

Abstract
Type 2 diabetes mellitus (T2DM) is a multifactorial disease, and its aetiology involves a complex interplay between genetic, epigenetic, and environmental factors. In recent years, evidences from both human and animal experiments have correlated early life factors with programming diabetes risk in adult life. Fetal and neonatal period is crucial for organ development. Many maternal factors during pregnancy may increase the risk of diabetes of offsprings in later life, which include malnutrition, healthy (hyperglycemia and obesity), behavior (smoking, drinking, and junk food diet), hormone administration, and even stress. In neonates, catch-up growth, lactation, glucocorticoids administration, and stress have all been found to increase the risk of insulin resistance or T2DM. Unfavorable environments (socioeconomic situation and famine) or obesity also has long-term negative effects on children by causing increased susceptibility to T2DM in adults. We also address the potential mechanisms that may underlie the developmental programming of T2DM. Therefore, it might be possible to prevent or delay the risk for T2DM by improving pre- and/or postnatal factors.
1. Introduction
Type 2 diabetes (T2DM) is a metabolic disease caused by genetic and multiple environmental factors. Epidemical and experimental studies have found that detrimental early life factors may predispose high incidence of cardiovascular disease and metabolic diseases in later life, which is also termed as “barker hypothesis.” Organs are under development and functional maturation from fetal stage to childhood; disturbance of the homeostasis during crucial periods might predispose increased risk of insulin resistance and even T2DM in late life.
2. Part I: Prenatal Factors (Figure 1)
Figure 1.
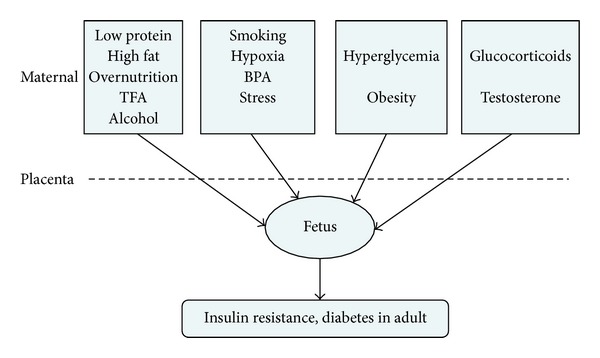
Prenatal factors mentioned in recent years that might correlate with insulin resistance and/or T2DM. Data from human and animal studies have shown that malnutrition or overnutrition, metabolic disorders, exposure to hypoxia, some chemicals and hormones, and unhealthy lifestyle such as smoking and alcohol drinking during pregnancy might predispose detrimental long-term effects on offspring, leading to increased risk of insulin resistance or T2DM. TFA: transfatty acids; BPA: biophenol A.
2.1. Diet and Nutrition
It has been suggested that the quality and quantity of the nutrition during pregnancy may cause strong and permanent effects on the fetus. The altered structure of chromosome during this procedure might be the cause of cell dysfunction and increased susceptibility to diseases through altered gene expression [1].
2.1.1. Malnutrition and Low Protein Diet
The associations between maternal malnutrition, low protein diet, and T2DM have been widely studied. Typical epidemical studies from the population born during the Dutch famine period [2] or in some poor countries [3] have found that those who had been exposed to maternal malnutrition may have increased morbidity of metabolic diseases including T2DM in adult life.
The mechanisms responsible for the prenatal malnutrition programming insulin resistance or T2DM remain unclear. Orozco-Solís et al. [4] have found that low protein diet during pregnancy and lactating may cause permanent altered hypothalamic expression of genes in rat offspring involved in insulin signaling and lipid and glucose metabolism, which may programme metabolic diseases.
In addition, the effect of low protein diet during pregnancy on postnatal β cell has also been noticed recently. Increased oxidative stress and fibrosis [5], decreased HNF4a expression with increased DNA methylation in P2 promoters [6], defected mitochondriogenesis and mitochondria dysfunction [7], and increased cell differentiation instead of proliferation [8] were found in β cell of adult animal offspring whose mothers were under low protein diet during pregnancy. These may cause β-cell dysfunction and consequently increase the incidence of T2DM in postnatal life (Figure 2).
Figure 2.
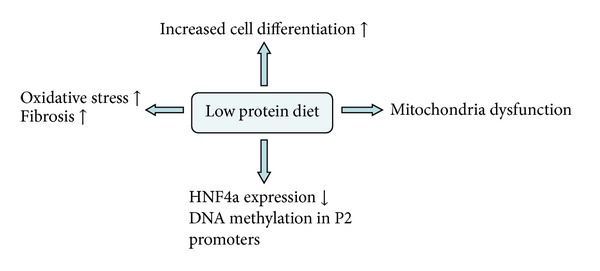
The effect of low protein diet during pregnancy on postnatal β cell. Low protein diet during pregnancy may lead to increased oxidative stress, fibrosis, decreased HNF4a expression, defected mitochondriogenesis, and mitochondria dysfunction, and increased cell differentiation instead of proliferation was found in β cell of adult animal offspring, which may participate in β-cell dysfunction and consequently increase the incidence of T2DM.
2.1.2. Overnutrition
High Protein. A study from Maurer and Reimer [9] in Wistar rats has found that high protein diet during pregnancy and lactating may cause increased resistin and IL-6 mRNA levels in brown fat tissue in 35-day-old offspring; both factors were included in the pathogenesis of insulin resistance [10, 11].
High Fat Diet. Both human and animal studies have identified that fat diet may cause obesity and insulin resistance [12, 13]. Intriguingly, the effect of high fat diet on metabolic disorders seemed to be programmative. The prenatal period is a key developmental window for nutrition status. Masuyama and Hiramatsu [14] found that mice offspring exposed to high fat diet during pregnancy developed insulin resistance and hyperlipidemia at 24 wks of age, which was associated with altered levels of leptin in adipose tissue. The experiment conducted in C57BL/6 mice by Liang et al. [15] has also showed that high saturated fatty acids diet during pregnancy led to insulin resistance, hyperglycemia in adult offspring under normal diet condition. The mechanisms underlying are still under investigation. Evidences from animal study have indicated that overexposure to high fat diet in utero may lead to elevated mRNA level of hypothalamic signal transducer and activator of transcription-3 and suppressor of cytokine signalling-3 in the offspring [16]. Both of these two factors are found to participate in obese and insulin resistance cases [17]. In addition, prenatal exposure to high saturated fats may cause increased hepatic phosphoenolpyruvate carboxykinase expression, fatty liver, reduced basal acetyl CoA carboxylase phosphorylation, and insulin signalling [18]. Impaired Wnt/β-catenin signaling pathway in skeletal muscle has also been found [19], which may also participate in pathogenesis of insulin resistance in adult life, since the insulin sensitivity can be improved by activating Wnt/β-catenin [20].
2.1.3. Transfatty Acids and Junk Food
Transfatty acids are unsaturated fatty acids that contain nonconjugated double bond in the trans-configuration. So far, data that correlated transfatty acids diet with insulin resistance or diabetes is weak and inconsistent [21–23]. However, it is still worth noting that prenatal exposure to transfatty acids might cause impaired insulin resistance and increased content of abdominal fat after birth [24]. No similar effects can be observed in mice exposed to transfatty acid during lactating.
Data about long-term effects of prenatal junk food taking is quite limited; experiment from Bayol et al. [25] has indicated that junk food taking during prenatal and lactating period may cause reduced insulin sensitivity in female offspring rats. More intensive studies still need to be performed for the convincing conclusion.
2.1.4. Alcohol
Studies have already correlated chronic alcohol intake with insulin resistance even T2DM [26, 27]. In the series of studies performed by Chen and Nyomba they have found that SD rats with alcohol intake (4 g/kg/day) during pregnancy may have hyperglycemia and reduced glucose transporter type 4 (GLUT4) content in muscle in adult offspring after a reduced birth weight and then catch up growth [28]. In addition, in this animal model, impaired inhibition effects of insulin on hepatic gene expression of phosphoenolpyruvate carboxykinase and peroxisome proliferator activated receptor gamma coactivator-1 mRNA [29] and reduced phosphorylation of protein kinase C zeta isoform [30] were exhibited in the offspring. Yao et al. [31] have found prenatal alcohol intake elevated expression of Tribbles 3 and phosphatase and tensin homolog deleted on chromosome 10 in both liver and muscle [32, 33], leading to impaired insulin sensitivity [34, 35]. In addition, increased 11beta-hydroxysteroid dehydrogenase type-1 level in liver and adipose tissue [36] may also partly contribute to the insulin resistance caused by prenatal alcohol taking though elevating local glucocorticoid levels.
2.2. Environmental Factors
2.2.1. Biophenol A
Biophenol A, a biochemical material used in plastic containers that are widely used in daily life [37], has been found that it may achieve similar effects with estrogen [38, 39]. Studies have supported that biophenol A might be correlated with the pathogenesis of T2DM [40, 41]. A human study performed by Lang et al. [42] has shown that the biophenol A concentration in urine positively correlated with cardiovascular diseases and diabetes.
In rats, 50 μg/kg·d biophenol A intake during pregnancy and lactating period may lead to insulin resistance in adult offspring, and this effect can be largely enhanced by high fat diet after birth [43]. Similar results have also been described by Alonso-Magdalena et al. [44] who further found that the altered Ca2+ signaling pathway and reduced cell numbers in pancreas might contribute to reduced insulin sensitivity. However, controversial conclusions have also been raised out by Ryan et al. [45] in CD-1 mice, which indicate that perinatal exposure to ecologically relevant dose of BPA could not impair the glucose tolerance in the offspring. Therefore, different biophenol A dosages applied in different animal models may vary the conclusion.
2.2.2. Maternal Hypoxia
Data from animal experiment has found that exposure to hypoxia during pregnancy leads to insulin resistance, impaired glucose homeostasis, and altered expression of genes involved in insulin-signaling pathways in the offspring [46]. Mechanisms underlying this relationship are unclear since intrauterine hypoxia may partly correlate with undernutrition. However, Camm et al. [47] found that, compared to prenatal undernutrition, prenatal hypoxia may cause different gene expression patterns in the liver and muscle in adult offspring, including reduced expression of hepatic insulin receptor substrate 1, phospho-Akt, and muscle Akt2, indicating that prenatal hypoxia may promote markers of insulin resistance independent of undernutrition.
2.2.3. Maternal Smoking
Studies have reported the unfavorable effects of smoking on diabetes in adult [48, 49]. However, a clearly causal relationship has only been found between maternal smoking and increased risk of T2DM in the offspring. A human study performed by Thiering et al. [50] had found increased insulin levels in 10-year-old children after prenatal smoking, and breast milk feeding made this alteration even more magnificent. This finding is consistent with the study performed previously by Bruin et al. [51] in animals which indicated that both conception and lactation periods were needed for nicotine exposure that may result in permanent β-cell loss and subsequent impaired glucose tolerance. In addition, Holloway et al. [52] found that fetal and neonatal exposure to nicotine has transgenerational effects and insulin resistance can be found in the F2 offspring. Exact mechanism still remains largely unknown; reduced pancreas cell numbers and size and reduced expression of β-cell marker genes such as pdx-1, Pax-1, and Nkx6.1 [53] all have been addressed. In addition, data from Chen et al. have indicated that nicotine may also downregulate gene expression of appetite regulators neuropeptide Y and pro-opiomelanocortin in the arcuate nucleus of the hypothalamus in fetal brain, which may consequently lead to unhealthy eating habits in the offspring and predispose high risk of obesity or diabetes [54].
2.3. Prenatal Psychological Stress
It is already known that exposure to high levels of maternal stress hormones during pregnancy may produce detrimental effects on the offspring [55]. The effect of prenatal stress in programming T2DM has been found in both human and animal studies [56–58]. A retrospective study has shown that children exposed to stress caused by bereavement during their prenatal life had more risk to T2DM later in life [57]. Another human data from Entringer et al. [58] found that maternal stressful life experiences may cause significantly elevated 2-hour insulin and C-peptide levels under glucose tolerance test in young adult offspring, indicating insulin resistance, which is independent of birth weight and family history of diabetes. The elucidation of the mechanism underlying this relationship is still not clear. A finding from a human study has shown that prenatal stress leads to shorter leukocyte telomere length in adult offspring [59], which has also been found to positively correlate with the pathogenesis diabetes [60] and children obese [61].
2.4. The Metabolic Situation during Pregnancy
2.4.1. Obesity
Maternal obesity has risen dramatically over the past 20 years. Evidences from human and animal studies suggest that maternal obesity in pregnancy predisposes hyperinsulinemia, insulin resistance, and T2DM in the offspring [62–65]. Shankar et al. [66] have found in mice that the male offspring with overweight mother may exhibit magnificent increase in body weight and adipose tissue content, which also combined with insulin resistance and increased levels of insulin, leptin, and resistin. The precise underlying mechanisms that contribute to increased susceptibility of offspring to develop insulin resistance in later life remain poorly understood. Both increased number of apoptosis of the fetal pancreas β cell [67] and accelerated fetal β-cell growth and cell proliferation (which was regarded as overload working and consequently end up to β cell failure) [68] were observed in animal offsprings with obese mother, which may all contribute to the increased blood glucose level after birth. In addition, increased hepatic lipogenesis and fatty liver disease [69, 70] found in the offspring exposed to maternal obesity also contribute to hepatic insulin resistance.
2.4.2. High Gestational Glucose Concentration
Exposure to elevated intrauterine glucose environment has been found to cause alterations in fetal growth patterns, which predispose these infants to developing obesity, insulin resistance, and diabetes later in life. So far the effects of intrauterine hyperglycemia on the offspring have been studied in human in pregnant mothers with T2DM or with gestational diabetes and in diabetic animal models mainly caused by streptozotocin treatment. Data accumulated from theses studies uniformly show glucose intolerance in the offspring. Human study performed by Boerschmann et al. [71] has indicated that, compared with those children with T1DM and normal glycemia mothers, children with mothers with gestational diabetes mellitus exhibit overweight and increased HOMA-IR. Another study from Bush et al. [72] in 5–10-year-old children also found that maternal gestational glucose concentration was inversely associated with offspring insulin sensitivity. Insulin resistance was also observed in rodent offspring prenatally under hyperglycemia environment caused by streptozotocin injection [73, 74]. It seems that in utero “diabetic” environment in which the fetus develops can increase the risk of diabetes in the child. In addition to genetic susceptibility, blunted insulin sensitivity in the offspring might largely contribute to this correlation. Relative gene expression was only explored in animal models which indicated that intrauterine hyperglycemia induced by streptozotocin injection resulted in increased hepatic gluconeogenic gene expression of glucose-6-phosphatase and phosphoenolpyruvate carboxykinase in the offspring [74] and the adult offspring of this cohort are prone to develop insulin resistance under high fat diet [73]. A human study found that maternal diabetes might cause an inherent defect in β-cell glucose sensitivity in the adult offspring [75].
2.5. Maternal Hormone Levels during Pregnancy
2.5.1. Prenatal Testosterone
Prenatal testosterone overexposure has been considered to be correlated with polycystic ovary syndrome in adult female and was widely studied [76, 77]. Animal experiments performed in sheep [78, 79], rodents [80–82], and even monkeys [83] have all confirmed that prenatal testosterone overexposure leads to insulin resistance in the offspring. Testosterone overexposure during fetal development may impair insulin sensitivity pathways in both liver and muscle [79], increase hepatic gluconeogenesis [84], and impair pancreas islet response to glucose [80] in the offspring.
2.5.2. Prenatal Glucocorticoids
Synthetic glucocorticoids have been used in pregnant women who are at risk of preterm delivery to promote fetal lung maturation. However, concerns have already emerged about the metabolic disorders caused by prenatal glucocorticoids excess. Studies from animal models have found that prenatal glucocorticoids treatment leads to increased hepatic gene expression of hepatocyte nuclear factor 4 alpha [85], phosphoenolpyruvate carboxykinase [86], and glucose-6-phosphatase [87] in the offspring, indicating elevated hepatic gluconeogenesis and hepatic insulin resistance. Nyirenda et al. also found that prenatal dexamethasone administration during late gestation may result in elevated 11 beta-HSD1 [88] and glucocorticoids receptor [86] expression in the liver, which may cause insulin resistance by increasing local glucocorticoids level [89] or activity.
Similar phenomenon has also been found when increased endogenous glucocorticoids pass through maternal to fetus. So far, maternal nicotine [90], food or energy restriction [91], and alcohol intake [92] have all been found to impair placental barrier and consequently cause increased endogenous glucocorticoids in utero.
3. Part II: Postnatal Factors
3.1. New Born
3.1.1. Catch-Up Growth
Catch-up growth, which appeared after lower birth body weight, is the issue that has been studied for years. Accumulated data suggest that low birth weight and catch-up growth are strongly associated with increased risk of insulin resistance and type 2 diabetes [93–96]. Intriguingly, different periods of catch-up growth seem to cause different effects on glucose tolerance and insulin sensitivity. Catch-up growth only in the first year after birth seems to have no effect on insulin sensitivity in 7-year-old child [97], while sustained catch-up growth (more than 1 year after birth) leads to higher insulin levels in 7-year-old child [97] or insulin resistance in 8-year-old child [98]. Compelling evidences raise the thrifty “catch-up fat” mechanisms, indicating that this growth trajectory is characterized by a disproportionately higher rate of fat gain and redistribution of glucose from skeletal muscle to adipose tissue, contributing to insulin resistance in skeletal muscle while hyperresponsiveness to insulin in adipose tissue [99–101].
3.1.2. Lactation
Lactation is also a sensitive period for the programming of later metabolic disorders (Figure 3).
Figure 3.
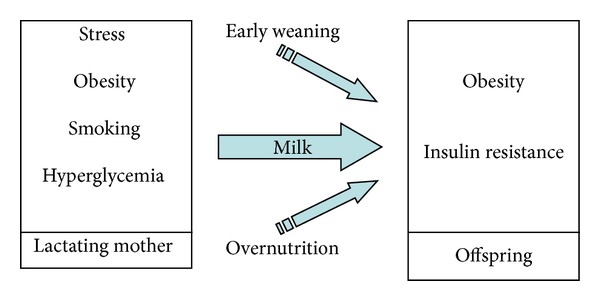
Lactation and insulin resistance. It has been found that both early weaning and overfeeding by more milk intake may lead to insulin resistance in later life. Maternal stress, obesity, hyperglycemia, and even smoking during lactation might also cause reduced insulin sensitivity in the offspring, which suggest that the breast milk can be the “agent,” transferring altered levels of hormones, insulin, or fatty acid contents from maternal circulation to neonate.
Early Weaning. Early weaning may lead to undernutrition, which consequently program metabolic disorders later in life. Hyperglycaemia, higher insulin resistance index, and hyperleptinemia were observed in 6-month-old rats that were weaned early, which were accompanied with central leptin resistance [102].
Overnutrition. Overnutrition during lactation period is associated with metabolic disorders in later life. An experiment conducted in male mice by Pentinat et al. [103] has found that overgrowth mice caused by reduced pups per dams during lactation may develop metabolic disorders at the age of 4 months, including obesity, insulin resistance, and glucose intolerance. Similar results have also been found by Plagemann et al. in their serial experiments performed on rats [104, 105]. Strikingly, the effect of neonatal overnutrition on diabetes risk can be “inherent” to subsequent generations. Impaired glucose tolerance was found in the adult male mice offspring with the father overfed neonatally, and peripheral insulin resistance was found in the grand offspring, although these two generations of animals were not exposed to overnutrition during the neonatal time [103]. Increased oxidative stress in liver and reduced hepatic insulin signaling pathways [106] may underlie effects of neonatal overfeeding. Moreover, early overfeeding leads to permanent dysregulation of hypothalamic circuits in animal models, including reduced negative feedback to the satiety signal insulin on medial arcuate neurons in juvenile as well as adult rats [104] and increased hypothalamic insulin receptor promoter methylation ratio [105]. This may lead to functional resistance to insulin and leptin, which may underlie permanently an increase in food intake, overweight, and insulin resistance.
Maternal Situation during Lactation. Maternal physical or pathological situation during lactation may imprint elevated risk of metabolic diseases in the offspring. Experiments in animals have showed that mother under stress [107], with obesity [108], and exposed to nicotine [109] during lactating period may lead to obesity and insulin resistance in adult offspring, which implicates that the postnatal maternal environment is a major effecter of metabolic outcome in the offspring. It is also found that fostering nondiabetic offspring to diabetic dams may produce smaller offspring with altered arcuate nucleus neuropeptide Y, agouti-related peptide, and pro-opiomelanocortin expression [108]. The underlying mechanism is still far more conclusive. Therefore, the breast milk can be the “agent”; altered levels of hormones, insulin, or fatty acid contents may enter the milk from maternal circulation and then can be transferred to neonate.
3.1.3. Neonatal Stress
Limited data from animal studies have found that stress caused by handling during the neonatal period may also be detrimental. Studies have found that neonatal mice, which were under maternal separation plus subcutaneous sham injection during the lactation period, developed hyperglycemia, hyperinsulinemia, hyperleptinemia, and hyperlipidemia in adult under fasting [110, 111]. Increased plasma corticosterone and adrenocorticotropin were found in these animals [110, 111] which might be responsible for the “diabetic” alteration.
3.1.4. Neonatal Hormone Exposure
There are evidences indicated that exposure to some hormones during neonatal life may predispose metabolic disorders in adult life. Glucocorticoids treatment in neonatal rats caused increased fasting and postprandial blood glucose, which is combined with magnificent insulin resistance and lipid disorder in later life [112]. In addition, in newborn female rats, one subcutaneous injection with 0.35 mg oestradiol benzoate led to reduced insulin sensitivity in adult life by inducing inflammation and disturbance glucose metabolism in skeletal muscle [113], while 1 mg testosterone injection to female neonatal rats caused insulin resistance and increased mesenteric adipose tissue content in adult life [114].
3.1.5. Neonatal Monosodium Glutamate Intake
Monosodium glutamate (MSG) is the sodium salt of glutamic acid, and it is a flavor enhancer that is widely used in Chinese food. The study of neonatal MSG treatment on neonatal animals has been performed since the 1970s and so far there are plenty of animal experiments that have evidenced detrimental effects of MSG administration during early life time, including growth retardation, retinal degeneration, and increased proinflammation in hippocampus [115–117]. In 1997, Hirata et al. found that MSG-treated animals developed central obesity, altered glucose tolerance, and hyperinsulinaemia [118]. More similar evidences have been documented in later experiments [119–121], indicating that neonatal MSG treatment may lead to increased risk of diabetes in adult life. MSG may cause obesity and nonfatty liver [120, 122] and increased mRNA level of IL-6, TNFα, resistin, and leptin in visceral fat tissue [119], which might all predispose insulin resistance in later life.
3.2. Childhood
3.2.1. Low Socioeconomic Status
Socioeconomic status has a notable impact on health disparities, including type 2 diabetes risk. Low childhood socioeconomic status was found linked to type 2 diabetes in some studies [123, 124] and the association remained even after being adjusted for adult socioeconomic status and obesity. Low childhood socioeconomic status was considered to be a robust independent factor of incidence of type 2 diabetes in adulthood and the risk was found greater when childhood socioeconomic status combined with adult obesity. Poor nutrition, unhealthy behaviors, and limited access to material goods and limited socioeconomic opportunities may contribute to altered body composition in later life, which might explain the relationship between childhood socioeconomic position and metabolic disorders in adult.
3.2.2. Famine
Undernutrition during childhood has been found to be associated with an increased type 2 diabetes risk in adulthood. Study in women who had experienced Dutch famine has shown that short period of moderate or severe undernutrition during childhood increases type 2 diabetes risk in adulthood [125].
3.2.3. Obesity
Childhood obesity is an issue of serious medical and social concern. Many studies have demonstrated the positive correlations between childhood obesity and adult metabolic disorders, including type 2 diabetes [61, 126, 127]. Obesity, which mostly caused by high caloric food intake, may always combine with insulin resistance [128]. An unfavorable programming of body composition could be one mechanism linking early childhood growth with later increased risk for type 2 diabetes. In addition, a study performed in 793 French children aged 2–17 yr has suggested that obese children have significantly shorter leukocyte telomeres than their nonobese counterparts [61]. Leukocyte telomere length (LTL), a marker of biological age, is associated with age-related conditions including cardiovascular disease and type 2 diabetes which highlights a potentially deleterious impact of early onset obesity on future health.
4. Conclusion
There is increasing recognition that the risk of type 2 diabetes can be influenced by prenatal, neonatal, and childhood exposures. In the present studies, we have reviewed nutritional, environmental, and physiological factors from prenatal to postnatal periods, which have been documented in studies that may correlate with insulin resistance or type 2 diabetes in adult life. Further investigations are still required. However, relative knowledge education might be successful in women of child-bearing age and ultimate to reduce the disease risk in their potential offspring.
Acknowledgments
This work is supported by the National Nature Science Foundation of China: 81100607, 30800440, and 81200070; Hebei Province Nature Science Foundation: H2012206069 and H2012206009; and the Scientific Foundation of the Department of Public Health of Hebei Province: 20100099 and 20100356.
References
- 1.Mathers JC. Early nutrition: impact on epigenetics. Forum of Nutrition. 2007;60:42–48. doi: 10.1159/000107066. [DOI] [PubMed] [Google Scholar]
- 2.Portrait F, Teeuwiszen E, Deeg D. Early life undernutrition and chronic diseases at older ages: the effects of the Dutch famine on cardiovascular diseases and diabetes. Social Science and Medicine. 2011;73(5):711–718. doi: 10.1016/j.socscimed.2011.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Yajnik CS. Early life origins of insulin resistance and type 2 diabetes in india and other Asian countries. Journal of Nutrition. 2004;134(1):205–210. doi: 10.1093/jn/134.1.205. [DOI] [PubMed] [Google Scholar]
- 4.Orozco-Solís R, Matos RJB, Guzmán-Quevedo O, et al. Nutritional programming in the rat is linked to long-lasting changes in nutrient sensing and energy homeostasis in the hypothalamus. PLoS ONE. 2010;5(10) doi: 10.1371/journal.pone.0013537.e13537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tarry-Adkins JL, Chen J, Jones RH, Smith NH, Ozanne SE. Poor maternal nutrition leads to alterations in oxidative stress, antioxidant defense capacity, and markers of fibrosis in rat islets: potential underlying mechanisms for development of the diabetic phenotype in later life. FASEB Journal. 2010;24(8):2762–2771. doi: 10.1096/fj.10-156075. [DOI] [PubMed] [Google Scholar]
- 6.Sandovici I, Smith NH, Nitert MD, et al. Maternal diet and aging alter the epigenetic control of a promoter-enhancer interaction at the Hnf4a gene in rat pancreatic islets. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(13):5449–5454. doi: 10.1073/pnas.1019007108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theys N, Ahn M, Bouckenooghe T, Reusens B, Remacle C. Maternal malnutrition programs pancreatic islet mitochondrial dysfunction in the adult offspring. Journal of Nutritional Biochemistry. 2011;22(10):985–994. doi: 10.1016/j.jnutbio.2010.08.015. [DOI] [PubMed] [Google Scholar]
- 8.Rodriguez-Trejo A, Ortiz-Lopez MG, Zambrano E, et al. Developmental programming of neonatal pancreatic β-cells by a maternal low-protein diet in rats involves a switch from proliferation to differentiation. The American Journal of Physiology—Endocrinology and Metabolism. 2012;302(11):E1431–E1439. doi: 10.1152/ajpendo.00619.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maurer AD, Reimer RA. Maternal consumption of high-prebiotic fibre or -protein diets during pregnancy and lactation differentially influences satiety hormones and expression of genes involved in glucose and lipid metabolism in offspring in rats. The British Journal of Nutrition. 2011;105(3):329–338. doi: 10.1017/S0007114510003533. [DOI] [PubMed] [Google Scholar]
- 10.Sadashiv, Tiwari S, Paul BN, et al. Resistin gene expression in visceral adipose tissue of postmenopausal women and its association with insulin resistance. Women's Health. 2012;8(5):521–528. doi: 10.2217/whe.12.46. [DOI] [PubMed] [Google Scholar]
- 11.Matsubara T, Mita A, Minami K, et al. PGRN is a key adipokine mediating high fat diet-induced insulin resistance and obesity through IL-6 in adipose tissue. Cell Metabolism. 2012;15(1):38–50. doi: 10.1016/j.cmet.2011.12.002. [DOI] [PubMed] [Google Scholar]
- 12.Lee KM, Yang SJ, Kim YD, et al. Disruption of the cereblon gene enhances hepatic AMPK activity and prevents high-fat diet-induced obesity and insulin resistance in mice. Diabetes. 2013;62(6):1855–1864. doi: 10.2337/db12-1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Black MH, Watanabe RM, Trigo E, et al. High-fat diet is associated with obesity-mediated insulin resistance and β-cell dysfunction in Mexican Americans. Journal of Nutrition. 2013;143(4):479–485. doi: 10.3945/jn.112.170449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Masuyama H, Hiramatsu Y. Effects of a high-fat diet exposure in utero on the metabolic syndrome-like phenomenon in mouse offspring through epigenetic changes in adipocytokine gene expression. Endocrinology. 2012;153(6):2823–2830. doi: 10.1210/en.2011-2161. [DOI] [PubMed] [Google Scholar]
- 15.Liang C, Oest ME, Prater MR. Intrauterine exposure to high saturated fat diet elevates risk of adult-onset chronic diseases in C57BL/6 mice. Birth Defects Research B. 2009;86(5):377–384. doi: 10.1002/bdrb.20206. [DOI] [PubMed] [Google Scholar]
- 16.Rajia S, Chen H, Morris MJ. Maternal overnutrition impacts offspring adiposity and brain appetite markers-modulation by postweaning diet. Journal of Neuroendocrinology. 2010;22(8):905–914. doi: 10.1111/j.1365-2826.2010.02005.x. [DOI] [PubMed] [Google Scholar]
- 17.Briancon N, McNay DE, Maratos-Flier E, Flier JS. Combined neural inactivation of suppressor of cytokine signaling-3 and protein-tyrosine phosphatase-1B reveals additive, synergistic, and factor-specific roles in the regulation of body energy balance. Diabetes. 2010;59(12):3074–3084. doi: 10.2337/db10-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ashino NG, Saito KN, Souza FD, et al. Maternal high-fat feeding through pregnancy and lactation predisposes mouse offspring to molecular insulin resistance and fatty liver. Journal of Nutritional Biochemistry. 2012;23(4):341–348. doi: 10.1016/j.jnutbio.2010.12.011. [DOI] [PubMed] [Google Scholar]
- 19.Yang KF, Shen XH, Cai W. Prenatal and early postnatal exposure to high-saturated-fat diet represses Wnt signaling and myogenic genes in offspring rats. Experimental Biology and Medicine. 2012;237(8):912–918. doi: 10.1258/ebm.2012.011395. [DOI] [PubMed] [Google Scholar]
- 20.Abiola M, Favier M, Christodoulou-Vafeiadou E, Pichard A, Martelly I, Guillet-Deniau I. Activation of Wnt/β-catenin signaling increases insulin sensitivity through a reciprocal regulation of Wnt10b and SREBP-1c in skeletal muscle cells. PLoS ONE. 2009;4(12) doi: 10.1371/journal.pone.0008509.e8509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson AK, Minihane A-M, Williams CM. Trans fatty acids, insulin resistance and diabetes. European Journal of Clinical Nutrition. 2011;65(5):553–564. doi: 10.1038/ejcn.2010.240. [DOI] [PubMed] [Google Scholar]
- 22.Micha R, Mozaffarian D. Trans fatty acids: effects on metabolic syndrome, heart disease and diabetes. Nature Reviews Endocrinology. 2009;5(6):335–344. doi: 10.1038/nrendo.2009.79. [DOI] [PubMed] [Google Scholar]
- 23.Dorfman SE, Laurent D, Gounarides JS, et al. Metabolic implications of dietary trans-fatty acids. Obesity. 2009;17(6):1200–1207. doi: 10.1038/oby.2008.662. [DOI] [PubMed] [Google Scholar]
- 24.Kavanagh K, Sajadian S, Jenkins KA, et al. Neonatal and fetal exposure to trans-fatty acids retards early growth and adiposity while adversely affecting glucose in mice. Nutrition Research. 2010;30(6):418–426. doi: 10.1016/j.nutres.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bayol SA, Simbi BH, Fowkes RC, Stickland NC. A maternal “junk food” diet in pregnancy and lactation promotes nonalcoholic fatty liver disease in rat offspring. Endocrinology. 2010;151(4):1451–1461. doi: 10.1210/en.2009-1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramirez T, Tong M, Chen WC, Nguyen QG, Wands JR, de la Monte SM. Chronic alcohol-induced hepatic insulin resistance and endoplasmic reticulum stress ameliorated by peroxisome-proliferator activated receptor-delta agonist treatment. Journal of Gastroenterology and Hepatology. 2013;28(1):179–187. doi: 10.1111/j.1440-1746.2012.07256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yue F, Zhang X, Zhang H, Jiang X, Gao L, Zhao J. Association of alcohol consumption with the impaired β-cell function independent of body mass index among Chinese men. Endocrine Journal. 2012;59(5):425–433. doi: 10.1507/endocrj.ej12-0003. [DOI] [PubMed] [Google Scholar]
- 28.Chen L, Nyomba BLG. Effects of prenatal alcohol exposure on glucose tolerance in the rat offspring. Metabolism. 2003;52(4):454–462. doi: 10.1053/meta.2003.50073. [DOI] [PubMed] [Google Scholar]
- 29.Chen L, Zhang T, Nyomba BLG. Insulin resistance of gluconeogenic pathways in neonatal rats after prenatal ethanol exposure. The American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2004;286(3):R554–R559. doi: 10.1152/ajpregu.00076.2003. [DOI] [PubMed] [Google Scholar]
- 30.Chen L, Yao XH, Nyomba BLG. In vivo insulin signaling through PI3-kinase is impaired in skeletal muscle of adult rat offspring exposed to ethanol in utero. Journal of Applied Physiology. 2005;99(2):528–534. doi: 10.1152/japplphysiol.01098.2004. [DOI] [PubMed] [Google Scholar]
- 31.Yao XH, Chen L, Nyomba BLG. Adult rats prenatally exposed to ethanol have increased gluconeogenesis and impaired insulin response of hepatic gluconeogenic genes. Journal of Applied Physiology. 2006;100(2):642–648. doi: 10.1152/japplphysiol.01115.2005. [DOI] [PubMed] [Google Scholar]
- 32.Yao XH, Nyomba BLG. Hepatic insulin resistance induced by prenatal alcohol exposure is associated with reduced PTEN and TRB3 acetylation in adult rat offspring. The American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2008;294(6):R1797–R1806. doi: 10.1152/ajpregu.00804.2007. [DOI] [PubMed] [Google Scholar]
- 33.Yao XH, Nyomba BLG. Abnormal glucose homeostasis in adult female rat offspring after intrauterine ethanol exposure. The American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2007;292(5):R1926–R1933. doi: 10.1152/ajpregu.00822.2006. [DOI] [PubMed] [Google Scholar]
- 34.Beguinot F. Tribbles homologue 3 (TRIB3) and the insulin-resistance genes in type 2 diabetes. Diabetologia. 2010;53(9):1831–1834. doi: 10.1007/s00125-010-1824-7. [DOI] [PubMed] [Google Scholar]
- 35.Gupta A, Dey CS. PTEN, a widely known negative regulator of insulin/PI3K signaling, positively regulates neuronal insulin resistance. Molecular Biology of the Cell. 2012;23(19):3882–3898. doi: 10.1091/mbc.E12-05-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nammi S, Dembele K, Nyomba BLG. Increased 11β-hydroxysteroid dehydrogenase type-1 and hexose-6-phosphate dehydrogenase in liver and adipose tissue of rat offspring exposed to alcohol in utero. The American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2007;292(3):R1101–R1109. doi: 10.1152/ajpregu.00255.2006. [DOI] [PubMed] [Google Scholar]
- 37.Groff T. Bisphenol A: invisible pollution. Current Opinion in Pediatrics. 2010;22(4):524–529. doi: 10.1097/MOP.0b013e32833b03f8. [DOI] [PubMed] [Google Scholar]
- 38.Alonso-Magdalena P, Morimoto S, Ripoll C, Fuentes E, Nadal A. The estrogenic effect of bisphenol a disrupts pancreatic β-cell function in vivo and induces insulin resistance. Environmental Health Perspectives. 2006;114(1):106–112. doi: 10.1289/ehp.8451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nadal A, Alonso-Magdalena P, Soriano S, Quesada I, Ropero AB. The pancreatic β-cell as a target of estrogens and xenoestrogens: implications for blood glucose homeostasis and diabetes. Molecular and Cellular Endocrinology. 2009;304(1-2):63–68. doi: 10.1016/j.mce.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 40.Alonso-Magdalena P, Ropero AB, Soriano S, Quesada I, Nadal A. Bisphenol-A: a new diabetogenic factor? Hormones. 2010;9(2):118–126. doi: 10.1007/BF03401277. [DOI] [PubMed] [Google Scholar]
- 41.Makaji E, Raha S, Wade MG, Holloway AC. Effect of environmental contaminants on β cell function. International Journal of Toxicology. 2011;30(4):410–418. doi: 10.1177/1091581811405544. [DOI] [PubMed] [Google Scholar]
- 42.Lang IA, Galloway TS, Scarlett A, et al. Association of Urinary Bisphenol A concentration with medical disorders and laboratory abnormalities in adults. The Journal of the American Medical Association. 2008;300(11):1303–1310. doi: 10.1001/jama.300.11.1303. [DOI] [PubMed] [Google Scholar]
- 43.Wei J, Lin Y, Li Y, et al. Perinatal exposure to bisphenol A at reference dose predisposes offspring to metabolic syndrome in adult rats on a high-fat diet. Endocrinology. 2011;152(8):3049–3061. doi: 10.1210/en.2011-0045. [DOI] [PubMed] [Google Scholar]
- 44.Alonso-Magdalena P, Vieira E, Soriano S, et al. Bisphenol a exposure during pregnancy disrupts glucose homeostasis in mothers and adult male offspring. Environmental Health Perspectives. 2010;118(9):1243–1250. doi: 10.1289/ehp.1001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryan KK, Haller AM, Sorrell JE, Woods SC, Jandacek RJ, Seeley RJ. Perinatal exposure to bisphenol-A and the development of metabolic syndrome in CD-1 mice. Endocrinology. 2010;151(6):2603–2612. doi: 10.1210/en.2009-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao L, Mao C, Li S, et al. Hepatic insulin signaling changes: possible mechanism in prenatal hypoxia-increased susceptibility of fatty liver in adulthood. Endocrinology. 2012;153(10):4955–4965. doi: 10.1210/en.2012-1349. [DOI] [PubMed] [Google Scholar]
- 47.Camm EJ, Martin-Gronert MS, Wright NL, Hansell JA, Ozanne SE, Giussani DA. Prenatal hypoxia independent of undernutrition promotes molecular markers of insulin resistance in adult offspring. FASEB Journal. 2011;25(1):420–427. doi: 10.1096/fj.10-158188. [DOI] [PubMed] [Google Scholar]
- 48.Chang SA. Smoking and type 2 diabetes mellitus. Diabetes and Metabolism Journal. 2012;36(6):399–403. doi: 10.4093/dmj.2012.36.6.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bergman BC, Perreault L, Hunerdosse D, et al. nd reversible mechanisms of smoking-induced insulin resistance in humans. Diabetes. 2012;61(12):3156–3166. doi: 10.2337/db12-0418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Thiering E, Brüske I, Kratzsch J, et al. Prenatal and postnatal tobacco smoke exposure and development of insulin resistance in 10 year old children. International Journal of Hygiene and Environmental Health. 2011;214(5):361–368. doi: 10.1016/j.ijheh.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 51.Bruin JE, Kellenberger LD, Gerstein HC, Morrison KM, Holloway AC. Fetal and neonatal nicotine exposure and postnatal glucose homeostasis: identifying critical windows of exposure. Journal of Endocrinology. 2007;194(1):171–178. doi: 10.1677/JOE-07-0050. [DOI] [PubMed] [Google Scholar]
- 52.Holloway AC, Cuu DQ, Morrison KM, Gerstein HC, Tarnopolsky MA. Transgenerational effects of fetal and neonatal exposure to nicotine. Endocrine. 2007;31(3):254–259. doi: 10.1007/s12020-007-0043-6. [DOI] [PubMed] [Google Scholar]
- 53.Somm E, Schwitzgebel VM, Vauthay DM, et al. Prenatal nicotine exposure alters early pancreatic islet and adipose tissue development with consequences on the control of body weight and glucose metabolism later in life. Endocrinology. 2008;149(12):6289–6299. doi: 10.1210/en.2008-0361. [DOI] [PubMed] [Google Scholar]
- 54.Chen H, Saad S, Sandow SL, Bertrand PP. Cigarette smoking and brain regulation of energy homeostasis. Frontiers in Pharmacology. 2012;3(article 147) doi: 10.3389/fphar.2012.00147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Coe CL, Lubach GR, Shirtcliff EA. Maternal stress during pregnancy predisposes for iron deficiency in infant monkeys impacting innate immunity. Pediatric Research. 2007;61(5, part 1):520–524. doi: 10.1203/pdr.0b013e318045be53. [DOI] [PubMed] [Google Scholar]
- 56.Lesage J, Del-Favero F, Leonhardt M, et al. Prenatal stress induces intrauterine growth restriction and programmes glucose intolerance and feeding behaviour disturbances in the aged rat. Journal of Endocrinology. 2004;181(2):291–296. doi: 10.1677/joe.0.1810291. [DOI] [PubMed] [Google Scholar]
- 57.Li J, Olsen J, Vestergaard M, Obel C, Kristensen JK, Virk J. Prenatal exposure to bereavement and type-2 diabetes: a Danish longitudinal population based study. PLoS ONE. 2012;7(8) doi: 10.1371/journal.pone.0043508.e43508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Entringer S, Wüst S, Kumsta R, et al. Prenatal psychosocial stress exposure is associated with insulin resistance in young adults. The American Journal of Obstetrics and Gynecology. 2008;199(5):498.e1–498.e7. doi: 10.1016/j.ajog.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Entringer S, Epel ES, Kumsta R, et al. Stress exposure in intrauterine life is associated with shorter telomere length in young adulthood. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(33):E513–E518. doi: 10.1073/pnas.1107759108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Salpea KD, Talmud PJ, Cooper JA, et al. Association of telomere length with type 2 diabetes, oxidative stress and UCP2 gene variation. Atherosclerosis. 2010;209(1):42–50. doi: 10.1016/j.atherosclerosis.2009.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Buxton JL, Walters RG, Visvikis-Siest S, Meyre D, Froguel P, Blakemore AIF. Childhood obesity is associated with shorter leukocyte telomere length. Journal of Clinical Endocrinology and Metabolism. 2011;96(5):1500–1505. doi: 10.1210/jc.2010-2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fernandez-Twinn DS, Blackmore HL, Siggens L, et al. The programming of cardiac hypertrophy in the offspring by maternal obesity is associated with hyperinsulinemia, AKT, ERK, and mTOR activation. Endocrinology. 2012;153(12):5961–5971. doi: 10.1210/en.2012-1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O'Reilly JR, Reynolds RM. The risk of maternal obesity to the long-term health of the offspring. Clinical Endocrinology. 2013;78(1):9–16. doi: 10.1111/cen.12055. [DOI] [PubMed] [Google Scholar]
- 64.Obregon M. Maternal obesity results in offspring prone to metabolic syndrome. Endocrinology. 2010;151(8):3475–3476. doi: 10.1210/en.2010-0611. [DOI] [PubMed] [Google Scholar]
- 65.Boney CM, Verma A, Tucker R, Vohr BR. Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus. Pediatrics. 2005;115(3):e290–e296. doi: 10.1542/peds.2004-1808. [DOI] [PubMed] [Google Scholar]
- 66.Shankar K, Kang P, Harrell A, et al. Maternal overweight programs insulin and adiponectin signaling in the offspring. Endocrinology. 2010;151(6):2577–2589. doi: 10.1210/en.2010-0017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang L, Long NM, Hein SM, Ma Y, Nathanielsz PW, Ford SP. Maternal obesity in ewes results in reduced fetal pancreatic β-cell numbers in late gestation and decreased circulating insulin concentration at term. Domestic Animal Endocrinology. 2011;40(1):30–39. doi: 10.1016/j.domaniend.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ford SP, Zhang L, Zhu M, et al. Maternal obesity accelerates fetal pancreatic β-cell but not α-cell development in sheep: prenatal consequences. The American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2009;297(3):R835–R843. doi: 10.1152/ajpregu.00072.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Oben JA, Mouralidarane A, Samuelsson A, et al. Maternal obesity during pregnancy and lactation programs the development of offspring non-alcoholic fatty liver disease in mice. Journal of Hepatology. 2010;52(6):913–920. doi: 10.1016/j.jhep.2009.12.042. [DOI] [PubMed] [Google Scholar]
- 70.Borengasser SJ, Lau F, Kang P, et al. Maternal obesity during gestation impairs fatty acid oxidation and mitochondrial SIRT3 expression in rat offspring at weaning. PLoS ONE. 2011;6(8) doi: 10.1371/journal.pone.0024068.e24068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Boerschmann H, Pflüger M, Henneberger L, Ziegler A, Hummel S. Prevalence and predictors of overweight and insulin resistance in offspring of mothers with gestational diabetes mellitus. Diabetes Care. 2010;33(8):1845–1849. doi: 10.2337/dc10-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bush NC, Chandler-Laney PC, Rouse DJ, Granger WM, Oster RA, Gower BA. Higher maternal gestational glucose concentration is associated with lower offspring insulin sensitivity and altered β-cell function. Journal of Clinical Endocrinology and Metabolism. 2011;96(5):E803–E809. doi: 10.1210/jc.2010-2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Song Y, Li J, Zhao Y, et al. Severe maternal hyperglycemia exacerbates the development of insulin resistance and fatty liver in the offspring on high fat diet. Experimental Diabetes Research. 2012;2012:8 pages. doi: 10.1155/2012/254976.254976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ma JM, Zeng CJ, Zhang L, Shou C, Yang HX. Increased hepatic peroxisome proliferator-activated receptor coactivator-1α expression precedes the development of insulin resistance in offspring of rats from severe hyperglycemic mothers. Chinese Medical Journal. 2012;125(7):1224–1229. [PubMed] [Google Scholar]
- 75.Natali A, Muscelli E, Mari A, et al. Insulin sensitivity and β-cell function in the offspring of type 2 diabetic patients: impact of line of inheritance. Journal of Clinical Endocrinology and Metabolism. 2010;95(10):4703–4711. doi: 10.1210/jc.2009-2721. [DOI] [PubMed] [Google Scholar]
- 76.King AJ, Olivier NB, Mohankumar PS, Lee JS, Padmanabhan V, Fink GD. Hypertension caused by prenatal testosterone excess in female sheep. The American Journal of Physiology—Endocrinology and Metabolism. 2007;292(6):E1837–E1841. doi: 10.1152/ajpendo.00668.2006. [DOI] [PubMed] [Google Scholar]
- 77.Recabarren SE, Padmanabhan V, Codner E, et al. Postnatal developmental consequences of altered insulin sensitivity in female sheep treated prenatally with testosterone. The American Journal of Physiology—Endocrinology and Metabolism. 2005;289(5):E801–E806. doi: 10.1152/ajpendo.00107.2005. [DOI] [PubMed] [Google Scholar]
- 78.Padmanabhan V, Veiga-Lopez A, Abbott DH, Recabarren SE, Herkimer C. Developmental programming: impact of prenatal testosterone excess and postnatal weight gain on insulin sensitivity index and transfer of traits to offspring of overweight females. Endocrinology. 2010;151(2):595–605. doi: 10.1210/en.2009-1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nada SE, Thompson RC, Padmanabhan V. Developmental programming: differential effects of prenatal testosterone excess on insulin target tissues. Endocrinology. 2010;151(11):5165–5173. doi: 10.1210/en.2010-0666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roland AV, Nunemaker CS, Keller SR, Moenter SM. Prenatal androgen exposure programs metabolic dysfunction in female mice. Journal of Endocrinology. 2010;207(2):213–223. doi: 10.1677/JOE-10-0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Amalfi S, Velez LM, Heber MF, et al. Prenatal hyperandrogenization induces metabolic and endocrine alterations which depend on the levels of testosterone exposure. PLoS ONE. 2012;7(5) doi: 10.1371/journal.pone.0037658.e37658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sun M, Maliqueo M, Benrick A, et al. Maternal androgen excess reduces placental and fetal weights, increases placental steroidogenesis, and leads to long-term health effects in their female offspring. The American Journal of Physiology—Endocrinology and Metabolism. 2012;303(11):E1373–E1385. doi: 10.1152/ajpendo.00421.2012. [DOI] [PubMed] [Google Scholar]
- 83.Bruns CM, Baum ST, Colman RJ, et al. Insulin resistance and impaired insulin secretion in prenatally androgenized male rhesus monkeys. Journal of Clinical Endocrinology and Metabolism. 2004;89(12):6218–6223. doi: 10.1210/jc.2004-0918. [DOI] [PubMed] [Google Scholar]
- 84.Hogg K, Wood C, McNeilly AS, Duncan WC. The in utero programming effect of increased maternal androgens and a direct fetal intervention on liver and metabolic function in adult sheep. PLoS ONE. 2011;6(9) doi: 10.1371/journal.pone.0024877.e24877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nyirenda MJ, Dean S, Lyons V, Chapman KE, Seckl JR. Prenatal programming of hepatocyte nuclear factor 4α in the rat: a key mechanism in the “foetal origins of hyperglycaemia”? Diabetologia. 2006;49(6):1412–1420. doi: 10.1007/s00125-006-0188-5. [DOI] [PubMed] [Google Scholar]
- 86.Nyirenda MJ, Lindsay RS, Kenyon CJ, Burchell A, Seckl JR. Glucocorticoid exposure in late gestation permanently programs rat hepatic phosphoenolpyruvate carboxykinase and glucocorticoid receptor expression and causes glucose intolerance in adult offspring. Journal of Clinical Investigation. 1998;101(10):2174–2181. doi: 10.1172/JCI1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Franko KL, Giussani DA, Forhead AJ, Fowden AL. Effects of dexamethasone on the glucogenic capacity of fetal, pregnant, and non-pregnant adult sheep. Journal of Endocrinology. 2007;192(1):67–73. doi: 10.1677/joe.1.07063. [DOI] [PubMed] [Google Scholar]
- 88.Nyirenda MJ, Carter R, Tang JI, et al. Prenatal programming of metabolic syndrome in the common marmoset is associated with increased expression of 11β-hydroxysteroid dehydrogenase type 1. Diabetes. 2009;58(12):2873–2879. doi: 10.2337/db09-0873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li G, Hernandez-Ono A, Crooke RM, Graham MJ, Ginsberg HN. Antisense reduction of 11β-hydroxysteroid dehydrogenase type 1 enhances energy expenditure and insulin sensitivity independent of food intake in C57BL/6J mice on a Western-type diet. Metabolism. 2012;61(6):823–835. doi: 10.1016/j.metabol.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu D, Liang G, Yan YE, et al. Nicotine-induced over-exposure to maternal glucocorticoid and activated glucocorticoid metabolism causes hypothalamic-pituitary-adrenal axis-associated neuroendocrine metabolic alterations in fetal rats. Toxicology Letters. 2012;209(3):282–290. doi: 10.1016/j.toxlet.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 91.Valtat B, Dupuis C, Zenaty D, et al. Genetic evidence of the programming of β cell mass and function by glucocorticoids in mice. Diabetologia. 2011;54(2):350–359. doi: 10.1007/s00125-010-1898-2. [DOI] [PubMed] [Google Scholar]
- 92.Liang G, Chen M, Pan XL, Zheng J, Wang H. Ethanol-induced inhibition of fetal hypothalamic-pituitary-adrenal axis due to prenatal overexposure to maternal glucocorticoid in mice. Experimental and Toxicologic Pathology. 2011;63(7-8):607–611. doi: 10.1016/j.etp.2010.04.015. [DOI] [PubMed] [Google Scholar]
- 93.Bruce KD, Hanson MA. The developmental origins, mechanisms, and implications of metabolic syndrome. Journal of Nutrition. 2010;140(3):648–652. doi: 10.3945/jn.109.111179. [DOI] [PubMed] [Google Scholar]
- 94.Fernandez-Twinn DS, Ozanne SE. Mechanisms by which poor early growth programs type-2 diabetes, obesity and the metabolic syndrome. Physiology and Behavior. 2006;88(3):234–243. doi: 10.1016/j.physbeh.2006.05.039. [DOI] [PubMed] [Google Scholar]
- 95.Martin-Gronert MS, Ozanne SE. Mechanisms linking suboptimal early nutrition and increased risk of type 2 diabetes and obesity. Journal of Nutrition. 2010;140(3):662–666. doi: 10.3945/jn.109.111237. [DOI] [PubMed] [Google Scholar]
- 96.Deng HZ, Deng H, Su Z, et al. Insulin resistance and adiponectin levels are associated with height catch-up growth in pre-pubertal Chinese individuals born small for gestational age. Nutrition and Metabolism. 2012;9(1, article 107) doi: 10.1186/1743-7075-9-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Crowther NJ, Cameron N, Trusler J, Toman M, Norris SA, Gray IP. Influence of catch-up growth on glucose tolerance and β-cell function in 7-year-old children: results from the birth to twenty study. Pediatrics. 2008;121(6):e1715–e1722. doi: 10.1542/peds.2007-3147. [DOI] [PubMed] [Google Scholar]
- 98.Ong KK, Petry CJ, Emmett PM, et al. Insulin sensitivity and secretion in normal children related to size at birth, postnatal growth, and plasma insulin-like growth factor-I levels. Diabetologia. 2004;47(6):1064–1070. doi: 10.1007/s00125-004-1405-8. [DOI] [PubMed] [Google Scholar]
- 99.Cettour-Rose P, Samec S, Russell AP, et al. Redistribution of glucose from skeletal muscle to adipose tissue during catch-up fat: a link between catch-up growth and later metabolic syndrome. Diabetes. 2005;54(3):751–756. doi: 10.2337/diabetes.54.3.751. [DOI] [PubMed] [Google Scholar]
- 100.Dulloo AG. Thrifty energy metabolism in catch-up growth trajectories to insulin and leptin resistance. Best Practice and Research in Clinical Endocrinology and Metabolism. 2008;22(1):155–171. doi: 10.1016/j.beem.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 101.Dulloo AG, Jacquet J, Seydoux J, Montani J- The thrifty “catch-up fat” phenotype: its impact on insulin sensitivity during growth trajectories to obesity and metabolic syndrome. International Journal of Obesity. 2006;30(supplement 4):S23–S35. doi: 10.1038/sj.ijo.0803516. [DOI] [PubMed] [Google Scholar]
- 102.Lima NS, de Moura EG, Passos MCF, et al. Early weaning causes undernutrition for a short period and programmes some metabolic syndrome components and leptin resistance in adult rat offspring. The British Journal of Nutrition. 2011;105(9):1405–1413. doi: 10.1017/S0007114510005064. [DOI] [PubMed] [Google Scholar]
- 103.Pentinat T, Ramon-Krauel M, Cebria J, Diaz R, Jimenez-Chillaron JC. Transgenerational inheritance of glucose intolerance in a mouse model of neonatal overnutrition. Endocrinology. 2010;151(12):5617–5623. doi: 10.1210/en.2010-0684. [DOI] [PubMed] [Google Scholar]
- 104.Davidowa H, Plagemann A. Insulin resistance of hypothalamic arcuate neurons in neonatally overfed rats. NeuroReport. 2007;18(5):521–524. doi: 10.1097/WNR.0b013e32805dfb93. [DOI] [PubMed] [Google Scholar]
- 105.Plagemann A, Roepke K, Harder T, et al. Epigenetic malprogramming of the insulin receptor promoter due to developmental overfeeding. Journal of Perinatal Medicine. 2010;38(4):393–400. doi: 10.1515/jpm.2010.051. [DOI] [PubMed] [Google Scholar]
- 106.Conceicao EP, Franco JG, Oliveira E, et al. Oxidative stress programming in a rat model of postnatal early overnutrition—role of insulin resistance. Journal of Nutritional Biochemistry. 2013;24(1):81–87. doi: 10.1016/j.jnutbio.2012.02.010. [DOI] [PubMed] [Google Scholar]
- 107.Kaufman D, Banerji MA, Shorman I, et al. Early-life stress and the development of obesity and insulin resistance in juvenile bonnet macaques. Diabetes. 2007;56(5):1382–1386. doi: 10.2337/db06-1409. [DOI] [PubMed] [Google Scholar]
- 108.Gorski JN, Dunn-Meynell AA, Hartman TG, Levin BE. Postnatal environment overrides genetic and prenatal factors influencing offspring obesity and insulin resistance. The American Journal of Physiology—Regulatory Integrative and Comparative Physiology. 2006;291(3):R768–R778. doi: 10.1152/ajpregu.00138.2006. [DOI] [PubMed] [Google Scholar]
- 109.de Oliveira E, Moura EG, Santos-Silva AP, et al. Neonatal nicotine exposure causes insulin and leptin resistance and inhibits hypothalamic leptin signaling in adult rat offspring. Journal of Endocrinology. 2010;206(1):55–63. doi: 10.1677/JOE-10-0104. [DOI] [PubMed] [Google Scholar]
- 110.Loizzo S, Campana G, Vella S, et al. Post-natal stress-induced endocrine and metabolic alterations in mice at adulthood involve different pro-opiomelanocortin-derived peptides. Peptides. 2010;31(11):2123–2129. doi: 10.1016/j.peptides.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 111.Loizzo A, Loizzo S, Galietta G, et al. Overweight and metabolic and hormonal parameter disruption are induced in adult male mice by manipulations during lactation period. Pediatric Research. 2006;59(1):111–115. doi: 10.1203/01.pdr.0000190575.12965.ce. [DOI] [PubMed] [Google Scholar]
- 112.Baxi DB, Singh PK, Vachhrajani KD, Ramachandran AV. Plasticity changes in adult metabolic homeostasis and tissue oxidative stress: neonatal programming by corticosterone and melatonin as deprogrammer. Journal of Maternal-Fetal and Neonatal Medicine. 2012;25(6):831–844. doi: 10.3109/14767058.2011.599456. [DOI] [PubMed] [Google Scholar]
- 113.Alexanderson C, Eriksson E, Stener-Victorin E, Lönn M, Holmáng A. Early postnatal oestradiol exposure causes insulin resistance and signs of inflammation in circulation and skeletal muscle. Journal of Endocrinology. 2009;201(1):49–58. doi: 10.1677/JOE-08-0534. [DOI] [PubMed] [Google Scholar]
- 114.Nilsson C, Niklasson M, Eriksson E, Björntorp P, Holmäng A. Imprinting of female offspring with testosterone results in insulin resistance and changes in body fat distribution at adult age in rats. Journal of Clinical Investigation. 1998;101(1):74–78. doi: 10.1172/JCI1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kuznetsova EG, Amstislavskaya TG, Bulygina VV, Il’nitskaya SI, Tibeikina MA, Skrinskaya YA. Effects of administration of sodium glutamate during the neonatal period on behavior and blood corticosterone levels in male mice. Neuroscience and Behavioral Physiology. 2007;37(8):827–833. doi: 10.1007/s11055-007-0088-2. [DOI] [PubMed] [Google Scholar]
- 116.Rácz B, Tamás A, Kiss P, et al. Involvement of ERK and CREB signaling pathways in the protective effect of PACAP in monosodium glutamate-induced retinal lesion. Annals of the New York Academy of Sciences. 2006;1070:507–511. doi: 10.1196/annals.1317.070. [DOI] [PubMed] [Google Scholar]
- 117.Chaparro-Huerta V, Rivera-Cervantes MC, Flores-Soto ME, Gómez-Pinedo U, Beas-Zárate C. Proinflammatory cytokines and apoptosis following glutamate-induced excitotoxicity mediated by p38 MAPK in the hippocampus of neonatal rats. Journal of Neuroimmunology. 2005;165(1-2):53–62. doi: 10.1016/j.jneuroim.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 118.Hirata AE, Andrade IS, Vaskevicius P, Dolnikoff MS. Monosodium glutamate (MSG)-obese rats develop glucose intolerance and insulin resistance to peripheral glucose uptake. Brazilian Journal of Medical and Biological Research. 1997;30(5):671–674. doi: 10.1590/s0100-879x1997000500016. [DOI] [PubMed] [Google Scholar]
- 119.Roman-Ramos R, Almanza-Perez JC, Garcia-Macedo R, et al. Monosodium glutamate neonatal intoxication associated with obesity in adult stage is characterized by chronic inflammation and increased mRNA expression of peroxisome proliferator-activated receptors in mice. Basic and Clinical Pharmacology and Toxicology. 2011;108(6):406–413. doi: 10.1111/j.1742-7843.2011.00671.x. [DOI] [PubMed] [Google Scholar]
- 120.Sasaki Y, Suzuki W, Shimada T, et al. Dose dependent development of diabetes mellitus and non-alcoholic steatohepatitis in monosodium glutamate-induced obese mice. Life Sciences. 2009;85(13-14):490–498. doi: 10.1016/j.lfs.2009.07.017. [DOI] [PubMed] [Google Scholar]
- 121.Andreazzi AE, Scomparin DX, Mesquita FP, et al. Swimming exercise at weaning improves glycemic control and inhibits the onset of monosodium L-glutamate-obesity in mice. Journal of Endocrinology. 2009;201(3):351–359. doi: 10.1677/JOE-08-0312. [DOI] [PubMed] [Google Scholar]
- 122.Sasaki Y, Shimada T, Iizuka S, et al. Effects of bezafibrate in nonalcoholic steatohepatitis model mice with monosodium glutamate-induced metabolic syndrome. European Journal of Pharmacology. 2011;662(1–3):1–8. doi: 10.1016/j.ejphar.2011.04.051. [DOI] [PubMed] [Google Scholar]
- 123.Maty SC, Lynch JW, Raghunathan TE, Kaplan GA. Childhood socioeconomic position, gender, adult body mass index, and incidence of type 2 diabetes mellitus over 34 years in the Alameda county study. The American Journal of Public Health. 2008;98(8):1486–1494. doi: 10.2105/AJPH.2007.123653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tamayo T, Christian H, Rathmann W. Impact of early psychosocial factors (childhood socioeconomic factors and adversities) on future risk of type 2 diabetes, metabolic disturbances and obesity: a systematic review. BMC Public Health. 2010;10, article 525 doi: 10.1186/1471-2458-10-525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.van Abeelen AF, Elias SG, Bossuyt PM, et al. Famine exposure in the young and the risk of type 2 diabetes in adulthood. Diabetes. 2012;61(9):2255–2260. doi: 10.2337/db11-1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Biro FM, Wien M. Childhood obesity and adult morbidities. The American Journal of Clinical Nutrition. 2010;91(5):1499S–1505S. doi: 10.3945/ajcn.2010.28701B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Franks PW, Hanson RL, Knowler WC, Sievers ML, Bennett PH, Looker HC. Childhood obesity, other cardiovascular risk factors, and premature death. The New England Journal of Medicine. 2010;362(6):485–493. doi: 10.1056/NEJMoa0904130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Caceres M, Teran CG, Rodriguez S, Medina M. Prevalence of insulin resistance and its association with metabolic syndrome criteria among Bolivian children and adolescents with obesity. BMC Pediatrics. 2008;8, article 31 doi: 10.1186/1471-2431-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]