

Abstract
The present study explored the neuroprotective effect of Coeloglossum viride var. bracteatum extract (CE) against oxidative stress in rat cortical neurons. The results demonstrated that administration of CE inhibited hydrogen peroxide-induced neurotoxicity tested by MTT, LDH release, and TUNEL assays. We further found that CE inhibited the activation of caspase-3 (Csp3) induced by hydrogen peroxide. Moreover, CE was found to reverse the hydrogen peroxide-induced downregulation of active AKT and Bcl-2. We then showed that the neuroprotective effect of CE was blocked by adding the AKT inhibitor, Ly294002. Thus, our data strongly indicated that CE played a neuroprotective role against oxidative stress-induced neurotoxicity.
1. Introduction
Coeloglossum viride var. bracteatum extract (CE) is extracted from a plant called Coeloglossum viride var. bracteatum, an orchidaceae family plant. CE is widely used as a traditional Chinese medicine in the Northwest of China, especially in Tibet, Qinghai, Gansu Inner Mongolia, and Shanxi provinces [1]. It has been described as a traditional Chinese medicine; CE increases body fluid production and vital energy is beneficiary to memory and tranquilization [1]. In 2004, CE has been identified as a mixture of four compounds [2]. More recently, CE has been shown to rescue learning and memory deficit induced by scopolamine in rodents [3].
Alzheimer's Disease (AD) is a progressive neurodegenerative disease characterized by extracellular senile plaques composed of amyloid beta and intracellular neurofibrillary tangles (NFTs) [4]. Neuronal loss in AD is thought to be contributed by amyloid beta toxicity [5], and growing evidence suggests that the amyloid beta toxicity is mediated by oxidative damage in neurons [4]. Numerous studies suggested that oxidative stress plays a major role in the pathogenesis of AD. It has been observed that oxidative stress is increased in brains from patients with AD [6]. Moreover, reduction of antioxidant enzyme activity including superoxide dismutase (SOD) and catalase has been reported in brains from patients with AD [7, 8]. In addition, deficiency of superoxide dismutase D1 (Sod1) in a mouse model of AD resulted in accelerated amyloid beta plaque formation and memory deficit, and the phenomena were mediated by oxidative damage [9]. Interestingly, a recent study found that an antioxidant MitoQ blocked increased oxidative stress, increased amyloid beta plaque formation, and elevated caspase activity and synaptic loss in an animal model of AD [10]. Thus oxidative stress is a promising therapeutic target for treating AD.
In our present study, we investigated the role and mechanism of action of CE in neuroprotection against oxidative stress. Our results for the first time suggested that CE might activate AKT signaling pathway to upregulate the expression of the antiapoptotic protein Bcl-2 to mediate neuroprotection during stress.
2. Material and Methods
2.1. Primary Cortical Neuronal Culture and Treatments
The cortical neuronal cultures were prepared as described previously. Tissues were dissected from the whole brain tissues of newborn SD rats (Experimental Animal Center of Peking University Health Science Center, Beijing, China) in Dulbecco's modified Eagle's medium (DMEM) (Invitrogen). The tissues were dissociated mechanically and then incubated with 0.25% trypsin (Invitrogen) for 30 minutes at 37°C. The mixture was then filtered through a nylon mesh to obtain homogenous suspension. After filtering the mixture through 70 μm sterilized filters, the flow-through was centrifuged to pellet cells. Cells were then resuspended in DMEM with 10% fetal bovine serum (FBS), 2 g/L HEPES, penicillin G (100 U/mL), and 100 ug/mL streptomycin (all from Invitrogen). Cells were plated in poly-L-lysine-coated plates or coverslips and maintained in a humidified incubator with 5% CO2 and 95% O2 at 37°C. 10 μM cytosine arabinoside (Sigma) was supplemented after plating for 2–4 days to inhibit glia cell growth. Cells were treated after 7-8 days in culture. CE (a gift from Dr. Li Tang, Department of Pharmacology, Minzu University of China) and AKT inhibitor Ly294002 (cell signaling) were added freshly into culture medium during treatments.
2.2. Measurement of Cell Death: MTT Assay, LDH Release Assay, and TUNNEL Assay
The viability of cells after various treatments was estimated by their ability to reduce the dye methyl thiazolyl tetrazolium (MTT, Sigma) to blue formazan crystal. Cells cultured in 96-well plate for 7 days were gently washed with 0.01 M PBS. After wash, 90 μL of medium with 10 μL of 5 mg/mL MTT solution was added to each well and the plate was maintained at 37°C for 2–4 hours. Then the reactions were dissolved in DMSO for quantification by measuring the absorption at 570 nm using a microplate spectrophotometer (Bio-Rad), representing relative cell viability.
The cytotoxicity of cells after various treatments was evaluated by lactate dehydrogenase (LDH) release. This was achieved with a CytoTox 96 Non-Radioactive Cytotoxicity Assay kit according to the manufacturer's instructions (Promega).
Cell death was examined after treatments by fixing cells in fresh 4% paraformaldehyde and 4% sucrose in PBS for 20 minutes at room temperature and permeabilized in 0.1% triton X-100 and 0.1% sodium citrate in PBS for 2 minutes on ice. Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining was performed using the in situ cell death detection kit I as described by the manufacturer (Roche). The coverslips were then washed once in distilled water for 5 minutes and mounted on glass slides to be observed under a fluorescence microscope. The percentage of cell death was determined by the ratio of the number of TUNEL-positive cells over the total of 100 cells in one count. The average of 5 counts was calculated as the percentage of neuronal cell death in a certain treatment.
2.3. Western Blots
The neurons cultured in 6-well plates were washed three times with 0.01 M PBS; 100 μL of cell lysis buffer with 1% phenylmethanesulfonyl fluoride (PMSF) was added into each well and cells were harvested with cell scrapers. The extracts were iced for 30 minutes and centrifuged at 14,800 g for 15 minutes, and the supernatant was harvested. Denatured protein samples diluted with loading buffer were loaded equally to each lane and separated by 10% SDS-PAGE and then blotted onto a polyvinylidene fluoride (PVDF, Millipore) membrane. The membrane was then incubated for 1 hour in blocking buffer (tris-buffered saline containing 5% no-fat milk powder) at room temperature. The membrane was incubated at 4°C with the primary antibodies, washed with trisbuffered saline Tween-20 (TBST), and incubated again with horseradish peroxidase (HRP)-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) followed by washing. The primary antibodies used include purified polyclonal anti-β-actin antibody (Santa Cruz), monoclonal antiactivated Csp3 antibody (cell signaling) and polyclonal anti-Bcl-2 antibody (Santa Cruz), and monoclonal anti-p-AKT antibody (cell signaling). Immunoblots were developed in the presence of enhanced chemiluminescence reagents, and the images detected in X-ray films were quantified by densitometric scanning using Gel Imaging Analysis System Gel-Pro 4400 (Media Cybernetics).
2.4. Statistical Evaluation
All data are presented as means ± S.E.M. Statistical significance (**P < 0.01 or ***P < 0.001) among groups was determined by two-tailed Student's t-test.
3. Results
To explore the neuroprotective effect of CE in primary cultured cortical neurons, we treated the cortical neurons with vehicle, 0.1 mg/mL CE, 1 mg/mL CE, or 10 mg/mL CE in the absence or presence of 100 μM H2O2. As shown in Figure 1(a), the cell viability of cultured cortical neurons significantly decreased after 24 h incubation with H2O2 compared to the control group as tested by MTT assay; however, 1 mg/mL CE or 10 mg/mL CE significantly inhibited the H2O2 induced decrease of cell viability, while CE alone had no marked influence on the cell viability in primary cultured cortical neurons (Figure 1(a)).
Figure 1.
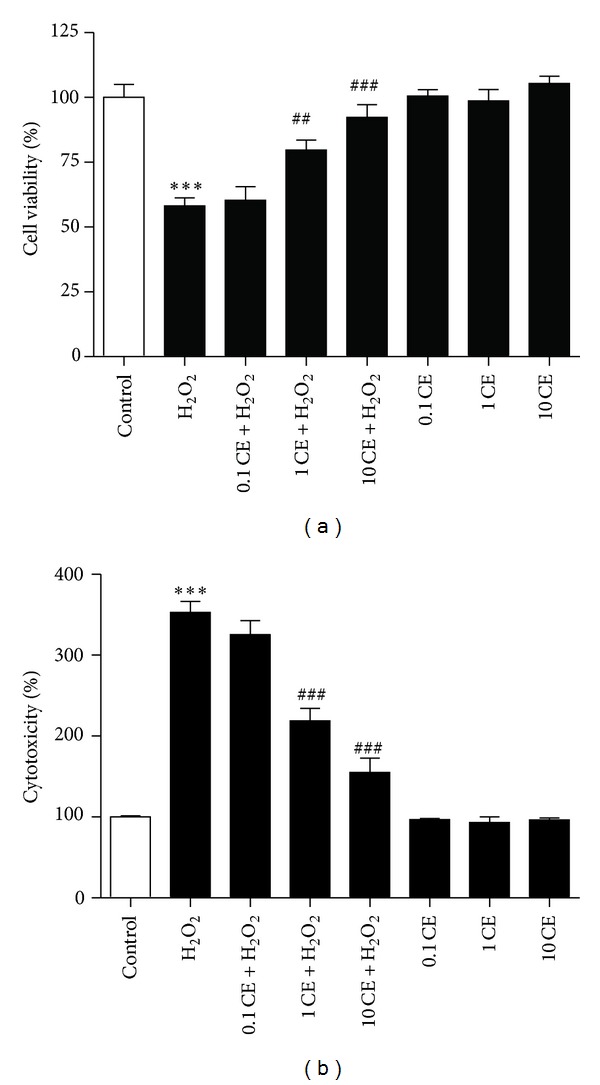
Protective effects of CE against oxidative stress in primary cultured cortical neurons. (a) Bar graphs showing the reduced cell viability after H2O2 treatment that was significantly attenuated by the treatment of the neurons with 1 mg/mL and 10 mg/mL CE as tested by MTT assay. (b) Bar graphs showing the H2O2 induced cytotoxicity that was significantly reduced by the treatment of the neurons with 1 mg/mL and 10 mg/mL CE as tested by LDH release assay. ∗ refers to a comparison of the control group and # refers to a comparison of the H2O2 group.
To confirm the neuroprotective effect of CE against oxidative stress, LDH release assay was used to measure the cytotoxicity. Results showed that H2O2 significantly increased the cytotoxicity in the neurons compared to the control group; however, treatment with 1 mg/mL CE or 10 mg/mL CE significantly reduced the cytotoxicity induced by H2O2, while CE alone had no marked influence on the cytotoxicity in the neurons (Figure 1(b)).
Since the above results suggested that the neuroprotective effect of CE peaks at 10 mg/mL CE, this concentration was used in all subsequent experiments. Results from TUNEL assay further confirmed the neuroprotective effect of CE in the primary cultured cortical neurons. As shown in Figure 2, the number of died cortical neurons significantly increased after treatment with H2O2 compared to the control group; the number of died cells significantly reduced by incubation of the neurons with CE. Moreover, CE alone had no marked influence on the cell death of the neurons.
Figure 2.
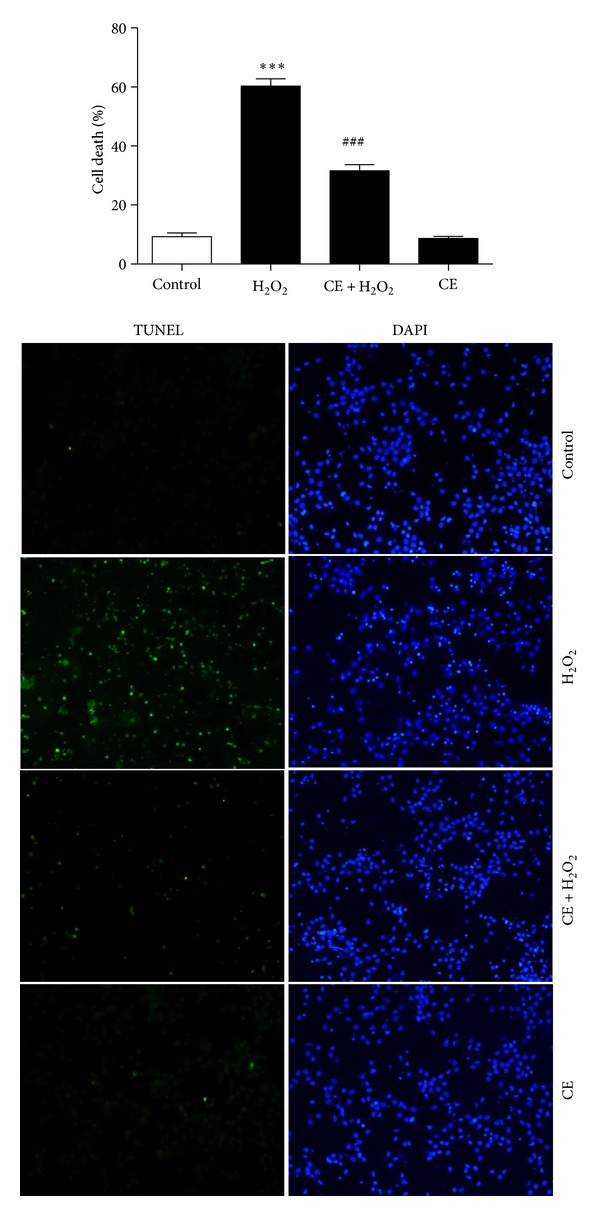
Photomicrographs and histograms showing that treatment with CE reduced H2O2 induced cell death assessed by TUNEL assay. ∗ refers to a comparison of the control group and # refers to a comparison of the H2O2 group.
To investigate the mechanism underlying the neuroprotective effect of CE, we tested the expression of apoptotic or antiapoptotic proteins kinase B (AKT), Bcl-2, and Csp3 after different treatments. Results from western blot showed that the phosphorylation of AKT was decreased significantly after H2O2 treatment compared to the control group; however, incubation of the neurons with CE reversed the downregulation of phosphorylated AKT induced by H2O2 (Figure 3(a)). Moreover, as shown in Figure 4(a), the antiapoptotic protein Bcl-2 was significantly decreased in the cortical neurons treated with H2O2 compared to the control group; however, incubation with CE significantly inhibited the H2O2 induced decrease of Bcl-2 in the neurons compared to the H2O2 treated group. Furthermore, we found that the H2O2 induced activation of Csp3 was blocked by incubation with CE.
Figure 3.
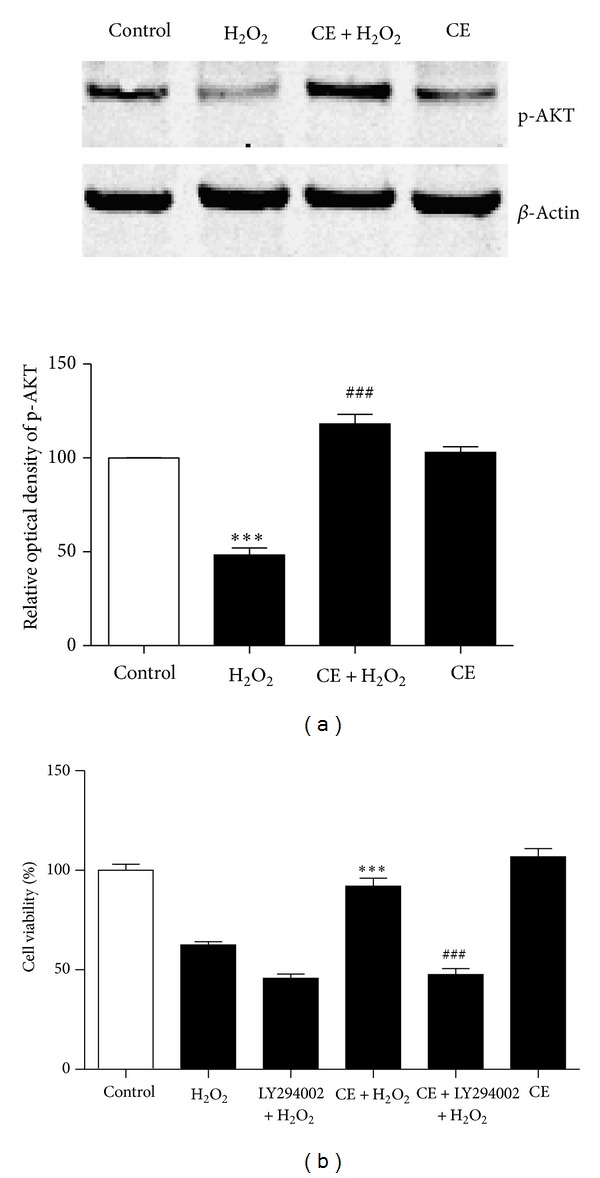
AKT signaling pathway is required for the neuroprotective effect of CE. (a) representative western blot probed with an antibody specific against p-AKT after various treatments in the neurons. Actin served as an internal control. Summary of the optical density of p-AKT. Results showed that CE significantly inhibited H2O2 induced decreases in the content of p-AKT. ∗ refers to a comparison of the control group; # refers to a comparison of the H2O2 group. (b) Bar graphs showing that the neuroprotective effect of CE was blocked by AKT inhibitor Ly294002, suggesting that AKT signaling pathway mediated the neuroprotective effect of CE in primary cultured cortical neurons.
Figure 4.
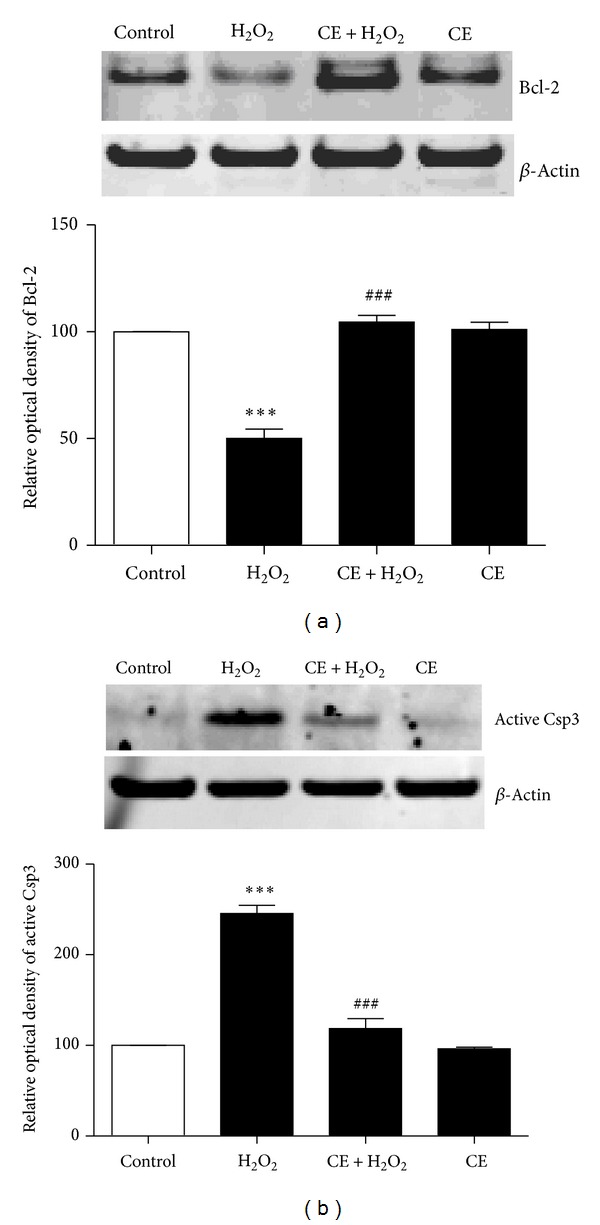
Neuroprotection by CE involves Bcl-2 and Csp3. (a) Representative western blot probed with an antibody specific against Bcl-2 after various treatments in the neurons. β-Actin served as an internal control. Summary of the optical density of Bcl-2. Results showed that CE significantly inhibited H2O2 induced decreases in the content of Bcl-2. (b) Representative western blot probed with an antibody specific against active Csp3 after Csp3 various treatments in the neurons. Actin served as an internal control. Summary of the optical density of active Csp3. Results showed that H2O2-induced activation of Csp3 was blocked by CE. ∗ refers to a comparison of the control group; # refers to a comparison of the H2O2 group.
To confirm the regulation of the AKT required for the neuroprotective effect of CE, we use the AKT inhibitor, Ly294002. Western blot showed that the phosphorylation of AKT was abolished after incubation with Ly294002 in the neurons (data not shown), demonstrating the specificity of the inhibitor. Next, the primary cultured cortical neurons were treated with vehicle or 10 μM Ly294002 for 30 min. Subsequently, H2O2 was added to the neurons in the absence or presence of CE for 24 h. As shown in Figure 3(a), Ly294002 completely blocked the neuroprotective effect of CE against oxidative stress in the cortical neurons.
4. Discussion
Neurodegenerative diseases are a wide class of hereditary and sporadic conditions characterized by progressive nervous system dysfunction. These disorders include Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease (HD), and prion diseases which are caused by a combination of genetic and environmental factors. With more than 200 billion dollars for studying and treating AD alone globally per year, neurodegenerative diseases remain without curative or preventive treatments, and the available therapies provide only symptom improvements. Recently, evidence is emerging that natural products are promising targets to treat neurodegenerative diseases such as AD and PD with less side effects. For example, studies have demonstrated that curcumin and green tea polyphenols protected against amyloid beta-induced neurotoxicity in vitro [11–13]. Moreover, curcumin suppressed oxidative damage and blocked cognitive deficits in an animal model of AD [14]. Here we demonstrated that CE protected against hydrogen peroxide-induced cell death in primary cultured neurons. This is consistent with our previous findings that CE inhibited amyloid beta-induced neurotoxicity. Moreover, CE rescued ischemia-induced neuronal death and cognitive impairment in rats [15]. Thus our data support the hypothesis that oxidative stress plays an important role in mediating amyloid beta-induced neuropathology, memory deficits, and the progression of AD.
AKT signaling pathway is a major pathway for survival and neuroprotection [16]. It has been reported that the p-AKT levels are decreased in brains from patients with AD and an animal model of AD [17, 18]. Here we showed that oxidative stress decreased the level of phosphorylated AKT and CE reversed the oxidative stress-induced downregulation of active AKT, which has antiapoptotic property. More importantly, we found that AKT inhibitor blocked the neuroprotective effect of CE against oxidative stress in primary cultured cortical neurons, suggesting the neuroprotective effect of CE was mediated by AKT signaling pathway. Hence our results are consistent with a role for AKT signaling pathway in stress and the progression of AD.
Bcl-2 family is a large family of proteins with pro- or antiapoptotic properties. These include Bax which is a proapoptotic protein and Bcl-2 which is an antiapoptotic protein. Upon apoptosis, Bax undergoes a conformational change which exposes its hydrophobic C terminal; then it is oligomerized and translocated to mitochondria from cytosol to form pores, which then leads to the release of cytochrome c and the activation of caspase, eventually leading to cell death [19, 20]. In contrast, Bcl-2 interacts with Bax to prevent the pore formation in mitochondria to mediate survival and neuroprotection [21]. Given the important role of Bcl-2 (Bax) to caspase signaling pathway in apoptosis, it is not surprising to find that the expression of Bcl-2 was altered in brains from patients with AD [22]. Furthermore, Csp3 has been observed to be increased and enriched in postsynaptic densities in AD [23]. Interestingly, a study found that the inhibition of Csp3 activity rescued the synaptic failure and cognitive dysfunction in an animal model of AD [24]. Here we demonstrated that the decrease in Bcl-2 protein caused by oxidative stress was prevented by treatment with CE. Moreover, our results showed that the activation of Csp3 caused by oxidative stress was blocked by treatment with CE. Based on our new findings, CE could be a potential therapeutic target for AD. Thus continued investigation into the function of CE is warranted.
Authors' Contribution
Zhe Guo and Rui-Yuan Pan contributed equally to this work.
Acknowledgments
The authors thank the support from Minzu University 985 fund (MUC98504-14), the Fundamental Research Funds for the Central Universities (1112KYZY41), and 111 Project (B08044).
References
- 1.Zhang D, Liu G, Shi J, Zhang J. Coeloglossum viride var. bracteatum extract attenuates D-galactose and NaNO2 induced memory impairment in mice. Journal of Ethnopharmacology. 2006;104(1-2):250–256. doi: 10.1016/j.jep.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 2.Huang S-Y, Li G-Q, Shi J-G, Mo S-Y. Chemical constituents of the rhizomes of Coeloglossum viride var. bracteatum. Journal of Asian Natural Products Research. 2004;6(1):49–61. doi: 10.1080/1028602031000119826. [DOI] [PubMed] [Google Scholar]
- 3.Li M, Wang Y-F, Ma B, Liu G-T, Zhang J-J. Effect and mechanism of Coeloglossum viride var. bracteatum extract on scopolamine-induced deficits of learning and memory behavior of rodents. Acta Pharmaceutica Sinica. 2009;44(5):468–472. [PubMed] [Google Scholar]
- 4.Quintanilla RA, Orellana JA, von Bernhardi R. Understanding risk factors for Alzheimer's disease: interplay of neuroinflammation, connexin-based communication and oxidative stress. Archives of Medical Research. 2012;43:632–644. doi: 10.1016/j.arcmed.2012.10.016. [DOI] [PubMed] [Google Scholar]
- 5.Cheng Y, Yu L-C. Galanin protects amyloid-β-induced neurotoxicity on primary cultured hippocampal neurons of rats. Journal of Alzheimer’s Disease. 2010;20(4):1143–1157. doi: 10.3233/JAD-2010-091234. [DOI] [PubMed] [Google Scholar]
- 6.Mecocci P, MacGarvey U, Beal MF. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Annals of Neurology. 1994;36(5):747–751. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- 7.Furuta A, Price DL, Pardo CA, et al. Localization of superoxide dismutases in Alzheimer’s disease and Down’s syndrome neocortex and hippocampus. The American Journal of Pathology. 1995;146(2):357–367. [PMC free article] [PubMed] [Google Scholar]
- 8.Omar RA, Chyan Y-J, Andorn AC, Poeggeler B, Robakis NK, Pappolla MA. Increased expression but reduced activity of antioxidant enzymes in Alzheimer’s disease. Journal of Alzheimer’s Disease. 1999;1(3):139–145. doi: 10.3233/jad-1999-1301. [DOI] [PubMed] [Google Scholar]
- 9.Murakami K, Murata N, Noda Y, et al. SOD1 (copper/zinc superoxide dismutase) deficiency drives amyloid β protein oligomerization and memory loss in mouse model of Alzheimer disease. The Journal of Biological Chemistry. 2011;286(52):44557–44568. doi: 10.1074/jbc.M111.279208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant mitoq prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. The Journal of Neuroscience. 2011;31(44):15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qin X-Y, Cheng Y, Cui J, Zhang Y, Yu L-C. Potential protection of curcumin against amyloid β-induced toxicity on cultured rat prefrontal cortical neurons. Neuroscience Letters. 2009;463(2):158–161. doi: 10.1016/j.neulet.2009.07.047. [DOI] [PubMed] [Google Scholar]
- 12.Qin X-Y, Cheng Y, Yu L-C. Potential protection of curcumin against intracellular amyloid β-induced toxicity in cultured rat prefrontal cortical neurons. Neuroscience Letters. 2010;480(1):21–24. doi: 10.1016/j.neulet.2010.05.062. [DOI] [PubMed] [Google Scholar]
- 13.Qin X-Y, Cheng Y, Yu L-C. Potential protection of green tea polyphenols against intracellular amyloid beta-induced toxicity on primary cultured prefrontal cortical neurons of rats. Neuroscience Letters. 2012;513(2):170–173. doi: 10.1016/j.neulet.2012.02.029. [DOI] [PubMed] [Google Scholar]
- 14.Frautschy SA, Hu W, Kim P, et al. Phenolic anti-inflammatory antioxidant reversal of Aβ-induced cognitive deficits and neuropathology. Neurobiology of Aging. 2001;22(6):993–1005. doi: 10.1016/s0197-4580(01)00300-1. [DOI] [PubMed] [Google Scholar]
- 15.Ma B, Li M, Nong H, Shi J, Liu G, Zhang J. Protective effects of extract of Coeloglossum viride var. bracteatum on ischemia-induced neuronal death and cognitive impairment in rats. Behavioural Pharmacology. 2008;19(4):325–333. doi: 10.1097/FBP.0b013e3282feb0ac. [DOI] [PubMed] [Google Scholar]
- 16.Cheng Y, Cawley NX, Loh YP. Carboxypeptidase E/NFalpha1: a new neurotrophic factor against oxidative stress-induced apoptotic cell death mediated by ERK and PI3-K/AKT pathways. PloS ONE. 2013;8 doi: 10.1371/journal.pone.0071578.e71578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee H-K, Kumar P, Fu Q, Rosen KM, Querfurth HW. The insulin/Akt signaling pathway is targeted by intracellular β-amyloid. Molecular Biology of the Cell. 2009;20(5):1533–1544. doi: 10.1091/mbc.E08-07-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Magrané J, Rosen KM, Smith RC, Walsh K, Gouras GK, Querfurth HW. Intraneuronal β-amyloid expression downregulates the Akt survival pathway and blunts the stress response. The Journal of Neuroscience. 2005;25(47):10960–10969. doi: 10.1523/JNEUROSCI.1723-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deng Y, Wu X. Peg3/Pw1 promotes p53-mediated apoptosis by inducing Bax translocation from cytosol to mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2000;97(22):12050–12055. doi: 10.1073/pnas.97.22.12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goping IS, Gross A, Lavoie JN, et al. Regulated targeting of BAX to mitochondria. The Journal of Cell Biology. 1998;143(1):207–215. doi: 10.1083/jcb.143.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends in Cell Biology. 2008;18(4):157–164. doi: 10.1016/j.tcb.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kitamura Y, Shimohama S, Kamoshima W, et al. Alteration of proteins regulating apoptosis, Bcl-2, Bcl-x, Bax, Bak, Bad, ICH-1 and CPP32, in Alzheimer’s disease. Brain Research. 1998;780(2):260–269. doi: 10.1016/s0006-8993(97)01202-x. [DOI] [PubMed] [Google Scholar]
- 23.Louneva N, Cohen JW, Han L-Y, et al. Caspase-3 is enriched in postsynaptic densities and increased in Alzheimer’s disease. The American Journal of Pathology. 2008;173(5):1488–1495. doi: 10.2353/ajpath.2008.080434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.D’Amelio M, Cavallucci V, Middei S, et al. Caspase-3 triggers early synaptic dysfunction in a mouse model of Alzheimer’s disease. Nature Neuroscience. 2011;14(1):69–79. doi: 10.1038/nn.2709. [DOI] [PubMed] [Google Scholar]