/). Boundaries of the pericardial area were traced and the computer determined pericardial area. Length of the heart was measured with a segmented line that started at the beginning of the inflow tract, traveled across the atrium to the atrioventricular (AV) junction, and then continued straight across the ventricle to the outflow attachment. The computer then calculated the length of this line. Three replicate experiments (n = 3) were conducted using groups of control or TCDD-treated fish with 50–100 fish per group. Statistical analysis was performed using Minitab 12. Significance was determined using t tests followed by Fisher’s least significant difference test and Levene’s test (P < 0.05).
Red Blood Cell Perfusion Rate.
Red blood cell (RBC) perfusion rates were measured at 96 hpf as previously described (Prasch et al., 2003; Carney et al., 2006b). Larvae were mounted in 3% methylcellulose and 10-second videos of the caudal end of the tail were taken using a MotionScope camera (DEL Imaging Systems, LLC., Cheshire, CT) mounted on a Nikon TE300 inverted microscope. For each fish, the number of RBCs moving through a reference point in each of the four most caudal intersegmental vessels was measured, and the average was calculated (1 fish = 1 n; n = 10). Statistical analysis was performed using Minitab 12. Significance was determined using one-way analysis of variance followed by Fisher’s least significant difference test and Levene’s test (P < 0.05).
Histology.
Zebrafish were fixed in 4% paraformaldehyde at 4°C overnight, dehydrated to ethanol, embedded in paraffin, and sectioned (8 μm). Fish sections were stained with hematoxylin and eosin (King Heiden et al., 2009) and imaged using a Zeiss Axiocam digital camera mounted on a Zeiss Axioplan microscope (Carl Zeiss AG, Oberkochen, Germany).
In Situ Hybridization.
Whole mount in situ hybridization was conducted as previously described (Plavicki et al., 2013). A 560–base pair fragment of sox9b was amplified from embryonic zebrafish cDNA and subcloned into a pCRII-TOPO vector (Invitrogen). Primers used were: 5′ -gtg cag taa agc gca tct gaa-3′ and 5′ -gcg caa gta tgt gtg tgt gtg-3′. Synthesis of the sox9b probe, restriction enzyme used, and detection were conducted as previously described (Hofsteen et al., 2013).
Immunohistochemistry and Confocal Microscopy.
Antibody staining was performed as previously described (Plavicki et al., 2013). The antibody against activated leukocyte cell adhesion molecule was used at a 1:50 dilution in phosphate buffered saline with 4% bovine serum albumin and 0.3% Triton (PBT). The antibody against DsRed (Anaspec, Fremont, CA) was used at a 1:200 dilution in PBT. Secondary antibodies (Alexa 488, Alexa 568; Invitrogen) were used at 1:100 dilution in PBT. Confocal images were collected on an Olympus Fluoview FV1000 microscope.
Proepicardium Imaging.
Live embryos were imaged in 3% methylcellulose using a Nikon TE300 inverted microscope attached to a Princeton Instruments Micromax CCD camera. Ten-second videos showing the presumptive PE site were captured for each embryo using MotionScope software and analyzed using Metamorph software. This technique allowed us to differentiate the PE from surrounding tissue due to the PE remaining relatively stationary while adhered to the pericardium adjacent to the AV junction.
Morpholinos.
All morpholino oligonucleotides (MOs) were from Gene Tools, LLC (Philomath, OR) and used as previously reported (Prasch et al., 2003; Antkiewicz et al., 2006; Xiong et al., 2008). MOs were fluorescein tagged at the 3′ ends to monitor injection success. The MO sequences were: sox9a, 5′ AAT GAA TTA CTC ACC TCC AAA GTT T 3′; sox9b, 5′ TGC AGT AAT TTA CCG GAG TGT TCT C 3′ (Yan et al., 2005). The standard Gene Tools Control MO (5′-CCT CTT ACC TCA GTT ACA ATT TAT A-3′) was used to control for nonspecific responses. All zebrafish were injected at the 1-4 cell stage with 3 nl of 1- or 2-nM solution containing the MO with Fast Green (40 μg/ml) to allow visualization of the injected liquid with a dissecting microscope. Injected embryos were examined during blastula formation using epifluorescence microscopy for incorporation of the fluorescent MO into cells.
Sox9b mRNA Injection.
Wild-type (AB) embryos were injected with sox9b mRNA (200 pg) at the 1-4 cell stage. Shortly following mRNA injection, fish were exposed to either DMSO as a control (n = 46) or TCDD (n = 73) as described above in Materials and Methods. As a control, a subset of fish were exposed to DMSO (n = 50) or TCDD (n = 50) in parallel but were not injected. Fish were raised in 0.003% phenylthiourea in the water to inhibit pigment formation and were analyzed for PE formation at 72 hpf as previously described. Sox9b mRNA was synthesized as described (Xiong, 2008). Briefly, a pCMV-Sport6ccd vector (Open Biosystems, Huntsville, AL) containing full-length sox9b was digested with Not1, and the sox9b mRNA was synthesized with SP6 polymerase following manufacturer’s instructions (SP6 mMessage mMachine Kit; Ambion, Austin, TX).
Results
Zebrafish sox9b Is Expressed in the Developing Heart Ventricle.
We first sought to determine if sox9b was expressed in the developing zebrafish heart. In situ hybridization experiments showed a specific signal in the developing heart at 72 hpf, which became even more distinct at 96 hpf (Fig. 1, A and B).
Fig. 1.

Sox9b is expressed in the zebrafish larval heart. Wild-type AB zebrafish carrying sox9b:EGFP were collected at the indicated times for either in situ hybridization with a sox9b probe (A and B) or confocal microscopy (C and D). Lateral views are shown in all images, with head extending leftward, and the yolk sac to the right, with n = 6 fish examined per group. For the in situ images, the arrowheads point to the heart. For the confocal images, the white outline indicates the border of the atrium, indicated as A; the ventricle is indicated as V. The sox9b-GFP signal is shown in green. Blue indicates immunostaining for activated leukocyte cell adhesion molecule (ALCAM).
A sox9b:EGFP reporter line (−2450/0sox9b:EGFP; J. Plvicki, F. Burns, T. Baker, P. Hofsteen, K. Xiong, R. Peterson, and W. Heideman, manuscript in preparation) showed a more distinct signal, allowing individual cells to be identified (Fig. 1, C and D). In these experiments, it was apparent that sox9b is expressed in myocardial cells, especially in the ventricle.
The in situ hybridization and reporter expression patterns showed consistent evidence for sox9b expression in the zebrafish heart during the period of sensitivity to TCDD-induced cardiotoxicity.
TCDD and sox9b Expression in the Larval Heart.
To determine if TCDD affects the expression of sox9b in the larval heart, we exposed the sox9b:EGFP reporter line to TCDD at fertilization and assessed sox9b expression in the heart at 72 hpf. Compared with the control, we consistently observed a noticeable decrease in the green fluorescent protein (GFP) signal in TCDD-exposed hearts (Fig. 2, A and B).
Fig. 2.

TCDD reduces sox9b expression in the zebrafish larval heart. Zebrafish embryos carrying the sox9b:EGFP reporter were exposed to TCDD as described in Materials and Methods. Embryos were examined at 72 hpf using confocal microscopy and representative lateral images are shown: (A) control heart, (B) TCDD-treated. White outline denotes ventricle (V); anterior is to the left. (C) Hearts were isolated at 72 hpf for quantitative reverse-transcription polymerase chain reaction measurement of sox9b expression, normalized to β-actin mRNA for each treatment group. Results are mean ± S.E., n = 3 replicate experiments. Asterisk denotes significantly different from controls (P < 0.05).
To directly measure downregulation of sox9b mRNA, fish were treated as before and hearts were isolated at 72 hpf for mRNA extraction and quantitative reverse-transcription polymerase chain reaction. TCDD caused an approximately 2-fold downregulation of sox9b mRNA in the TCDD-exposed hearts relative to the control hearts (Fig. 2C). We conclude that TCDD downregulates sox9b in the developing zebrafish heart.
Loss of sox9b Impairs Heart Development.
If sox9b downregulation by TCDD causes heart malformation, then other means of decreasing sox9b expression should also produce heart malformation. To test this, we examined developing hearts in sox9b deletion mutants. In these experiments, we crossed heterozygous sox9bb971 zebrafish and examined cardiac function in the homozygous sox9bb971 null offspring. The hallmark phenotype identifying sox9bb971 null homozygotes is a pronounced curlydown or “corkscrew” tail. We therefore examined heart development in the ∼25% offspring with this phenotype.
By 72 hpf, homozygous sox9bb971 nulls showed clear signs of heart malformation and pericardial edema (data not shown), which became more pronounced at 96 hpf (Fig. 3). The pericardial edema observed in the sox9bb971 null homozygotes was very similar to that produced by TCDD. Furthermore, the larval hearts showed an unlooping defect and heart elongation that resembled the response to TCDD. While both sox9bb971 nulls and TCDD-treated wild-type fish had unlooped and elongated hearts, the heart chambers of the sox9bb971 null fish appeared more functional. The atrium in the TCDD-treated hearts was uniformly string-like with little apparent lumen, and a constricted ventricle. In contrast, loss of sox9b produced elongated chambers with open lumens.
Fig. 3.

Loss of sox9b produces cardiac malformations that resemble those produced by TCDD. Representative brightfield photomicrographs of 96-hpf zebrafish. Lateral views are shown in all images, with head extending leftward, and the yolk sac to the right. Arrowheads denote location of the heart ventricle (V) and atrium (A). AB control and TCDD-treated fish are at left and right, respectively. The center panel shows a sox9bb971 homozygous mutant fish.
Hallmark characteristics of TCDD-induced heart malformation include an unlooped extended heart and pronounced pericardial edema, or effusion (Antkiewicz et al., 2005, 2006). These phenotypes can be compared quantitatively by measuring pericardial area and heart length from the sinus venosus (SV) to the bulbous arteriosus (BA), producing the SV-BA distance at 96 hpf. In our measurements, the pericardial edema in both sox9b null and TCDD-treated larvae were significantly different from control, but not from each other (Table 1). The SV-BA distance in homozygous sox9bb971 null mutant heart was significantly longer than wild-type hearts; however, the length in TCDD-exposed hearts was greater (Table 1), consistent with the more elongated appearance of the TCDD-treated heart in Fig. 3.
TABLE 1.
Effect of sox9b-null mutation and TCDD treatment on pericardial area, heart length, and blood flow
Control, homozygous sox9bb971 deletion mutants, and TCDD-treated wild-type fish were obtained by spawning and TCDD exposure as described in Materials and Methods. Pericardial area, heart length, and blood flow rate were measured using videomicroscopy as described in Materials and Methods. Results are presented as the mean ± S.E.M.
| Response | Control | sox9b Null | TCDD |
|---|---|---|---|
| Pericardial area (μm2) | 44 ± 0.36 | 59 ± 4.0* | 86 ± 1.0* |
| Heart length (μm) | 150 ± 5.8 | 220 ± 14.0* | 320 ± 8.0 ** |
| RBC perfusion (RBCs/10 sec) | 63 ± 3 | 33 ± 7* | 0** |
Significantly different from control; **significantly different from both control and the sox9b mutants (P ≤ 0.05).
We compared the effect of TCDD exposure and sox9b loss on RBC flow, using video capture microscopy to follow RBC movement at a set of intersegmental vessels in the tail. At 96 hpf, we found that loss of sox9b caused an approximately 2-fold decrease in the RBC perfusion rate relative to wild-type controls (Table 1). However, the decrease in RBC perfusion rate of sox9b null larvae did not approach the complete halt of RBC movement in the tail of TCDD-exposed larvae (Belair et al., 2001; Antkiewicz et al., 2005).
We also used MOs to specifically block sox9b mRNA maturation and subsequent Sox9b protein production (Yan et al., 2005). We found that injection of the sox9b MOs phenocopied the curled tail and cardiac malformations observed in the homozygous sox9bb971 null mutants. This response was dose-dependent: Injection of the 1-nM stock produced mild pericardial edema and heart defects, without consistently producing the curly-tail phenotype that characterizes complete loss of function (Fig. 4). Injection of the 2-nM stock produced more severe cardiac defects ranging from that seen in Fig. 4C to that shown in Fig. 4D. With this higher dose, the curly-tail phenotype was always evident. The control MO did not produce this phenotype at any concentration tested.
Fig. 4.

Graded doses of sox9b MO produce a range of cardiac malformation severity. Wild-type zebrafish embryos were injected with control and sox9b MOs and collected at 96 hpf for brightfield microscopy. Representative brightfield lateral images are shown with anterior to the left. (A) Control MO, (B) sox9b MO (1 nM), (C and D) sox9b MO (2 nM). The Fast Green used as part of the MO injection solution has colored the yolk green (n = 10 fish per treatment).
Overall, loss of sox9b produced substantial cardiac malformation with pronounced pericardial edema. This substantially overlapped the cardiotoxic effects of TCDD.
Loss of sox9b Prevents Zebrafish Epicardium Development.
Zebrafish embryos exposed to TCDD do not form the cluster of epicardial progenitor cells composing the PE or the epicardial layer surrounding the heart (Plavicki et al., 2013). We examined hearts from homozygous sox9bb971 mutants at 120 hpf, a time when the epicardium should envelop the ventricle. H&E sections showed normal epicardial cells, identified as flat, oblong cells on the periphery of myocardium in the wild-type sections (Fig. 5). In contrast, these cells were not visible in sections from the sox9bb971 mutants.
Fig. 5.

Sox9b is required for zebrafish epicardium development. (A and B) Brightfield images of hematoxylin and eosin–stained hearts from representative wild-type larva (A) and homozygous sox9bb971 null mutant larva (B) at 120 hpf. Black arrowheads indicate epicardial cells. (C and D) Embryos from the tcf21:DsRed epicardial cell reporter line were injected with the control (CMO) or sox9b MO (sox9bMO), and examined using confocal microscopy at 96 hpf for epicardium formation. Red indicates expression of tcf21, with white arrowheads indicating epicardial cells. Green indicates immunostaining for activated leukocyte cell adhesion molecule (ALCAM), marking cell boundaries, and the blue is 4′,6-diamidino-2-phenylindole (DAPI) staining, revealing nuclei. (E and F) Eggs from the pard3-GFP epicardial cell reporter line were injected with the control or sox9b MO, and examined using confocal microscopy at 96 hpf for epicardium formation. Green indicates expression of pard3 in epicardial cells which are indicated with white arrowheads. The blue is DAPI staining, revealing nuclei. For all panels representative images are shown with a minimum of n = 6 per group. Scale bar, 50 μm.
We confirmed this finding by injecting the sox9b-specific MO into zebrafish lines carrying reporters marking epicardial cells. In the tcf21:DsRed reporter line (Kikuchi et al., 2011), epicardial cells are marked with a DsRed+ signal. In the pard3:EGFP line, the GFP is expressed in the epicardium (Poon et al., 2010). Epicardial cells marked with DsRed or GFP were consistently observed in the control MO fish, lying along the outermost layer of the heart ventricle to form a sheath of epithelial cells (Fig. 5, C and E). However, the sox9b morphants, displaying the hallmark curly tail, lacked expression of either epicardial marker at 96 hpf (Fig. 5, D and F). We conclude that knockdown of sox9b prevents formation of the epicardium.
Sox9b Is Required for PE Formation.
We used video microscopy to identify the PE cluster forming at the region of the pericardium adjacent to the AV junction, where the PE forms (Serluca, 2008; Liu and Stainier, 2010). This allowed us to visualize the difference between the PE cluster, held stationary against the pericardial wall, and other clumps of cells associated with the moving heart. By 72 hpf the PE was clearly visible in the control fish but not in fish failing to express sox9b (Fig. 6). We were unable to find signs of PE formation in any homozygous sox9bb971 null mutants (Fig. 6B), nor in fish injected with the sox9b MO (Fig. 6C).
Fig. 6.
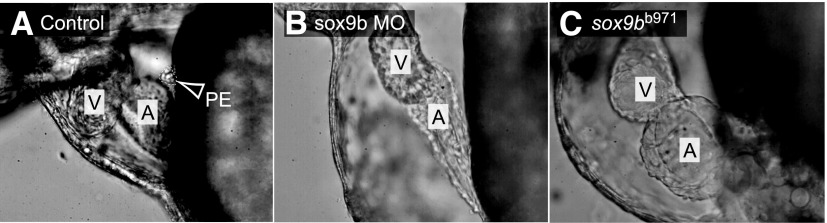
Formation of the proepicardium is dependent on sox9b expression. Representative lateral images of hearts within the pericardium of 72-hpf zebrafish. (A) Wild-type control; (B) wild-type zebrafish injected with sox9b MO; (C) homozygous sox9bb971 mutant (n = 10 per group; anterior to the left). Arrow and white outline indicate the “grape-like” PE in the wild-type control, and not found in the others. The atria and ventricles are marked as A and V, respectively.
Sox9b Is Expressed in Zebrafish Myocardial Cells.
We crossed the sox9b GFP reporter line with the tcf21 reporter line to determine where sox9b is expressed during heart and epicardium development. We examined these fish at 3, 7, and 21 days postfertilization (Fig. 7). The development of the epicardium was clearly marked by DsRed fluorescence. However, the GFP signal from the sox9b reporter, while clearly evident in the myocardial cells, did not overlap the DsRed signal from the PE and epicardial cells.
Fig. 7.

Sox9b is not expressed in proepicardial or epicardial cells. Representative ventral-lateral images of the zebrafish heart in reporter fish expressing both sox9b:EGFP and tcf21:DsRed with anterior to the left (n = minimum of 8 per group). The top panel shows the heart at 3 days postfertilization (A), the middle shows a heart at 7 days (B), and the bottom panel shows the heart at 21 days (C). The yellow arrowhead indicates the proepicardium, while white arrowheads denote epicardial cells. The ventricle and atrium are marked as V and A, respectively.
Endocardial Valve Cushions Do Not Form in sox9b Morphants.
After formation of the epicardial layer, epicardial-derived cells migrate into the underlying myocardium and assist development of the cardiac valves (Lie-Venema et al., 2007, 2008). Given that TCDD prevents formation of the valve cushions (Mehta et al., 2008), and reduced levels of sox9b prevent epicardium development, we hypothesized that sox9b may be needed for zebrafish valve development. During the normal progression of valve development, a ring of endothelial cells forms marking the presumptive valve sites at the AV junction and the outflow junction between the ventricle and bulbus arteriosus (Keegan et al., 2002; Bartman et al., 2004). These rings of endothelial cells thicken to form cushions that mature into valve leaflets at the AV junction and outflow tract. We used a cardiac endothelial cell reporter line (flk1:GFP) to follow valve development. For these images, the cell adhesion molecule was visualized in red by immunostaining to show the surrounding heart cells. The formation of valve cushions and nascent valve leaflets at the AV junction can be seen at 96 hpf in the control heart shown in Fig. 8, and more clearly at higher magnification in the panel at lower left. In contrast, the sox9b morphants lacked the cushion and leaflet and had no accumulation of the GFP-labeled endothelial cells at the valve sites. Instead, the endothelial cells in the sox9b-deficient fish were distributed throughout the ventricle and atrium. These results indicate that sox9b is necessary for valve cushion development.
Fig. 8.

Endocardial valve cushions fail to form following loss of sox9b. Representative 96-hpf ventral images of flk1:GFP zebrafish that were injected with sox9b MO or control MO (CMO) (n = 5 per group). Samples were immunostained with activated leukocyte cell adhesion molecule (ALCAM) in red to show cell boundaries. The ventricles and atria are marked as A and V, respectively. (A and B) 10× magnification; (C and D) 40× magnification. The green arrows indicate the valve cushions and nascent leaflets forming in the AV junction; the red arrows indicate the sites at the AV junction where valve cushions failed to form. Scale bar, 50 μm.
Ectopic sox9b Expression Rescues PE Formation in TCDD-Treated Embryos.
The finding that loss of sox9b partially phenocopies TCDD toxicity suggests that TCDD causes cardiotoxicity by reducing sox9b expression. To test this we injected sox9b mRNA into embryos at the 1–4 cell stage and then treated the embryos with TCDD as described in Materials and Methods. Expression of injected mRNA tends to follow a mosaic pattern, so not all of the injected embryos would be expected to express sox9b in the region of the heart, and might not be rescued. However, we found that a significant fraction of injected fish (19/73 = 26%) developed PEs, even in the presence of TCDD (Fig. 9).
Fig. 9.

Sox9b mRNA injection can restore PE formation in fish treated with TCDD. (C and D) Wild-type (AB) embryos were injected with sox9b mRNA (200 pg) at the 1-4 cell stage, or left uninjected as controls (A and B). The embryos were then exposed to TCDD (B and D) or vehicle (A and C) (DMSO) as in previous experiments, as indicated. Images were collected with differential interference contrast (DIC) microscopy at 72 hpf. The A and V indicate the atrium and ventricle, respectively. Where present the PE is indicated by white arrowheads. Fish were raised in 0.003% propylthiourea in the water to inhibit pigment formation. Representative images are shown, with n = 46–73 individuals examined.
We found that while sox9b mRNA injection frequently restored PE formation, it never restored normal heart morphology, nor did we observe epicardial cell migration onto the heart. While rescue experiments are difficult to interpret, this indicates that decreased sox9b produced by TCDD is responsible for the failure of PE formation.
Zebrafish sox9a Morphants Lack Notable Cardiac Defects.
Zebrafish have two copies of the mammalian Sox9 gene, sox9a and sox9b (Chiang et al., 2001). It is not clear how much the functions of the two genes have diverged. Therefore, we injected sox9a MOs into the pard3:EGFP epicardial reporter line to determine whether loss of sox9a expression would also affect epicardium development.
The sox9a morphants exhibited the jaw phenotype reported by Yan et al. (2005) (Supplemental Fig. 1), indicating that the sox9a MO had been effective in reducing sox9a expression. As with the previous work, we sometimes observed mild pericardial edema; the sox9a morphants did not have notable defects in the heart structure and, in particular, the epicardium developed normally (Supplemental Fig. 1). These data indicate a divergent role between sox9a and sox9b during development of the zebrafish heart.
Discussion
Sox9b and TCDD-Induced Heart Malformation.
As with the developing jaw, we found that TCDD downregulates sox9b in the embryonic zebrafish heart. From this, we hypothesized that the downregulation of sox9b expression might contribute to TCDD-induced heart malformation. We found that indeed, decreased sox9b expression largely phenocopied the cardiac malformations caused by TCDD. Loss of sox9b expression produced most, but not all, signs of TCDD-induced cardiotoxicity. Loss of sox9b expression was associated with pericardial edema, unlooping, loss of the PE, and failure to form the epicardium and endocardial cushions.
In previous work we used microarrays to search for AHR gene targets responsible for cardiotoxicity (Carney et al., 2006a). At the time, we were expecting to find genes upregulated by TCDD, and even though sox9b was found in the list of significantly altered genes, it was downregulated only by approximately 2-fold in the embryonic hearts examined in this experiment. It was not until we found a 14-fold downregulation of sox9b in the jaw that we began to consider genes downregulated by activated AHR as important (Xiong et al., 2008).
However, loss of sox9b did not produce the typical TCDD-induced compacted ventricle and elongated string-like atrium. Additionally, while TCDD treatment produced a complete halt in circulation, loss of sox9b slowed but did not completely stop circulation. Homozygous sox9bb971 mutants are completely lacking sox9b, while TCDD produces an approximately 50% decrease in sox9b mRNA in the heart. Therefore, we conclude that TCDD-induced downregulation of sox9b can account for some, but not all of the cardiotoxic effects of TCDD.
The overlap between the effects of TCDD and loss of sox9b suggested that downregulation of sox9b by TCDD exposure leads to some of the cardiotoxicity observed. Our attempts to rescue the cardiotoxicity by injection of extra sox9b mRNA sheds some light on this. The restoration of the PE in TCDD-treated fish by sox9b addition supports the idea that downregulation of sox9b plays a role in the loss of the PE and subsequent failure of epicardium formation. However, sox9b mRNA did not prevent other forms of TCDD cardiotoxicity.
Rescue experiments are generally difficult because it is unlikely that whatever manipulation is chosen will precisely reverse the loss of the biologic molecule in question. For example, bulk injection of sox9b mRNA cannot be expected to undo cell-specific losses of endogenous sox9b mRNA caused by TCDD. The situation is even harder in cases such as ours, in which we already know that loss of sox9b cannot account for all of the cardiotoxicity produced by TCDD. In this case, complete rescue would be very surprising, while no rescue at all would not be unexpected: adding back sox9b cannot reverse sox9b-independent TCDD effects. Despite this perhaps pessimistic view, we observed some rescue. The failure to rescue heart malformation could be attributed to a number of factors including the possibility that the mRNA persisted only long enough to ensure PE specification but not for 3 days of heart development, the possibility that expression in the heart was somehow inadequate, and the simple possibility that the heart malformation is due to some factor other than sox9b loss. More specific and sophisticated experiments will be needed to distinguish between these and other possibilities.
It is possible that TCDD affects both the heart and the vasculature to produce the observed circulation collapse. TCDD activates AHR in the vascular endothelium of fish, birds, and mammals, and suppresses vascular remodeling of the rat placenta, and coronary vasculogenesis in the chicken embryo (Ivnitski-Steele et al., 2005; Ishimura et al., 2006). In lake trout and zebrafish larvae TCDD induces cytochrome P4501A in the vascular endothelium (Andreasen et al., 2002; Cook et al., 2003). Furthermore, TCDD induces a rearrangement of the zebrafish proencephalic artery, and a reduction in mesencephalic blood flow (Teraoka et al., 2010; Kubota et al., 2011).
Sox9 and the Vertebrate Heart.
In the mouse heart, Sox9 has been implicated in endocardial cushion formation and valve leaflet remodeling (Montero et al., 2002; Akiyama et al., 2004; Lincoln et al., 2004). Furthermore, loss of Sox9 expression in Sox9flox/flox:Tie2-cre mice resulted in embryonic death between E11.5 and E14.5 days postconception. These mice exhibited pericardial edema and increased blood pooling (Lincoln et al., 2007). Akiyama and colleagues also documented embryonic death at E11.5 and 12 days postconception in Sox9-null mutants. These Sox9 null mice had severe blood vessel dilation, suggesting congestive heart failure (Akiyama et al., 2004). In both cases, death occurred during the temporal developmental window of the murine epicardium. Furthermore, the cardiac phenotype seen in Sox9 null mice is strikingly similar to the phenotype that we found in sox9b null zebrafish larvae.
Although the functional role for Sox9 may be conserved in the zebrafish, we found an interesting divergence in expression pattern. In mice, most myocardial cells do not express Sox9. Murine Sox9 is expressed in epicardial cells undergoing epithelial-to-mesenchymal transition as they invade the myocardium (Smith et al., 2011). Sox9 is expressed in the developing chick heart around and in the valve cushion mesenchyme, but not broadly across the myocardium. In the zebrafish heart, we found sox9b expressed in myocardial cells, but not in epicardial cells.
Sox9b and the Epicardium.
Although loss of sox9b was ultimately devastating, sox9b expression was dispensable for initial heart chamber formation. A similar pattern was observed in fish exposed to TCDD. In both cases, the appearance of cardiac malformation coincided with the normal timing of PE and epicardium formation. Thus, we speculate that most of the cardiac malformations observed in response to TCDD exposure or loss of sox9b can be attributed to loss of the PE and epicardium (Plavicki et al., 2013).
In recent experiments, we demonstrated that ectopic expression of a constitutively active AHR from the cmlc2 promoter causes cardiac malformations characterized by loss of the epicardium (W. Heideman and R. E. Peterson, unpublished data). Therefore, events downstream of AHR in myocardial cells disrupt the formation and migration of epicardial cells. Furthermore, while sox9b expression was critical for the formation of the epicardium, we found sox9b expressed in ventricular cardiomyocytes but not in cells of the PE or epicardium. The genes regulated by the Sox9 transcription factor are primarily proteins that make up the extracellular matrix (Rahkonen et al., 2003; Lincoln et al., 2007). Since the epicardium is derived from an extracardiac source of progenitor cells that migrate to and differentiate at the heart (Lie-Venema et al., 2007; Serluca, 2008), we speculate that sox9b in cardiomyocytes provides extracellular signals that guide PE development and epicardium migration. In our model, this signal is disrupted by TCDD.
Conclusion.
TCDD and related compounds have been known for some time to disrupt development by activating a specific receptor, AHR. However, the identities of genes regulated by AHR that mediate TCDD-induced heart malformation remain largely unknown. Here we show that, as in the developing jaw, sox9b is a downstream target of AHR in the heart. We may find that misregulation of sox9b becomes a common thread in AHR-mediated developmental toxicity.
While it seems obvious that the outer layer of the heart is needed for normal function, we do not understand how it forms. We show here that expression of sox9b is required for assembly of progenitor cells on the pericardium wall, and subsequent formation of the epicardium. We note that sox9b is found in cardiomyocytes awaiting the outer layer, not in the epicardial cells that fail to form, and plays an important role in regulating the production of proteins needed for extracellular matrices such as collagen. We speculate that sox9b is involved in extracellular signaling to nascent epicardial cells.
Supplementary Material
Acknowledgments
The authors thank Dorothy Nesbit for expert assistance. The authors also thank K. Poss, V. Korzh, J. Postlethwait, and D. Graham for zebrafish lines and advice.
Abbreviations
- AHR
aryl hydrocarbon receptor
- AV
atrioventricular
- BA
bulbous arteriosus
- DMSO
dimethyl sulfoxide
- GFP
green fluorescent protein
- hpf
hours postfertilization
- MO
morpholino oligonucleotide
- PBT
phosphate buffered saline with 0.3% Triton
- PE
proepicardium
- RBC
red blood cell
- SV
sinus venosus
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
Authorship Contributions
Participated in research design: Hofsteen, Plavicki, Johnson, Heideman.
Conducted experiments: Hofsteen, Plavicki, Johnson.
Performed data analysis: Hofsteen, Johnson.
Wrote or contributed to the writing of the manuscript: Hofsteen, Peterson, Heideman.
Footnotes
This work was supported by the National Institutes of Health National Institute of Environmental Health Sciences [Grant R01 ES012716]; and the University of Wisconsin Sea Grant Institute, National Sea Grant College Program, National Oceanic and Atmospheric Administration, U.S. Department of Commerce [Grant NA 16RG2257], Sea Grant Project R/BT-25.
The contents are solely the responsibility of the authors and do not necessarily represent the official view of the National Institutes of Health National Institute of Environmental Health Sciences. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Akiyama H, Chaboissier MC, Behringer RR, Rowitch DH, Schedl A, Epstein JA, de Crombrugghe B. (2004) Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc Natl Acad Sci USA 101:6502–6507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen EA, Spitsbergen JM, Tanguay RL, Stegeman JJ, Heideman W, Peterson RE. (2002) Tissue-specific expression of AHR2, ARNT2, and CYP1A in zebrafish embryos and larvae: effects of developmental stage and 2,3,7,8-tetrachlorodibenzo-p-dioxin exposure. Toxicol Sci 68:403–419 [DOI] [PubMed] [Google Scholar]
- Antkiewicz DS, Burns CG, Carney SA, Peterson RE, Heideman W. (2005) Heart malformation is an early response to TCDD in embryonic zebrafish. Toxicol Sci 84:368–377 [DOI] [PubMed] [Google Scholar]
- Antkiewicz DS, Peterson RE, Heideman W. (2006) Blocking expression of AHR2 and ARNT1 in zebrafish larvae protects against cardiac toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci 94:175–182 [DOI] [PubMed] [Google Scholar]
- Bartman T, Walsh EC, Wen KK, McKane M, Ren J, Alexander J, Rubenstein PA, Stainier DY. (2004) Early myocardial function affects endocardial cushion development in zebrafish. PLoS Biol 2:E129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belair CD, Peterson RE, Heideman W. (2001) Disruption of erythropoiesis by dioxin in the zebrafish. Dev Dyn 222:581–594 [DOI] [PubMed] [Google Scholar]
- Burns CG, MacRae CA. (2006) Purification of hearts from zebrafish embryos. Biotechniques 40:274–, 276, 278 passim. [PubMed] [Google Scholar]
- Carney SA, Chen J, Burns CG, Xiong KM, Peterson RE, Heideman W. (2006a) Aryl hydrocarbon receptor activation produces heart-specific transcriptional and toxic responses in developing zebrafish. Mol Pharmacol 70:549–561 [DOI] [PubMed] [Google Scholar]
- Carney SA, Prasch AL, Heideman W and Peterson RE (2006b) Understanding dioxin developmental toxicity using the zebrafish model. Birth Defects Res A Clin Mol Teratol 76:7–18. [DOI] [PubMed]
- Chang CY, Puga A. (1998) Constitutive activation of the aromatic hydrocarbon receptor. Mol Cell Biol 18:525–535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang EF, Pai CI, Wyatt M, Yan YL, Postlethwait J, Chung B. (2001) Two sox9 genes on duplicated zebrafish chromosomes: expression of similar transcription activators in distinct sites. Dev Biol 231:149–163 [DOI] [PubMed] [Google Scholar]
- Cook PM, Robbins JA, Endicott DD, Lodge KB, Guiney PD, Walker MK, Zabel EW, Peterson RE. (2003) Effects of aryl hydrocarbon receptor-mediated early life stage toxicity on lake trout populations in Lake Ontario during the 20th century. Environ Sci Technol 37:3864–3877 [DOI] [PubMed] [Google Scholar]
- Cross LM, Cook MA, Lin S, Chen JN, Rubinstein AL. (2003) Rapid analysis of angiogenesis drugs in a live fluorescent zebrafish assay. Arterioscler Thromb Vasc Biol 23:911–912 [DOI] [PubMed] [Google Scholar]
- Heideman W, Antkiewicz DS, Carney SA, Peterson RE. (2005) Zebrafish and cardiac toxicology. Cardiovasc Toxicol 5:203–214 [DOI] [PubMed] [Google Scholar]
- Henry TR, Spitsbergen JM, Hornung MW, Abnet CC, Peterson RE. (1997) Early life stage toxicity of 2,3,7,8-tetrachlorodibenzo-p-dioxin in zebrafish (Danio rerio). Toxicol Appl Pharmacol 142:56–68 [DOI] [PubMed] [Google Scholar]
- Hofsteen P, Mehta V, Kim MS, Peterson RE, Heideman W. (2013) TCDD inhibits heart regeneration in adult zebrafish. Toxicol Sci 132:211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston CS, Opitz JM, Spranger JW, Macpherson RI, Reed MH, Gilbert EF, Herrmann J, Schinzel A. (1983) The campomelic syndrome: review, report of 17 cases, and follow-up on the currently 17-year-old boy first reported by Maroteaux et al in 1971. Am J Med Genet 15:3–28 [DOI] [PubMed] [Google Scholar]
- Ishimura R, Kawakami T, Ohsako S, Nohara K, Tohyama C. (2006) Suppressive effect of 2,3,7,8-tetrachlorodibenzo-p-dioxin on vascular remodeling that takes place in the normal labyrinth zone of rat placenta during late gestation. Toxicol Sci 91:265–274 [DOI] [PubMed] [Google Scholar]
- Ivnitski-Steele ID, Friggens M, Chavez M and Walker MK (2005) 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) inhibition of coronary vasculogenesis is mediated, in part, by reduced responsiveness to endogenous angiogenic stimuli, including vascular endothelial growth factor A (VEGF-A). Birth Defects Res A Clin Mol Teratol 73:440–446. [DOI] [PubMed]
- Keegan BR, Feldman JL, Lee DH, Koos DS, Ho RK, Stainier DY, Yelon D. (2002) The elongation factors Pandora/Spt6 and Foggy/Spt5 promote transcription in the zebrafish embryo. Development 129:1623–1632 [DOI] [PubMed] [Google Scholar]
- Kikuchi K, Gupta V, Wang J, Holdway JE, Wills AA, Fang Y, Poss KD. (2011) tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 138:2895–2902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- King Heiden TC, Spitsbergen J, Heideman W, Peterson RE. (2009) Persistent adverse effects on health and reproduction caused by exposure of zebrafish to 2,3,7,8-tetrachlorodibenzo-p-dioxin during early development and gonad differentiation. Toxicol Sci 109:75–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota A, Stegeman JJ, Woodin BR, Iwanaga T, Harano R, Peterson RE, Hiraga T, Teraoka H. (2011) Role of zebrafish cytochrome P450 CYP1C genes in the reduced mesencephalic vein blood flow caused by activation of AHR2. Toxicol Appl Pharmacol 253:244–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanham KA, Peterson RE, Heideman W. (2012) Sensitivity to dioxin decreases as zebrafish mature. Toxicol Sci 127:360–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG, Poss KD. (2006) A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127:607–619 [DOI] [PubMed] [Google Scholar]
- Lie-Venema H, van den Akker NM, Bax NA, Winter EM, Maas S, Kekarainen T, Hoeben RC, deRuiter MC, Poelmann RE, Gittenberger-de Groot AC. (2007) Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. ScientificWorldJournal 7:1777–1798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie-Venema H, Eralp I, Markwald RR, van den Akker NM, Wijffels MC, Kolditz DP, van der Laarse A, Schalij MJ, Poelmann RE, Bogers AJ, et al. (2008) Periostin expression by epicardium-derived cells is involved in the development of the atrioventricular valves and fibrous heart skeleton. Differentiation 76:809–819 [DOI] [PubMed] [Google Scholar]
- Lincoln J, Alfieri CM, Yutzey KE. (2004) Development of heart valve leaflets and supporting apparatus in chicken and mouse embryos. Dev Dyn 230:239–250 [DOI] [PubMed] [Google Scholar]
- Lincoln J, Kist R, Scherer G, Yutzey KE. (2007) Sox9 is required for precursor cell expansion and extracellular matrix organization during mouse heart valve development. Dev Biol 305:120–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Stainier DY. (2010) Tbx5 and Bmp signaling are essential for proepicardium specification in zebrafish. Circ Res 106:1818–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta V, Peterson RE, Heideman W. (2008) 2,3,7,8-Tetrachlorodibenzo-p-dioxin exposure prevents cardiac valve formation in developing zebrafish. Toxicol Sci 104:303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero JA, Giron B, Arrechedera H, Cheng YC, Scotting P, Chimal-Monroy J, Garcia-Porrero JA, Hurle JM. (2002) Expression of Sox8, Sox9 and Sox10 in the developing valves and autonomic nerves of the embryonic heart. Mech Dev 118:199–202 [DOI] [PubMed] [Google Scholar]
- Olivey HE, Svensson EC. (2010) Epicardial-myocardial signaling directing coronary vasculogenesis. Circ Res 106:818–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plavicki J, Hofsteen P, Peterson RE, Heideman W. (2013) Dioxin inhibits zebrafish epicardium and proepicardium development. Toxicol Sci 131:558–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon KL, Liebling M, Kondrychyn I, Garcia-Lecea M, Korzh V. (2010) Zebrafish cardiac enhancer trap lines: new tools for in vivo studies of cardiovascular development and disease. Dev Dyn 239:914–926 [DOI] [PubMed] [Google Scholar]
- Prasch AL, Teraoka H, Carney SA, Dong W, Hiraga T, Stegeman JJ, Heideman W, Peterson RE. (2003) Aryl hydrocarbon receptor 2 mediates 2,3,7,8-tetrachlorodibenzo-p-dioxin developmental toxicity in zebrafish. Toxicol Sci 76:138–150 [DOI] [PubMed] [Google Scholar]
- Rahkonen O, Savontaus M, Abdelwahid E, Vuorio E, Jokinen E. (2003) Expression patterns of cartilage collagens and Sox9 during mouse heart development. Histochem Cell Biol 120:103–110 [DOI] [PubMed] [Google Scholar]
- Schmidt JV, Bradfield CA. (1996) Ah receptor signaling pathways. Annu Rev Cell Dev Biol 12:55–89 [DOI] [PubMed] [Google Scholar]
- Serluca FC. (2008) Development of the proepicardial organ in the zebrafish. Dev Biol 315:18–27 [DOI] [PubMed] [Google Scholar]
- Smith CL, Baek ST, Sung CY, Tallquist MD. (2011) Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ Res 108:e15–e26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson EC. (2010) Deciphering the signals specifying the proepicardium. Circ Res 106:1789–1790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teraoka H, Ogawa A, Kubota A, Stegeman JJ, Peterson RE, Hiraga T. (2010) Malformation of certain brain blood vessels caused by TCDD activation of Ahr2/Arnt1 signaling in developing zebrafish. Aquat Toxicol 99:241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong KM, Peterson RE, Heideman W. (2008) AHR-Mediated Downregulation of Sox9b Causes Jaw Malformation in Zebrafish Embryos. Mol Pharmacol 74:1544–1553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan YL, Willoughby J, Liu D, Crump JG, Wilson C, Miller CT, Singer A, Kimmel C, Westerfield M, Postlethwait JH. (2005) A pair of Sox: distinct and overlapping functions of zebrafish sox9 co-orthologs in craniofacial and pectoral fin development. Development 132:1069–1083 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.