

Abstract
Chemoprevention of lung carcinogenesis is one approach to controlling the epidemic of lung cancer caused by cigarette smoking. The target for chemoprevention should be the activities of the multiple carcinogens, toxicants, co-carcinogens, tumor promoters and inflammatory compounds in cigarette smoke. There are presently many agents both synthetic and naturally occurring that prevent lung tumor development in well established animal models. It seems likely that logically constructed mixtures of these agents, developed from the ground up, will be necessary for prevention of lung carcinogenesis
Introduction
Lung cancer kills more than 3000 people every day in the world and is its leading cause of cancer death. About 90% of this incredible toll is due to cigarette smoking. Clearly, we must continue our successful efforts in tobacco control which have resulted in a significant reduction in smoking prevalence in many countries. But there are still 1.3 billion smokers in the world and wealthy multinational tobacco companies continue to introduce cancer causing products designed to entice teenagers into a lifetime of nicotine addiction. While 70% of smokers attempt to quit each year, less than 5% succeed1, and the average success rates at six months post-quit, even with the most advanced smoking cessation programs, hover around 25%2.
In recent years, the rate of decrease in the prevalence of U.S. adult smoking has slowed significantly3 and remaining at about 20% from 2004 to 20074. This plateau has been observed even in some countries such as Ireland which has a significant tobacco control program (e.g., comprehensive smoke-free worksite policies, high cigarette prices and bans on tobacco advertising and promotion)5. This reduced rate of decline in smoking has been attributed to a plateau in smoking cessation success3, leading some researchers to believe that the remaining population of smokers is hardcore and are either unwilling or unable to quit6. An appreciable number of these smokers may be experiencing mental health disorders5.
The addicted smokers who fail as well as the ex-smokers who have succeeded in quitting are at high risk for lung cancer, and we must do something to help prevent this devastating disease with a 5 year survival rate of only 15%. Chemoprevention of lung carcinogenesis is one way forward. While the cardiovascular community has identified high risk individuals with biomarkers such as cholesterol and C reactive protein, and successfully treated them with preventive statins7, we in cancer research have yet to succeed in developing an effective lung carcinogenesis chemopreventive agent or strategy. The theme of this article is that a successful lung carcinogenesis chemopreventive agent will target tobacco smoke carcinogens and toxicants, the cause of lung cancer in smokers and ex-smokers, and that a successful strategy will integrate chemoprevention into the treatment portfolio of the addicted smoker as well as being available for the confirmed ex-smoker.
Treat lung carcinogenesis, not lung cancer
In chemoprevention, we aim to treat lung carcinogenesis, not lung cancer8. Lung carcinogenesis is barely in the vocabulary of the cancer research community, and certainly not in that of the lay community. The distinction is crucial. Lung cancer is the end result of lung carcinogenesis. Treatment of lung cancer is usually ineffective because a malignant tumor is discovered at a late stage. Treating lung carcinogenesis has the potential to prevent this disease.
How can we treat lung carcinogenesis? Since 90% of lung carcinogenesis is due to tobacco smoke exposure, our target must be the carcinogenic activity of tobacco smoke. In previous articles, we have presented a conceptual model for tobacco smoke-induced lung carcinogenesis9,10 (Box 1). This model indicates that, in our treatment of lung carcinogenesis, we would be wrong to focus on a single molecular pathway, because multiple pathways are altered. Others have come to similar conclusions11. We need to focus on the cause of the multiple aberrant biological pathways in lung carcinogenesis: the activities of tobacco smoke. Of course, removing tobacco smoke exposure is the ideal method for preventing lung carcinogenesis, but for reasons discussed above, this is only partially successful.
Box 1. A conceptual model for tobacco smoke-induced lung carcinogenesis.
In this widely accepted model, people become addicted to nicotine in cigarette smoke, usually at a relatively young age when they experiment with cigarettes due to peer pressure and advertising. Nicotine is not a carcinogen, but each puff of each cigarette delivers a mixture of over 60 established carcinogens, along with toxicants, tumor promoters, co-carcinogens, oxidants, free radicals, and inflammatory agents. The carcinogens and their metabolites bind to DNA resulting in DNA adducts and subsequent somatic mutations. When these mutations occur in critical genes such as oncogenes and tumor suppressor genes, the result is loss of normal cellular growth control mechanisms, genomic instability, and cancer. A recent study validates this model. DNA sequencing of 623 cancer related genes revealed more than 1000 somatic mutations in 188 human lung adenocarcinomas, and 26 of these genes, including the tumor suppressor gene TP53 and the oncogene KRAS, were mutated at significantly high frequencies. Alterations were commonly observed in genes of the MAPK signaling, TP53 signaling, Wnt signaling, cell cycle and mTOR pathways144. The multiple mutations caused by tobacco smoke carcinogens are also consistent with the concept of field cancerization.
What are the activities of tobacco smoke that are critical in lung carcinogenesis? First and foremost are the lung carcinogens. Of the over 60 established carcinogens in cigarette smoke, there are at least 20 credible lung carcinogens9,10,12. These occur in both the gas phase and the particulate phase of tobacco smoke. The gas phase constituents include 1,3-butadiene, ethylene oxide, benzene, and aldehydes. The particulate phase constituents include polycyclic aromatic hydrocarbons (PAH), the best known of which is benzo[a]pyrene (BaP), and tobacco-specific nitrosamines such as the potent lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Consistent with the presence of these carcinogens, both the gas phase and the particulate phase of tobacco smoke can induce lung tumors in rodents upon exposure by inhalation13.
Second are the tumor promoters, co-carcinogens, and toxicants which have a variety of deleterious activities. Tumor promoters are not carcinogenic themselves, but enhance the activity of carcinogens when administered subsequently. The tumor promoting activities of tobacco smoke and its condensate have been clearly demonstrated by inhalation and mouse skin application studies14,15. These tumor promoters are only partially characterized, but extensive data indicate that they are found mainly in the weakly acidic fraction of tobacco smoke condensate16. Co-carcinogens are also not carcinogenic themselves, but enhance the activity of carcinogens when administered concurrently. Catechol, methyl catechols, and certain PAH are well established co-carcinogens in tobacco smoke, based on mouse skin studies15. One of the major toxicants in cigarette smoke, with a demonstrated relationship to lung carcinogenesis, is acrolein. While not strongly carcinogenic itself, acrolein is highly toxic to cilia of the lung, thus impeding clearance of tobacco smoke constituents17. Acrolein also reacts directly with DNA and protein to produce adducts with potentially important consequences18–20. Other toxicants in tobacco smoke include nitric oxide and poorly characterized free radicals, which may contribute to tumor promotion or co-carcinogenesis by causing oxidative damage.
Third are the inflammatory agents. A number of pro-inflammatory changes have been observed in smokers’ lungs, and inflammation is closely associated with tumor promotion and activation of factors such as NFκB21–24. Inflammation has a role in COPD associated with smoking25, and COPD (especially emphysema) in turn is an independent risk factor for lung cancer26. The specific agents in cigarette smoke responsible for inflammation are poorly defined, but the potential roles of oxidants and reactive aldehydes such as acrolein have been discussed20,25. It is important to keep in mind that exposure to all agents in cigarette smoke is simultaneous, thus concepts such as tumor initiation and tumor promotion may be artificial, or even irrelevant.
It is apparent to us that a mixture of chemopreventive agents will be necessary to counteract these three complex activities. This mixture should be developed from the ground up, by first determining the efficacy of individual agents, and then assessing their chemopreventive activities when tested as a mixture. Wattenberg has classified chemopreventive agents into two broad groups: blocking agents which prevent the interaction of carcinogens with DNA, and suppressing agents which prevent post-carcinogen treatment downstream effects27. These definitions are still useful, and it is likely that an effective mixture would contain as a minimum an agent with each type of activity. Two crucial requirements for any chemopreventive agent or mixture are efficacy and lack of toxicity. One must demonstrate efficacy in an animal model, or preferably models, before chemopreventive agents should be seriously considered for use in people. Lack of toxicity is also critical, or clinical utility will be compromised.
Animal models for pre-clinical evaluation of chemopreventive agents
In keeping with the theme of inhibiting the carcinogenic and toxic activities of tobacco smoke, the ideal animal model would use tobacco smoke itself as the carcinogen. Unfortunately, this is not so simple. Rodent models for inhalation of tobacco smoke pose many difficulties13 (Box 2). In spite of the limitations, one relatively practical model using strain A/J mice has been described by Witschi and co-workers28. A/J mice develop lung tumors with age. Lung tumor multiplicity is significantly and reproducibly increased by carcinogen treatment29,30. The cigarette smoke inhalation protocol leads to a reproducible increase in lung tumor multiplicity in these mice, and in some cases, lung tumor incidence28. However, the increase in lung tumor multiplicity, from about 0.5–1 lung tumors per mouse in controls to 1.1–2.8 lung tumors per mouse in the mice treated with cigarette smoke, although significant, is relatively small. This creates severe practical problems when using this model for chemoprevention studies. A number of chemoprevention experiments using relatively small groups of animals have been reported using this smoke inhalation assay, but most of the results were statistically insignificant, with the exception of a mixture of dexamethasone and myo-inositol28. Several other mouse strains have been used in experiments of similar design, but the tumorigenic response to cigarette smoke was generally quite weak31.
Box 2. Problems with rodent models of smoke inhalation.
Rodents are obligatory nose breathers with complex nasal structures different from those in humans, leading to different deposition patterns in rodents versus humans. Rodents will not inhale tobacco smoke voluntarily the way humans do, but rather adopt shallow breathing patterns and avoidance reactions. The exposure systems that have been used are problematic. Nose only exposure systems require extensive handling while whole body exposure systems result in deposition of particles on the pelt and oral exposure through grooming. Exposure in these systems can cause stress and lack of weight gain. Lung tumors have been induced by cigarette smoke exposure in both rats and mice, but lengthy whole body exposures are required, and the experiments used highly specialized inhalation facilities which are not widely available145,146.
By far the most commonly employed model for evaluating chemopreventive agents is the carcinogen-treated A/J mouse. The tumors induced by carcinogens have morphologic, histogenic, and molecular features similar to human lung adenocarcinoma32. The susceptibility of the A/J mouse to lung tumor development has been attributed to the pulmonary adenoma susceptibility (Pas1) gene, which is tightly linked to the Kras2 oncogene33. Four carcinogens - BaP, NNK, ethyl carbamate (urethane), and vinyl carbamate – have been extensively used for tumor induction in chemoprevention experiments (Box 3). BaP and NNK are widely viewed as important lung carcinogens in cigarette smoke. Urethane is the classic carcinogen used for lung tumor induction in A/J mice34, while vinyl carbamate is its proximate carcinogenic metabolite. Urethane has been reported as a constituent of cigarette smoke, but only sporadically, while vinyl carbamate has not been analyzed in cigarette smoke or in smokers as a metabolite. The doses of pure carcinogens used in these studies are thousands of times higher than the amounts present in cigarette smoke.
Box 3. Typical procedures for inducing lung tumors in A/J mice.
BaP: A/J mice were maintained on a semi-synthetic diet and, at age 9 weeks, were treated with 2 mg (7.9 μmol) in cottonseed oil, by gavage 56. This was repeated 4 and 7 days after the initial dose. The study was terminated 21 weeks after the last dose, giving about 13 lung tumors per mouse. A disadvantage is the induction of forestomach tumors which become large after 21 weeks and may kill the animals before the scheduled termination.
NNK: A/J mice were maintained on a semi-synthetic diet and, at 7 weeks of age, were treated with a single dose of 2 mg (10 μmol) by intraperitoneal injection. The experiment was terminated 16 weeks later, producing 8 – 12 lung tumors per mouse, and. no forestomach tumors. Use of an “open formula” diet significantly decreases tumor multiplicity147,148. The lung tumors observed at 16 weeks are all adenomas; adenocarcinoma are observed 40–50 weeks after treatment149.
Urethane: 6 week old mice were given a single i.p. injection (1 mg, about 225 μmol) per gram body weight, in saline. About 30 – 50 lung adenomas per mouse were observed 15 weeks after injection. Lung adenocarcinoma appeared 32 weeks after injection77.
Vinyl carbamate: Mice (7–8 weeks old) were injected i.p. with two doses (0.32 mg, 3.6 μmol) in saline, one week apart. The mice were sacrificed 16 weeks later and had 16 lung tumors per mouse, all of which were described as invasive carcinoma90, although in another study the carcinoma yield was apparently much lower79.
In studies with these carcinogens, statistically meaningful results can be obtained with only 15 mice per group. This approach is highly practical for examining potential chemopreventive efficacy. The chemopreventive agent can be given during carcinogen treatment, after carcinogen treatment, or throughout the experiment to decipher its potential at different stages of the carcinogenic process. These assays are relatively rapid and inexpensive. A variation on the use of single carcinogens is the use of BaP and NNK, given in multiple doses35. The object of this design is to more closely approximate the effects of cigarette smoke by using a mixture of two of its important carcinogens, and also to allow intervention with chemopreventive agents at various points during carcinogen treatment to reflect to some extent the situation in smokers who are transitioning to quitting36. This aspect has been virtually completely overlooked in previous efficacy studies. A typical design is illustrated in Figure 1. Treatment with chemopreventive agents in the diet could begin one day after the 4th carcinogen treatment (or at other intervals if desired) to approximate the transitioning smoker, or one week after the last carcinogen treatment, to mimic the situation in ex-smokers37.
Figure 1.

A design for evaluating chemopreventive agents against lung tumorigenesis in A/J mice. Lung tumors are induced by weekly gavage doses of NNK + BaP.
Intervention can begin during the carcinogen treatment period (shown here at week 4) or afterward. Adenoma can be scored at week 27 and adenocarcinoma at week 44.
While the A/J mouse is a widely used and convenient model for the induction of adenocarcinoma and investigation of the effects of chemopreventive agents, a similar model for induction of squamous cell carcinoma became available only fairly recently. Lijinsky and Reuber reported that application of N-nitroso-tris-chloroethylurea (NTCU) to the skin of Swiss mice produced various tumors including squamous cell carcinoma of the lung38. Wang et al treated 8 inbred strains of mice with NTCU by skin painting and observed that squamous cell carcinoma of the lung were produced in a strain-specific manner, with A/J, NIH Swiss and SWR/J being the most susceptible (tumor incidence 75 – 100%)39. This model should be useful for investigating chemoprevention of squamous cell carcinoma of the lung. However, it should be noted that NTCU is a synthetic carcinogen that is not present in cigarette smoke.
Treatment of F-344 rats with NNK results in the production of lung adenoma and adenocarcinoma, and this model has been used for investigating chemopreventive agents, although less frequently than the A/J mouse40. The rat studies are more expensive than the mouse experiments because 2 years are required for the development of lung tumors. The F-344 rat is far less susceptible to lung tumor induction than the A/J mouse, and there is virtually no background incidence of lung tumors. The rat model is an attractive one for confirming lung chemoprevention activity observed in mice40. In another approach, the induction and modification by chemopreventive agents of preneoplastic lesions of the lung induced by intratracheal instillation of NNK in Wistar rats has been described41. A hamster model of neuroendocrine lung carcinogenesis involving hyperoxic lung injury and treatment with NNK has also been used, as has an adenocarcinoma model initiated by NNK treatment of hamsters without hyperoxia42.
Preclinical studies identify effective chemopreventive agents
Naturally occurring and synthetic agents that prevent lung carcinogenesis in laboratory animals are summarized in Tables 1 and 2, and structures of individual compounds are shown in Figure 2. Our purpose here is to present an overview of current effective agents without a detailed evaluation of efficacy and potential toxicity which is beyond the scope of this review. We focus on agents that have been the subject of relatively recent investigations, mainly in this century. Previous reviews have summarized data on earlier studies40,43,44. The diversity of chemical structures in Figure 2 reflects the multiple targets that have been investigated for chemoprevention of lung carcinogenesis. This is appropriate because cigarette smoke causes multiple alterations in critical growth control pathways. Ultimately, rationally constructed mixtures of some of these agents will undoubtedly be needed for successful chemoprevention.
Table 1.
Some naturally occurring agents tested for chemoprevention of lung carcinogenesis.
| Compound | Administration to A/J Mice relative to Carcinogen | Percent Reduction in Lung Tumors per Mouse | Comments | References | |||
|---|---|---|---|---|---|---|---|
| Before/During | After | Throughout | Partially During, then After | ||||
| PEITC or PEITC-NAC | ✓ |
✓ |
✓ | ✓ | 30 to >70 <30 to 70 |
Highly effective against NNK, but not BaP; most effective when given during carcinogen treatment; also effective in NNK-treated rats, but not in tobacco smoke-treated A/J mice | 36,45–48,132 |
| 8-methoxypsoralen | ✓ |
✓ |
30 to >70 none |
52–54 | |||
| myo-inositol | ✓ | ✓ | ✓ | ✓ | 30 to 70 | 55–57,133 | |
| PEITC-NAC + myo-inositol | ✓ | ✓ | ✓ |
✓ |
30 to 70 30 to >70 |
Significant inhibition of adenocarcinoma observed | 36,37 |
| Indole-3-carbinol | ✓ | ✓ | ✓ | ✓ | 30 to 70 | 63–65 | |
| Tea, Polyphenon E, EGCG | ✓ | ✓ |
✓ |
30 to 70 30 to >70 |
Polyphenon E inhibits progression of adenoma to adenocarcinoma in mice; caffeine and black tea inhibit in NNK-treated rats; divergent results in hamsters; no effect in tobacco smoke-treated A/J mice | 42,68–71,75,81,134,135 | |
| Kava | ✓ | ✓ | ✓ | 30 to 70 | 73 | ||
| Anti-tumor B | ✓ | 30 to 70 | 74 | ||||
| Red ginseng | ✓ | 30 to 70 | 75 | ||||
| Pomegranate fruit extract | ✓ | 30 to 70 | 76 | ||||
| Silibinin | ✓ |
✓ |
none 30 to 70 |
77,78,136 | |||
| Rapamycin | ✓ | >70 (tumor load only) | anti-progression protocol only | 75 | |||
| Perillyl alcohol | ✓ | 30 to 70 | 103 | ||||
| Deguelin | ✓ | ✓ | ✓ | 30 to 70 | Potential toxic effects; new analogues investigated | 78,111,112 | |
Table 2.
Some synthetic agents tested for chemoprevention of lung carcinogenesis.
| Compound | Administration to A/J Mice relative to Carcinogen | Percent Reduction in Lung Tumors per Mouse | Comments | References | ||
|---|---|---|---|---|---|---|
| Before/During | After | Throughout | ||||
| FAS inhibitors | ✓ | 30 to 70 | 67 | |||
| Polyhenon E + atorvastatin | ✓ | 30 to 70 | 72 | |||
| Dexamethasone | ✓ |
✓ |
✓ |
<30 30 to 70 |
Some activity in tobacco smoke treated A/J mice, but not significant | 56,79,137 |
| Dexamethasone + myo-inositol | ✓ | >70 | Significant inhibition in tobacco smoke treated A/J mice | 56,80,81 | ||
| Budesonide | ✓ |
✓ |
30 to >70 30 to 70 |
Effective by inhalation or dietary administration; inhibited progression to adenocarcinoma | 82–86,138 | |
| DMFO | 30 to 70 | Inhibited lung tumorigenesis in a hamster model, post-carcinogen | 87 | |||
| CDDO-methyl ester | ✓ | 30 to 70 | Similar activity for ethyl amide | 90,139 | ||
| NRX194204 (rexinoid) | ✓ | 30 to 70 | 91 | |||
| Targretin (rexinoid) | ✓ | <30 to 70 | 92 | |||
| Sulindac and sulindac sulfone | ✓ | 30 to >70 | Only effective against chronic NNK; inhibition observed by other NSAIDS also | 95–97,140–142 | ||
| A79175, MK-886 (Lipoxygenase inhibitors) | ✓ |
✓ |
<30 to 70 0 to >70 |
100,101 | ||
| R115777 (farnesyltransferase inhibitor) | ✓ | 30 to 70 | 104 | |||
| FTI-276 (farnesyltransferase inhibitor) | ✓ | 30 to 70 | 103 | |||
| 1,4-phenylene-bis(methylene)-selenocyanate (XSC) | ✓ |
✓ |
>70 30 to 70 |
No effect in tobacco smoke-treated A/J mice | 81,105,106,108 | |
| Pro-drugs of selenocystine | ✓ | ✓ |
✓ |
30 to 70 30 to >70 |
109,110 | |
| 5-aza-2′-deoxycytidine | ✓ | ✓ | 30 to 70 | 113,114 | ||
| Erlotinib | ✓ | none | 143 | |||
Figure 2.

Structures of diverse inhibitors of lung carcinogenesis discussed in the paper
Multiple studies carried out over the past three decades clearly demonstrate that isothiocyanates inhibit lung carcinogenesis in animal models45. PEITC and its metabolite PEITC-NAC have been investigated in the most detail and, among isothiocyanates, overall have the best properties consistent with chemoprevention of lung carcinogenesis36,45–48. PEITC and PEITC-NAC are particularly effective against carcinogenesis by NNK, as shown in studies in both rats and mice, but they are less effective against lung carcinogenesis by PAH, or in the post-carcinogen treatment period. Benzyl isothiocyanate (BITC) is a highly effective inhibitor of PAH carcinogenesis49. In smokers, and in those transitioning to quitting, PEITC or PEITC-NAC could potentially neutralize the lung carcinogenic effects of NNK, at least based on animal studies in which these agents inhibit the metabolic activation of NNK. PEITC is a strong inhibitor of cytochrome P450 2A13 (Ki 30 nM), the most effective catalyst of NNK metabolic activation in the human respiratory tract50. BITC has the potential to neutralize carcinogenesis by PAH. Thus, tobacco smoke carcinogens are targets of isothiocyanates, but these compounds also have some favorable downstream effects on pathways involved in apoptosis and proliferation of transformed cells51. Similar to PEITC, 8-methoxypsoralen is an inhibitor of P450 2A enzymes and an effective inhibitor of NNK induced mouse lung tumorigenesis52–54.
Wattenberg was the first to demonstrate that myo-inositol is an effective inhibitor of lung carcinogenesis by both NNK and BaP55,56. It inhibits lung carcinogenesis by a mixture of BaP plus NNK when given either during the carcinogen treatment period or afterwards, thus suggesting potential efficacy in smokers and ex-smokers57. There appear to be virtually no toxic effects associated with myo-inositol treatment, as recently confirmed in a Phase I clinical trial in which the maximum tolerated dose was 18 g per day58. Although the major mechanism(s) by which myo-inositol inhibits lung carcinogenesis are not clear, a recent study demonstrates that it inhibits activation of Akt37. Tobacco smoke carcinogens and their post-carcinogen treatment activities are targets of myo-inositol.
Many epidemiologic studies demonstrate that consumption of cruciferous vegetables is associated with lower lung cancer risk, and this effect appears to be particularly strong in people with GSTM1 and GSTT1 null genotypes, indicating a diet-gene interaction59. The unique property of cruciferous vegetables is the presence of glucosinolates which, upon consumption of the raw vegetable (or to a lesser extent, the cooked vegetable), yield isothiocyanates and indole-3-carbinol among other products60. The chemopreventive properties of isothiocyanates as noted above are consistent with these observations, but among the major products to which humans are exposed when they consume common cruciferous vegetables are indole-3-carbinol and its dimer, di-indolyl methane (DIM) which forms in the stomach due to the low pH60–62. Indole-3-carbinol and DIM are both effective inhibitors of lung carcinogenesis by BaP plus NNK, and the effects of indole-3-carbinol have been observed both in the carcinogen treatment and post-carcinogen treatment phases63–65. Indole-3-carbinol enhances the hepatic clearance of NNK, and decreases levels of some critical proteins such as hypoxia inducible factor 1α (HIF-1α) and fatty acid synthase (FAS) in mouse lung tumors66. While indole-3-carbinol appears to have multiple targets, specific inhibitors of FAS such as C75 have chemopreventive activity against mouse lung tumorigenesis67.
A large body of experimental data demonstrates that tea and its constituents inhibit lung carcinogenesis in laboratory animals68,69. Green tea, popular in Asia, contains 30–40% by weight catechins such as (−)-epigallocatechin-3-gallate (EGCG) and others, whereas black tea, more popular in Western nations, is processed in such a way as to release phenol oxidase, thus oxidizing the catechins to oligomers such as theaflavins and to polymers called thearubigins68,69. A standardized green tea polyphenol preparation called “Polyphenon E” has also been used for chemoprevention studies. In the NNK lung carcinogenesis model, and in other models, green tea, black tea, and their decaffeinated versions, as well as Polyphenon E significantly inhibited tumor development68,69. Inhibition has also been seen in models using a variety of other lung carcinogens including BaP68,69. Both black tea and Polyphenon E inhibited the progression of adenoma to adenocarcinoma in mice treated with NNK70, and Polyphenon E inhibited progression to large carcinoma in BaP-treated mice71. Multiple mechanisms have been reported for the inhibitory properties of tea and its constituents including induction of phase II enzymes, decreased oxidative damage, induction of apoptosis, inhibition of cell proliferation, and others68,69. Synergistic inhibition was observed with a combination of Polyphenon E and atorvastatin72. Another beverage which has shown chemopreventive activity against lung carcinogenesis is kava, a root extract consumed widely by South Pacific islanders. Kava inhibited lung tumorigenesis when given in the carcinogen treatment or post-carcinogen treatment phases73.
A Chinese herbal mixture called Antitumor B, also known as Zeng Sheng Ping, is comprised of six plants, and has a history of safety in clinical use. Antitumor B significantly decreased tumor multiplicity and tumor load in mice treated with BaP74. Ginseng is also a traditional medicine used in Asia. It had suppressing activity against lung tumor multiplicity in mice treated with BaP75. Pomegranate fruit extract is another plant-based agent with considerable inhibitory activity against lung tumorigenesis76.
Silibinin, a flavonone from milk thistle, is structurally related to tea polyphenols. It has been used as a dietary supplement to improve liver function and as an anti-hepatotoxic drug77. It apparently has very low toxicity. Silibinin added to the diet, at concentrations of 0.033 – 1%, of mice treated with urethane significantly decreased lung tumor incidence recorded 20 weeks later77. Silibinin treatment decreased proliferation markers and tumor microvessel density, as well as lung tumor expression of vascular endothelial growth factor, inducible nitric oxide synthase, and cyclooxygenase-2, all believed to be involved in inflammation and tumor progression77. Silibinin (0.05 – 0.1% in the diet) given prior to BaP had no effect on tumor multiplicity or tumor load78. Thus, tumor promotion and inflammation are targets of silibinin.
Dexamethasone and budesonide are glucocorticoids, which bind to and activate the cytosolic glucocorticoid receptor. Dexamethasone had been shown to inhibit the promotion stage of carcinogenesis in various models and was first applied in lung carcinogenesis studies by Wattenberg55. In further studies, it demonstrated good activity in various mouse models, and particularly in combination with myo-inositol56,79–81. This combination is the only one reported to successfully inhibit lung tumorigenesis in the tobacco smoke inhalation model described by Witschi81. Similarly, budesonide shows good activity in multiple mouse models82–85. A potentially important approach to chemoprevention uses inhaled budesonide, greatly decreasing the risk of systemic side effects, while maintaining excellent efficacy at low doses86. This approach was also successful when combined with dietary myo-inositol in mouse models86. Difluoromethylornithine (DFMO), an inhibitor of ornithine decarboxylase involved in tumor promotion, is also effective as a chemopreventive agent against squamous cell carcinoma when given by inhalation to hamsters87.
Oleanane and ursane triterpenoids are pentacyclic compounds derived biosynthetically from squalene. Sporn, Gribble and co-workers have targeted inflammation with diverse structural analogues which inhibit inducible nitric oxide synthase and cyclooxygenase-2, and are also phase II enzyme inducers88,89. CDDO-methyl ester as well as CDDO-ethyl amide are potent inhibitors of vinyl carbamate-induced mouse lung carcinogenesis in the post-carcinogen phase90. Rexinoids, selective ligands for the retinoid X receptors RXRα, RXRβ, and RXRγ, with anti-inflammatory activity, are also effective. Targretin and NRX194204 have shown activity in the post carcinogen phase91,92.
Rapamycin, a natural product isolated from Streptomyces hygroscopicus, is an inhibitor of mTOR (mammalian target of rapamycin), which is downstream from Akt and PI3K, a pathway commonly activated in lung carcinogenesis. Rapamycin decreased lung tumor load, but not tumor multiplicity, in a mouse anti-progression protocol in which BaP was the carcinogen75.
Inflammation has also been targeted by non-steroidal anti-inflammatory drugs (NSAIDS) such as sulindac. Cyclooxygenase (COX) enzymes play a key role in the synthesis of prostanoids involved in inflammation. COX-1 is constitutive while COX-2 is inducible. COX-2 is induced and becomes constitutively expressed as tumors progress. COX-2 expression is observed in human lung non-small cell lung cancer and expression of both forms has been observed in normal mouse lung and lung tumors22,93,94. Sulindac, its sulfone metabolite, and aspirin, as well as several other COX inhibitors, are effective chemopreventive agents in NNK treated mice95–99. However, the specific COX-2 inhibitor celecoxib, while reducing pulmonary inflammation, had no effect on lung tumor multiplicity in A/J mice99. Interest in COX-2 inhibitors has been affected by cardiovascular toxicity22. Lipoxygenase inhibitors, which inhibit the formation of leukotrienes involved in inflammation, have also been effective. A-79175 and MK-866 are examples which inhibit lung carcinogenesis100,101.
Human lung adenocarcinoma commonly have a mutated ras oncogene102. The ras proteins are GTPases involved in regulation of signal transduction pathways controlling proliferation and apoptosis. Ras proteins are typically farnesylated to become active, so farnesyltransferase inhibitors are natural targets for chemoprevention. Several farnesyltransferase inhibitors including R115777, FTI-276, and perillyl alcohol, have shown activity in BaP or NNK induced mouse lung tumor models103,104.
Organoselenium compounds have emerged as an interesting class of agents. 1,4-Phenylenebis(methylene)selenocyanate (XSC), a relatively non-toxic organoselenium compound, inhibits BaP plus NNK induced mouse lung tumorigenesis when given either during or after carcinogen administration, and has some favorable effects on phase I and phase II enzymes105,106. XSC reduced the expression of COX-2, NF-κB, and cyclin D1 in lung cells107. In contrast to XSC, selenium enriched yeast had no effect on NNK induced mouse lung tumorigenesis108. Another interesting class of organoselenium compounds is the selenazolidine carboxylic acids, prodrugs of selenocystine, which inhibit NNK induced lung tumorigenesis109,110.
Deguelin is an inhibitor of the PI3K/Akt pathway and decreases the expression of COX-2. It was an effective inhibitor of BaP plus NNK mouse lung tumorigenesis in both the carcinogen administration and post-carcinogen administration phases78,111. There is concern about potential toxic effects of deguelin, and structural variants are being examined112.
Inhibition of endogenous DNA hypermethylation, which can inhibit transcription of tumor suppressor genes, is another chemoprevention target. 5-Aza-2′-deoxycytidine (DAC) inhibits DNA methylation by reducing cytosine-DNA methyltransferase 1 activity. DAC inhibited NNK induced lung tumorigenesis in two different mouse models, and its effects were potentiated by the histone deacetylase inhibitor phenylbutyrate113,114.
Collectively, the data reviewed here demonstrate that effective agents exist targeting the main types of activities responsible for lung carcinogenesis: tobacco smoke carcinogens, their multiple associated and post-carcinogen proliferative activities (e.g., tumor promotion and co-carcinogenesis), and inflammation. Rationally constructed mixtures of selected agents should logically be effective in antagonizing lung carcinogenesis.
Development of a mixture for chemoprevention of lung carcinogenesis: PEITC-NAC plus myo-inositol as an example
The solid efficacy and low toxicity of PEITC-NAC and myo-inositol, along with evidence that they have different targets in lung carcinogenesis, suggested that a combination of these agents might be useful for chemoprevention of lung carcinogenesis in smokers transitioning to quitting and in ex-smokers.
The first goal of this study was to test the agents alone in different temporal sequences that reflect to some extent the situation in a smoker transitioning to quitting. No smoker would begin using chemopreventive agents at the same time as initiating smoking, yet most of the experiments described in the previous section, in which agents were tested during or before the carcinogen administration phase, reflected that unlikely situation. Therefore, we tested PEITC-NAC and myo-inositol, individually and in combination, starting 24h after the 4th or 6th carcinogen administration (see Figure 1) and continued their administration until the end of the experiment, 19 weeks after the final carcinogen administration. The results were compared to those obtained when the compounds were given for the entire experiment, or only after carcinogen administration, the latter mimicking their use in ex-smokers. All treatments led to significant reductions in lung tumor multiplicity, except PEITC-NAC starting after the 6th carcinogen treatment, or given post-carcinogen. For both agents, there was a significant trend for increased reduction in lung tumor multiplicity with increased duration of treatment. Combinations of PEITC-NAC and myo-inositol were tested, using non-toxic doses at which the individual compounds significantly reduced lung tumor multiplicity. In general, the mixture of PEITC-NAC plus myo-inositol was more effective than either agent alone, and when all results were combined, the combination was significantly more effective, with the combined efficacy being roughly additive36.
These positive results set the stage for a more detailed investigation of the mixture of PEITC-NAC plus myo-inositol. Toxicity studies were carried out which established non-toxic doses of PEITC-NAC with the exception of the presence of eosinophilic granules in the bladder mucosa. The mixture of PEITC-NAC plus MI, when given from the 50% point of carcinogen administration until termination at 44 weeks, inhibited lung tumor multiplicity by 46 – 72% (depending on the dose), and by 32% when given in the post-carcinogen phase alone. All of these decreases were significant. There was also a significant reduction of up to 75% in adenocarcinoma formation by PEITC-NAC plus myo-inositol given from the 50% time point, and a significant 53% reduction when given post-carcinogen only. A photograph of typical mouse lungs from this study is shown in Figure 337. Parallel mechanistic studies demonstrated that the observed inhibition of lung tumorigenesis was attributable in part to inhibition of cell proliferation and induction of apoptosis. While NNK plus BaP treatment caused increased phosphorylation of Akt and BAD (resulting in loss of its proapoptotic function), these were inhibited by both PEITC-NAC and myo-inositol. Further, proteomic analysis demonstrated that PEITC-NAC plus myo-inositol altered levels of multiple critical proteins in lung tumors from these mice66. Collectively, these results demonstrate that the mixture of PEITC-NAC and myo-inositol is effective and can be advanced to the next stage of development.
Figure 3.
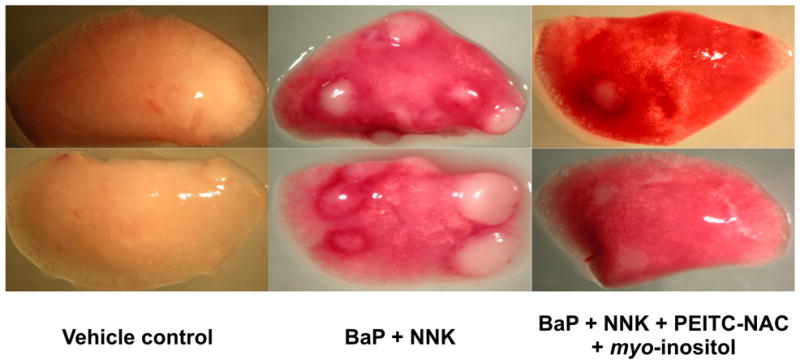
Photographs of lungs from two mice, each treated with vehicle (cottonseed oil) only, or a mixture of BaP plus NNK (2 μmol of each, once weekly for 8 weeks, as in Figure 1), or BaP plus NNK and a mixture of PEITC-NAC and myo-inositol in the diet, starting 24h after the 4th administration of BaP plus NNK. The mice were sacrificed 44 weeks after the beginning of the experiment, as in Figure 1.
From Animal Models to Clinical Trials
There are presently no chemopreventive agents that have demonstrated efficacy against lung cancer in clinical trials. All trials to date have yielded negative or even damaging results, as reviewed previously22,44,115–120. While potential reasons for these negative results have been discussed extensively in previous reviews, one major explanation is a violation in some cases of rule number one: efficacy in laboratory animal models of lung carcinogenesis. In this section we summarize some current clinical trials which are based at least partially on the efficacy studies summarized in Table 1 and discussed above. These trials are described on the National Cancer Institute web site121. Trial designs for chemoprevention have also been reviewed122.
A phase II trial of PEITC is designed to determine, as the primary endpoint, whether PEITC has the same inhibitory properties on the metabolic activation of NNK in smokers as it does in rats, in a randomized, placebo controlled trial. As a secondary endpoint, the effects of GSTM1 plus GSTT1 null status on the inhibitory activity of PEITC will be determined. In an associated longer term study, the effects of PEITC on biomarkers of bronchial epithelial cell apoptosis and proliferation will be assessed. This trial finds further support from the results of two nested case control studies demonstrating a significant relationship of the NNK biomarker total NNAL to lung cancer123,124.
Lam and co-workers obtained some evidence for regression of pulmonary dysplasia in subjects enrolled in a Phase I trial of myo-inositol58. This observation, together with the multiple efficacy studies described above and the established low toxicity of myo-inositol, led to a Phase II study to compare myo-inositol vs. placebo in the reversion of bronchial dysplasia in current or former smokers, as the primary endpoint. Secondary endpoints include biomarkers of proliferation, apoptosis, and angiogenesis in bronchial biopsy samples and biomarkers of inflammation in bronchial lavage and plasma samples.
A Phase II study with green tea will examine the effects of high dose green tea (four 12 oz. servings per day) or Polyphenon E (4 capsules per day) on biomarkers of oxidative damage in former smokers with COPD, as the primary endpoint. Secondary endpoints include body antioxidant status and antioxidant enzymes, and markers of apoptosis and proliferation in induced sputum. A second Phase II study of Polyphenon E will examine efficacy and safety in current or former smokers with bronchial dysplasia and increased inflammatory load as measured by C-reactive protein. Secondary endpoints in this trial include biomarkers of oxidative stress, inflammation, apoptosis, aberrant methylation, phase I and II enzyme expression, and proliferation.
A phase II trial of sulindac will examine the effects of sulindac vs. placebo on histologic grade of bronchial dysplasia determined in bronchoscopy exams in smokers or former smokers with bronchial dysplasia. Secondary endpoints include determination of the number of dysplastic lesions before and after treatment, and changes in biomarkers of the arachidonic acid pathway, as well as biomarkers of apoptosis and proliferation.
It is notable that there are no ongoing trials of mixtures of chemopreventive agents. This contrasts with a major theme of this article. The principal that mixtures can be effective in chemoprevention clinical trials has recently been established in a study of sulindac and DFMO for chemoprevention of recurrence of colon adenomas, without serious toxicity125.
The role of chemoprevention in treatment of smokers
The primary concern over the use of a chemopreventive agent against lung carcinogenesis is that it may give smokers a false sense of security. They may feel that smoking is “safe” or is significantly “safer,” which will result in their continuing to smoke, relapsing to smoking or even initiating smoking. However, providing treatments to those individuals who continue to practice behaviors that put them at high risk for disease is not uncommon. For example, statins or anithypertensives are not withheld from patients with poor eating habits and a sedentary lifestyle because the health care providers are concerned the use of these agents might contribute to the obesity epidemic. A similar analogy can be made with chemopreventive agents for smoking.
What is clear is that safeguards need to be in place so that smokers are not misled or have the misconception that using a chemopreventive agent is the solution and makes smoking safe. A chemopreventive agent may reduce the risk of one disease, such as lung cancer, however, there are other diseases that are associated with cigarette smoking including other cancers, heart disease and lung disease126. Therefore, smoking cessation must be the primary goal for and message to the patient. Although some researchers have advocated for the use of chemopreventive agents for only those who want to quit smoking, the rate of success is low and smokers often transition in and out of quitting127 and in and out of being motivated to quit128, making it difficult to determine who should and should not receive chemopreventive treatment if the prescription criterion is based only on whether or not the smoker is ready to quit. Therefore, we believe that smokers uninterested in quitting or unable to quit should be considered for chemopreventive therapy, although this approach has been barely recognized by those interested in tobacco harm reduction strategies129. In addition to smokers, based on the mechanism of action, successful quitters or former smokers potentially can benefit from chemopreventive agents.
According to the principles that are used to guide proposed public health interventions, it is critical that the intervention reduces rather than increases morbidity and mortality on a population level, that it results in no more harm than already exists, that the risks and benefits are distributed equitably across different populations (no population benefited at the expense of another), and that the autonomous choices of individuals and communities are respected130,131. These are the criteria by which chemopreventive therapies for tobacco-related diseases should be evaluated.
Conclusions
In spite of the lack of success in chemoprevention of lung carcinogenesis so far, there is reason to be optimistic. The data summarized here clearly demonstrate that there are multiple agents that are effective inhibitors of lung carcinogenesis in animal models, and these agents operate by diverse mechanisms. It is likely that success will depend on judicious use of a combination of these agents because cellular damage from years of cigarette smoking is both complex and extensive. Single agents that target single pathways or carcinogens are not likely to be successful. We need to target the multiple activities of cigarette smoke: its carcinogens and toxicants and their downstream, tumor promoting, and inflammatory effects. The successful mixture will be assembled stepwise and driven by efficacy testing in one or more of the animal models described here. This chemopreventive mixture will have minimal toxicity in animal models and humans, which might be achievable by using naturally occurring compounds in doses no greater than those present in common foods such as vegetables. All smokers should be considered for chemoprevention of lung carcinogenesis but with the strong message that no chemopreventive agent makes smoking safe. In addition, chemoprevention should be given in the context of providing smoking cessation advice and assistance. Ex-smokers should also benefit from chemoprevention. Although not discussed here, genetic, molecular and phenotypic biomarkers could be used to select those subjects at highest risk for lung cancer, and treatment should be promptly delivered to such individuals. While avoidance of tobacco products is the surest way to decrease lung cancer risk, chemoprevention promises to be a useful adjunct strategy.
At a Glance.
Lung cancer kills more than 3000 people every day in the world, and most of this toll is due to cigarette smoking. Although tobacco control is clearly the most desirable way to prevent lung cancer, cigarette smoking is addictive and despite considerable success to date, there are still over a billion smokers in the world who, along with ex-smokers, are at high risk for lung cancer. Chemoprevention of lung carcinogenesis is one way forward in control of this devastating disease.
In considering chemoprevention, it is crucial that we focus on treating lung carcinogenesis, not lung cancer. The disease process is carcinogenesis.
Lung carcinogenesis is caused by multiple carcinogens in cigarette smoke, along with tumor promoters, co-carcinogens, toxicants, and inflammatory agents. In devising chemoprevention strategies, these multiple agents should be our targets. Targeting a single pathway in lung carcinogenesis is not likely to be successful.
Because there are multiple carcinogenic and toxic constituents of tobacco smoke, we will need to develop a mixture of chemopreventive agents to counteract them. This mixture should be developed from the ground up, using animal models to demonstrate efficacy without appreciable toxicity.
Well established animal models are available for evaluating chemopreventive efficacy against lung carcinogenesis. The most commonly used model by far is the carcinogen treated A/J mouse, which develop adenocarcinoma similar to those seen in humans.
Many agents have shown chemopreventive efficacy against lung carcinogenesis in animal models. Examples include phenethyl isothiocyanate, indole-3-carbinol, myo-inositol, green and black tea and its constituents, silibinin, glucocorticoids, difluoromethylornithine, oleanane and ursane triterpenoids, non-steroidal anti-inflammatory drugs, farnesyltrasferase inhibitors, organoselenium compounds, and others. Some mixtures of these agents also demonstrate efficacy.
There have been no successful lung carcinogenesis clinical trials. Current trials include examinations of some of the agents listed above, but no mixtures.
In chemoprevention of lung carcinogenesis, we must target current smokers, smokers transitioning to quitting, and ex-smokers. While cessation is clearly the best way to decrease the probability of getting lung cancer, most smokers cannot quit, even after many tries. It would be unethical not to offer these people effective agents.
Biographies
Stephen S. Hecht
Stephen S. Hecht, Ph.D., is Wallin Professor of Cancer Prevention and head of the Carcinogenesis and Chemoprevention Program, Masonic Comprehensive Cancer Center, University of Minnesota. Hecht laboratory research focuses on identifying individuals susceptible to the cancer causing effects of tobacco. Tobacco carcinogen biomarkers are developed for integration into a predictive algorithm to identify susceptible smokers, who can then be targeted for preventive strategies. Naturally occurring chemopreventive agents against lung carcinogenesis are identified through efficacy and toxicity testing in relevant animal models. Mixtures of these agents are tested for efficacy and parallel studies are performed to elucidate their mechanisms of action.
Fekadu Kassie
Fekadu Kassie is a member of the Carcinogenesis and Chemoprevention Program at the Masonic Comprehensive Cancer Center of the University of Minnesota, and an Assistant Professor of Oncology and Comparative Medicine in the College of Veterinary Medicine, University of Minnesota. He received his DVM degree from Addis Ababa University, Ethiopia, and MSc and Ph.D. in Toxicology from the University of Vienna, Austria. In 2004, he joined the laboratory of Stephen S. Hecht as a research associate. He assumed his current position in 2008. His research interests are cancer chemoprevention and identification of cancer biomarkers.
Dorothy Hatsukami
Dorothy Hatsukami, Ph.D. is currently Forster Family Professor in Cancer Prevention and Professor of Psychiatry at the University of Minnesota, and Director of the Tobacco Use Research Center. She is Associate Director for Cancer Prevention and Control at the University of Minnesota Masonic Comprehensive Cancer Center. She is recognized for her work in the areas of nicotine addiction and its treatment (cigarettes and smokeless tobacco) in adult and adolescent smokers. She is currently a Principal Investigator of an NIH funded Transdisciplinary Tobacco Use Research Center (TTURC) which focuses on methods to reduce tobacco toxicant exposure and toxicity.
References
- 1.Gilpin EA, Pierce JP. Demographic differences in patterns in the incidence of smoking cessation: United States 1950–1990. Ann Epidemiol. 2002;12:141–150. doi: 10.1016/s1047-2797(01)00266-6. [DOI] [PubMed] [Google Scholar]
- 2.Fiore MC, et al. Treating tobacco use and dependence: 2008 update. Rockville, MD: U.S. Department of Health and Human Services, Public Health Service; 2008. Clinical Practice Guideline. [Google Scholar]
- 3.Giovino GA. Epidemiology of tobacco use in the United States. Oncogene. 2002;21:7326–7340. doi: 10.1038/sj.onc.1205808. [DOI] [PubMed] [Google Scholar]
- 4.Giovino GA. The tobacco epidemic in the United States. Am J Prev Med. 2007;33:S318–S326. doi: 10.1016/j.amepre.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 5.Warner KE. Charting the science of the future where tobacco-control research must go. Am J Prev Med. 2007;33:S314–S317. doi: 10.1016/j.amepre.2007.09.010. [DOI] [PubMed] [Google Scholar]
- 6.Warner KE, Burns DM. Hardening and the hard-core smoker: concepts, evidence, and implications. Nicotine Tob Res. 2003;5:37–48. doi: 10.1080/1462220021000060428. [DOI] [PubMed] [Google Scholar]
- 7.Ridker PM, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–2207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]
- 8.Sporn MB, Dunlop NM, Newton DL, Smith JM. Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids) Fed Proc. 1976;35:1332–1338. [PubMed] [Google Scholar]
- 9.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–1210. doi: 10.1093/jnci/91.14.1194. This paper summarizes the role of individual tobacco smoke carcinogens as causative agents for lung cancer in smokers. [DOI] [PubMed] [Google Scholar]
- 10.Hecht SS. Tobacco carcinogens, their biomarkers, and tobacco-induced cancer. Nature Rev Cancer. 2003;3:733–744. doi: 10.1038/nrc1190. This paper discusses a conceptual model for understanding tobacco carcinogenesis and reviews tobacco carcinogen biomarkers. [DOI] [PubMed] [Google Scholar]
- 11.Vogel G. Breakthrough of the year. Reprogramming cells. Science. 2008;322:1766–1767. doi: 10.1126/science.322.5909.1766. [DOI] [PubMed] [Google Scholar]
- 12.International Agency for Research on Cancer. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 83. IARC; Lyon, FR: 2004. pp. 53–119. This monograph provides a comprehensive review of tobacco smoke carcinogens and other relevant information. [PMC free article] [PubMed] [Google Scholar]
- 13.Hecht SS. Carcinogenicity studies of inhaled cigarette smoke in laboratory animals: old and new. Carcinogenesis. 2005;26:1488–1492. doi: 10.1093/carcin/bgi148. This review discusses progress and problems in inhalation studies of tobacco smoke carcinogenesis. [DOI] [PubMed] [Google Scholar]
- 14.International Agency for Research on Cancer. IARC Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Humans. Vol. 38. IARC; Lyon, FR: 1986. pp. 127–198. [Google Scholar]
- 15.Hoffmann D, Schmeltz I, Hecht SS, Wynder EL. In: Polycyclic Hydrocarbons and Cancer. Gelboin H, Ts’o POP, editors. Academic Press; New York: 1978. pp. 85–117. [Google Scholar]
- 16.Hecht SS, Thorne RL, Maronpot RR, Hoffmann D. Tumor-promoting subfractions of the weakly acidic fraction. J Natl Cancer Inst. 1975;55:1329–1336. doi: 10.1093/jnci/55.6.1329. [DOI] [PubMed] [Google Scholar]
- 17.Kensler CJ, Battista SP. Components of cigarette smoke with ciliary-depressant activity. Their selective removal by filters containing activated charcoal granules. N Engl J Med. 1963;269:1161–1166. doi: 10.1056/NEJM196311282692202. [DOI] [PubMed] [Google Scholar]
- 18.Chung FL, Young R, Hecht SS. Formation of cyclic 1, N2-propanodeoxyguanosine adducts in DNA upon reaction with acrolein or crotonaldehyde. Cancer Res. 1984;44:990–995. [PubMed] [Google Scholar]
- 19.Feng Z, Hu W, Hu Y, Tang MS. Acrolein is a major cigarette-related lung cancer agent. Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc Natl Acad Sci USA. 2006;103:15404–15409. doi: 10.1073/pnas.0607031103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thompson CA, Burcham PC. Genome-wide transcriptional responses to acrolein. Chem Res Toxicol. 2008 doi: 10.1021/tx8001934. [DOI] [PubMed] [Google Scholar]
- 21.Smith CJ, Perfetti TA, King JA. Perspectives on pulmonary inflammation and lung cancer risk in cigarette smokers. Inhal Toxicol. 2006;18:667–677. doi: 10.1080/08958370600742821. [DOI] [PubMed] [Google Scholar]
- 22.Lee JM, et al. Inflammation in lung carcinogenesis: new targets for lung cancer chemoprevention and treatment. Crit Rev Oncol Hematol. 2008;66:208–217. doi: 10.1016/j.critrevonc.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Malkinson AM. Role of inflammation in mouse lung tumorigenesis: a review. Exp Lung Res. 2005;31:57–82. doi: 10.1080/01902140490495020. This review discusses the role of inflammation in lung tumorigenesis in mice. [DOI] [PubMed] [Google Scholar]
- 24.Fischer SM. Comprehensive Toxicology. In: Bowden GT, Fischer SM, editors. Chemical Carcinogens and Anticarcinogens. Vol. 12. Elsevier Science; New York: 1997. pp. 349–381. [Google Scholar]
- 25.Kim V, Rogers TJ, Criner GJ. Frontiers in emphysema research. Semin Thorac Cardiovasc Surg. 2007;19:135–141. doi: 10.1053/j.semtcvs.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 26.Turner MC, Chen Y, Krewski D, Calle EE, Thun MJ. Chronic obstructive pulmonary disease is associated with lung cancer mortality in a prospective study of never smokers. Am J Respir Crit Care Med. 2007;176:285–290. doi: 10.1164/rccm.200612-1792OC. [DOI] [PubMed] [Google Scholar]
- 27.Wattenberg LW. Chemoprevention of cancer. Cancer Res. 1985;45:1–8. [PubMed] [Google Scholar]
- 28.Witschi H. A/J mouse as a model for lung tumorigenesis caused by tobacco smoke: strengths and weaknesses. Exp Lung Res. 2005;31:3–18. doi: 10.1080/01902140490494959. [DOI] [PubMed] [Google Scholar]
- 29.Stoner GD, Shimkin MB. Strain A mouse lung tumor bioassay. J Amer Coll Toxicol. 1982;1:145–169. [Google Scholar]
- 30.Stoner GD. Lung tumors in strain A mice as a bioassay for carcinogenicity of environmental chemicals. Exp Lung Res. 1991;17:405–423. doi: 10.3109/01902149109064428. [DOI] [PubMed] [Google Scholar]
- 31.De Flora S, et al. Induction and modulation of lung tumors: genomic and transcriptional alterations in cigarette smoke-exposed mice. Exp Lung Res. 2005;31:19–35. doi: 10.1080/01902140490494986. [DOI] [PubMed] [Google Scholar]
- 32.Malkinson AM. Primary lung tumors in mice as an aid for understanding, preventing, and treating human adenocarcinoma of the lung. Lung Cancer. 2001;32:265–279. doi: 10.1016/s0169-5002(00)00232-4. [DOI] [PubMed] [Google Scholar]
- 33.O’Donnell EP, Zerbe LK, Dwyer-Nield LD, Kisley LR, Malkinson AM. Quantitative analysis of early chemically-induced pulmonary lesions in mice of varying susceptibilities to lung tumorigenesis. Cancer Lett. 2006;241:197–202. doi: 10.1016/j.canlet.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 34.Shimkin MB, Stoner GD. Lung tumors in mice: application to carcinogenesis bioassay. Adv Cancer Res. 1975;21:1–58. doi: 10.1016/s0065-230x(08)60970-7. This classic paper reviews the older literature on the A/J mouse lung tumor bioassay. [DOI] [PubMed] [Google Scholar]
- 35.Hecht SS, Isaacs S, Trushin N. Lung tumor induction in A/J mice by the tobacco smoke carcinogens 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and benzo[a]pyrene: a potentially useful model for evaluation of chemopreventive agents. Carcinogenesis. 1994;15:2721–2725. doi: 10.1093/carcin/15.12.2721. [DOI] [PubMed] [Google Scholar]
- 36.Hecht SS, et al. Inhibition of lung tumorigenesis in A/J mice by N-acetyl-S-(N-2-phenethylthiocarbamoyl)-L-cysteine and myo-inositiol, individually and in combination. Carcinogenesis. 2002;23:1455–1461. doi: 10.1093/carcin/23.9.1455. [DOI] [PubMed] [Google Scholar]
- 37.Kassie F, et al. Combinations of N-acetyl-S-(N-2-phenethylthiocarbamoyl)-L-cysteine and myo-inositol inhibit tobacco smoke carcinogen-induced lung adenocarcinoma in A/J mice. Cancer Prev Res. 2008;1:285–297. doi: 10.1158/1940-6207.CAPR-08-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lijinsky W, Reuber MD. Neoplasms of the skin and other organs observed in Swiss mice treated with nitrosoalkylureas. J Cancer Res Clin Oncol. 1988;114:245–249. doi: 10.1007/BF00405829. [DOI] [PubMed] [Google Scholar]
- 39.Wang Y, et al. A chemically induced model for squamous cell carcinoma of the lung in mice: histopathology and strain susceptibility. Cancer Res. 2004;64:1647–1654. doi: 10.1158/0008-5472.can-03-3273. [DOI] [PubMed] [Google Scholar]
- 40.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11:559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 41.Ye B, et al. Induction of lung lesions in Wistar rats by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and its inhibition by aspirin and phenethyl isothiocyanate. BMC Cancer. 2007;7:90. doi: 10.1186/1471-2407-7-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schuller HM, Porter B, Riechert A, Walker K, Schmoyer R. Neuroendocrine lung carcinogenesis in hamsters is inhibited by green tea or theophylline while the development of adenocarcinomas is promoted: implications for chemoprevention in smokers. Lung Cancer. 2004;45:11–18. doi: 10.1016/j.lungcan.2003.12.007. [DOI] [PubMed] [Google Scholar]
- 43.Hecht SS. Approaches to chemoprevention of lung cancer based on carcinogens in tobacco smoke. Environ Health Perspect. 1997;105(Suppl 4):955–963. doi: 10.1289/ehp.97105s4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Zandwijk N. Chemoprevention in lung carcinogenesis - an overview. Eur J Cancer. 2005;41:1990–2002. doi: 10.1016/j.ejca.2005.05.011. This paper provides a useful overview of chemoprevention of lung carcinogenesis. [DOI] [PubMed] [Google Scholar]
- 45.Hecht SS. In: Cancer Chemoprevention Volume 1: Promising Cancer Chemopreventive Agents. Kelloff GJ, Hawk ET, Sigman CC, editors. The Humana Press; Totowa, N.J: 2004. pp. 21–35. [Google Scholar]
- 46.Morse MA, et al. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced DNA adduct formation and tumorigenicity in lung of F344 rats by dietary phenethyl isothiocyanate. Cancer Res. 1989;49:549–553. [PubMed] [Google Scholar]
- 47.Jiao D, et al. Chemopreventive activity of thiol conjugates of isothiocyanates for lung tumorigenesis. Carcinogenesis. 1997;18:2143–2147. doi: 10.1093/carcin/18.11.2143. [DOI] [PubMed] [Google Scholar]
- 48.Hecht SS, et al. Complete inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induced rat lung tumorigenesis and favorable modification of biomarkers by phenethyl isothiocyanate. Cancer Epidemiol Biomarkers & Prev. 1996;5:645–652. [PubMed] [Google Scholar]
- 49.Hecht SS, Kenney PMJ, Wang M, Upadhyaya P. Benzyl isothiocyanate: an effective inhibitor of polycyclic aromatic hydrocarbon tumorigenesis in A/J mouse lung. Cancer Lett. 2002;187:87–94. doi: 10.1016/s0304-3835(02)00410-x. [DOI] [PubMed] [Google Scholar]
- 50.von Weymarn LB, Chun JA, Hollenberg PF. Effects of benzyl and phenethyl isothiocyanate on P450s 2A6 and 2A13: potential for chemoprevention in smokers. Carcinogenesis. 2006;27:782–790. doi: 10.1093/carcin/bgi301. [DOI] [PubMed] [Google Scholar]
- 51.Xiao D, et al. Caspase-dependent apoptosis induction by phenethyl isothiocyanate, a cruciferous vegetable-derived cancer chemopreventive agent, is mediated by Bak and Bax. Clin Cancer Res. 2005;11:2670–2679. doi: 10.1158/1078-0432.CCR-04-1545. [DOI] [PubMed] [Google Scholar]
- 52.Takeuchi H, et al. Pretreatment with 8-methoxypsoralen, a potent human CYP2A6 inhibitor, strongly inhibits lung tumorigenesis induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in female A/J mice. Cancer Res. 2003;63:7581–7583. [PubMed] [Google Scholar]
- 53.Miyazaki M, et al. Mechanisms of chemopreventive effects of 8-methoxypsoralen against 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced mouse lung adenomas. Carcinogenesis. 2005;26:1947–1955. doi: 10.1093/carcin/bgi156. [DOI] [PubMed] [Google Scholar]
- 54.Takeuchi H, et al. Dose dependent inhibitory effects of dietary 8-methoxypsoralen on NNK-induced lung tumorigenesis in female A/J mice. Cancer Lett. 2006;234:232–238. doi: 10.1016/j.canlet.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 55.Estensen RD, Wattenberg LW. Studies of chemopreventive effects of myo-inositol on benzo[a]pyrene-induced neoplasia of the lung and forestomach of female A/J mice. Carcinogenesis. 1993;14:1975–1977. doi: 10.1093/carcin/14.9.1975. [DOI] [PubMed] [Google Scholar]
- 56.Wattenberg LW, Estensen RD. Chemopreventive effects of myo-mositol and dexamethasone on benzo[a]pyrene and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced pulmonary carcinogenesis in female A/J mice. Cancer Res. 1996;56:5132–5135. [PubMed] [Google Scholar]
- 57.Hecht SS, Kenney PMJ, Wang M, Upadhyaya P. Dose-response study of myo-inositol as an inhibitor of lung tumorigenesis induced in A/J mice by benzo[a]pyrene and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Lett. 2000;167:1–6. doi: 10.1016/s0304-3835(01)00454-2. [DOI] [PubMed] [Google Scholar]
- 58.Lam S, et al. A phase I study of myo-inositol for lung cancer chemoprevention. Cancer Epidemiol Biomarkers Prev. 2006;15:1526–1531. doi: 10.1158/1055-9965.EPI-06-0128. [DOI] [PubMed] [Google Scholar]
- 59.Lam TK, et al. Cruciferous vegetable consumption and lung cancer risk: a systematic review. Cancer Epidemiol Biomarkers Prev. 2009;18:184–195. doi: 10.1158/1055-9965.EPI-08-0710. This paper gives a current update on vegetable consumption and lung cancer risk. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fahey JW, Zalcmann AT, Talalay P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry. 2001;56:5–51. doi: 10.1016/s0031-9422(00)00316-2. [DOI] [PubMed] [Google Scholar]
- 61.Hecht SS, et al. Effects of cruciferous vegetable consumption on urinary metabolites of the tobacco-specific lung carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in Singapore Chinese. Cancer Epidemiol Biomarkers & Prev. 2004;13:997–1004. [PubMed] [Google Scholar]
- 62.International Agency for Research on Cancer. IARC Handbooks of Cancer Prevention. Vol. 9. IARC; Lyon, FR: 2004. pp. 25–42. [Google Scholar]
- 63.Morse MA, LaGreca SD, Amin SG, Chung FL. Effects of indole-3-carbinol on lung tumorigenesis and DNA methylation induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and on the metabolism and disposition of NNK in A/J mice. Cancer Res. 1990;50:2613–2617. [PubMed] [Google Scholar]
- 64.Kassie F, et al. Indole-3-carbinol inhibits 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone plus benzo[a]pyrene-induced lung tumorigenesis in A/J mice and modulates carcinogen-induced alterations in protein levels. Cancer Res. 2007;67:6502–6511. doi: 10.1158/0008-5472.CAN-06-4438. [DOI] [PubMed] [Google Scholar]
- 65.Kassie F, Matise I, Negia M, Upadhyaya P, Hecht SS. Dose-dependent inhibition of tobacco smoke carcinogen-induced lung tumorigenesis in A/J mice by indole-3-carbinol. Cancer Prev Res. 2008;1:568–576. doi: 10.1158/1940-6207.CAPR-08-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kassie F, et al. Chemopreventive agents modulate the protein expression profile of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone plus benzo[a]pyrene-induced lung tumors in A/J mice. Carcinogenesis. 2008;29:610–619. doi: 10.1093/carcin/bgn014. [DOI] [PubMed] [Google Scholar]
- 67.Orita H, Coulter J, Tully E, Kuhajda FP, Gabrielson E. Inhibiting fatty acid synthase for chemoprevention of chemically induced lung tumors. Clin Cancer Res. 2008;14:2458–2464. doi: 10.1158/1078-0432.CCR-07-4177. [DOI] [PubMed] [Google Scholar]
- 68.Yang CS, Liao J, Yang GY, Lu G. Inhibition of lung tumorigenesis by tea. Exp Lung Res. 2005;31:135–144. doi: 10.1080/01902140490495525. [DOI] [PubMed] [Google Scholar]
- 69.Clark J, You M. Chemoprevention of lung cancer by tea. Mol Nutr Food Res. 2006;50:144–151. doi: 10.1002/mnfr.200500135. [DOI] [PubMed] [Google Scholar]
- 70.Lu G, et al. Inhibition of adenoma progression to adenocarcinoma in a 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced lung tumorigenesis model in A/J mice by tea polyphenols and caffeine. Cancer Res. 2006;66:11494–11501. doi: 10.1158/0008-5472.CAN-06-1497. [DOI] [PubMed] [Google Scholar]
- 71.Anderson MW, et al. Effect of dietary green tea extract and aerosolized difluoromethylornithine during lung tumor progression in A/J strain mice. Carcinogenesis. 2008;29:1594–1600. doi: 10.1093/carcin/bgn129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lu G, et al. Synergistic inhibition of lung tumorigenesis by a combination of green tea polyphenols and atorvastatin. Clin Cancer Res. 2008;14:4981–4988. doi: 10.1158/1078-0432.CCR-07-1860. [DOI] [PubMed] [Google Scholar]
- 73.Johnson TE, et al. Chemopreventive effect of kava on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone plus benzo[a]pyrene-induced lung tumorigenesis in A/J mice. Cancer Prev Res. 2008;1:430–438. doi: 10.1158/1940-6207.CAPR-08-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang Z, et al. Cancer chemopreventive activity of a mixture of Chinese herbs (antitumor B) in mouse lung tumor models. Oncogene. 2004;23:3841–3850. doi: 10.1038/sj.onc.1207496. [DOI] [PubMed] [Google Scholar]
- 75.Yan Y, et al. Efficacy of polyphenon E, red ginseng, and rapamycin on benzo(a)pyrene-induced lung tumorigenesis in A/J mice. Neoplasia. 2006;8:52–58. doi: 10.1593/neo.05652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khan N, Afaq F, Kweon MH, Kim K, Mukhtar H. Oral consumption of pomegranate fruit extract inhibits growth and progression of primary lung tumors in mice. Cancer Res. 2007;67:3475–3482. doi: 10.1158/0008-5472.CAN-06-3941. [DOI] [PubMed] [Google Scholar]
- 77.Singh RP, et al. Effect of silibinin on the growth and progression of primary lung tumors in mice. J Natl Cancer Inst. 2006;98:846–855. doi: 10.1093/jnci/djj231. [DOI] [PubMed] [Google Scholar]
- 78.Yan Y, Wang Y, Tan Q, Lubet RA, You M. Efficacy of deguelin and silibinin on benzo(a)pyrene-induced lung tumorigenesis in A/J mice. Neoplasia. 2005;7:1053–1057. doi: 10.1593/neo.05532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gunning WT, Kramer PM, Lubet RA, Steele VE, Pereira MA. Chemoprevention of vinyl carbamate-induced lung tumors in strain A mice. Exp Lung Res. 2000;26:757–772. doi: 10.1080/01902140150216800. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Z, et al. A germ-line p53 mutation accelerates pulmonary tumorigenesis: p53- independent efficacy of chemopreventive agents green tea or dexamethasone/myo-inositol and chemotherapeutic agents taxol or adriamycin. Cancer Res. 2000;60:901–907. [PubMed] [Google Scholar]
- 81.Witschi H. Successful and not so successful chemoprevention of tobacco smoke-induced lung tumors. Exp Lung Res. 2000;26:743–755. doi: 10.1080/01902140150216792. [DOI] [PubMed] [Google Scholar]
- 82.Pereira MA, et al. Prevention of mouse lung tumors by budesonide and its modulation of biomarkers. Carcinogenesis. 2002;23:1185–1192. doi: 10.1093/carcin/23.7.1185. [DOI] [PubMed] [Google Scholar]
- 83.Wang Y, Zhang Z, Kastens E, Lubet RA, You M. Mice with alterations in both p53 and Ink4a/Arf display a striking increase in lung tumor multiplicity and progression: differential chemopreventive effect of budesonide in wild-type and mutant A/J mice. Cancer Res. 2003;63:4389–4395. [PubMed] [Google Scholar]
- 84.Lubet R, Wang Y, Zhang Z, You M. Mouse models incorporating alterations in the major tumor suppressor genes P53 and P16: their use in screening for potential carcinogens, developing further relevant mouse models, and screening for potential chemopreventive and chemotherapetutic agents. Exp Lung Res. 2005;31:117–133. doi: 10.1080/01902140490495499. [DOI] [PubMed] [Google Scholar]
- 85.Alyaqoub FS, et al. Prevention of mouse lung tumors and modulation of DNA methylation by combined treatment with budesonide and R115777 (Zarnestra MT) Carcinogenesis. 2007;28:124–129. doi: 10.1093/carcin/bgl136. [DOI] [PubMed] [Google Scholar]
- 86.Wattenberg LW, et al. Chemoprevention of pulmonary carcinogenesis by brief exposures to aerosolized budesonide or beclomethasone dipropionate and by the combination of aerosolized budesonide and dietary myo-inositol. Carcinogenesis. 2000;21:179–182. doi: 10.1093/carcin/21.2.179. [DOI] [PubMed] [Google Scholar]
- 87.Wattenberg LW, Wiedmann TS, Estensen RD. Chemoprevention of cancer of the upper respiratory tract of the Syrian golden hamster by aerosol administration of difluoromethylornithine and 5-fluorouracil. Cancer Res. 2004;64:2347–2349. doi: 10.1158/0008-5472.can-03-4032. [DOI] [PubMed] [Google Scholar]
- 88.Honda T, et al. Synthetic oleanane and ursane triterpenoids with modified rings A and C: a series of highly active inhibitors of nitric oxide production in mouse macrophages. J Med Chem. 2000;43:4233–4246. doi: 10.1021/jm0002230. [DOI] [PubMed] [Google Scholar]
- 89.Liby KT, Yore MM, Sporn MB. Triterpenoids and rexinoids as multifunctional agents for the prevention and treatment of cancer. Nat Rev Cancer. 2007;7:357–369. doi: 10.1038/nrc2129. [DOI] [PubMed] [Google Scholar]
- 90.Liby K, et al. The synthetic triterpenoids CDDO-methyl ester and CDDO-ethyl amide prevent lung cancer induced by vinyl carbamate in A/J mice. Cancer Res. 2007;67:2414–2419. doi: 10.1158/0008-5472.CAN-06-4534. [DOI] [PubMed] [Google Scholar]
- 91.Liby K, et al. A new rexinoid, NRX194204, prevents carcinogenesis in both the lung and mammary gland. Clin Cancer Res. 2007;13:6237–6243. doi: 10.1158/1078-0432.CCR-07-1342. [DOI] [PubMed] [Google Scholar]
- 92.Pereira MA, et al. Prevention of mouse lung tumors by targretin. Int J Cancer. 2006;118:2359–2362. doi: 10.1002/ijc.21618. [DOI] [PubMed] [Google Scholar]
- 93.Bauer AK, Dwyer-Nield LD, Malkinson AM. High cyclooxygenase 1 (COX-1) and cyclooxygenase 2 (COX-2) contents in mouse lung tumors. Carcinogenesis. 2000;21:543–550. doi: 10.1093/carcin/21.4.543. [DOI] [PubMed] [Google Scholar]
- 94.Wardlaw SA, March TH, Belinsky SA. Cyclooxygenase-2 expression is abundant in alveolar type II cells in lung cancer-sensitive mouse strains and in premalignant lesions. Carcinogenesis. 2000;21:1371–1377. [PubMed] [Google Scholar]
- 95.Rioux N, Castonguay A. Prevention of NNK-induced lung tumorigenesis in A/J mice by acetylsalicyclic acid and NS-398. Cancer Research. 1998;58:5354–6360. [PubMed] [Google Scholar]
- 96.Duperron C, Castonguay A. Chemoprevention efficacies of aspirin and sulindac against lung tumorigenesis in A/J mice. Carcinogenesis. 1997;18:1001–1006. doi: 10.1093/carcin/18.5.1001. [DOI] [PubMed] [Google Scholar]
- 97.Malkinson AM, et al. Inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced mouse lung tumor formation by FGN-1 (sulindac sulfone) Carcinogenesis. 1979;19:1353–1356. doi: 10.1093/carcin/19.8.1353. [DOI] [PubMed] [Google Scholar]
- 98.Jalbert G, Castonguay A. Effects of NSAIDs on NNK-induced pulmonary and gastric tumorigenesis in A/J mice. Cancer Lett. 1992;66:21–28. doi: 10.1016/0304-3835(92)90275-z. [DOI] [PubMed] [Google Scholar]
- 99.Kisley LR, et al. Celecoxib reduces pulmonary inflammation but not lung tumorigenesis in mice. Carcinogenesis. 2002;23:1653–1660. doi: 10.1093/carcin/23.10.1653. [DOI] [PubMed] [Google Scholar]
- 100.Rioux N, Castonguay A. Inhibitors of lipoxygenase: a new class of cancer chemopreventive. Carcinogenesis. 1998;19:1393–1400. doi: 10.1093/carcin/19.8.1393. [DOI] [PubMed] [Google Scholar]
- 101.Gunning WT, Kramer PM, Steele VE, Pereira MA. Chemoprevention by lipoxygenase and leukotriene pathway inhibitors of vinyl carbamate-induced lung tumors in mice. Cancer Res. 2002;62:4199–4201. [PubMed] [Google Scholar]
- 102.Ahrendt SA, et al. Cigarette smoking is strongly associated with mutation of the K-ras gene in patients with primary adenocarcinoma of the lung. Cancer. 2001;92:1525–1530. doi: 10.1002/1097-0142(20010915)92:6<1525::aid-cncr1478>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 103.Lantry LE, et al. Chemopreventive efficacy of promising farnesyltransferase inhibitors. Exp Lung Res. 2000;26:773–790. doi: 10.1080/01902140150216819. [DOI] [PubMed] [Google Scholar]
- 104.Gunning WT, et al. Chemoprevention of benzo(a)pyrene-induced lung tumors in mice by the farnesyltransferase inhibitor R115777. Clin Cancer Res. 2003;9:1927–1930. [PubMed] [Google Scholar]
- 105.Prokopczyk B, et al. Chemoprevention of lung tumorigenesis induced by a mixture of benzo(a)pyrene and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone by the organoselenium compound 1,4-phenylenebis(methylene)selenocyanate. Cancer Lett. 2000;161:35–46. doi: 10.1016/s0304-3835(00)00590-5. [DOI] [PubMed] [Google Scholar]
- 106.Richie JP, Jr, et al. The organoselenium compound 1,4-phenylenebis(methylene)selenocyanate inhibits 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced tumorgenesis and enhances glutathione-related antioxidant levels in A/J mouse lung. Chem Biol Interact. 2006;161:93–103. doi: 10.1016/j.cbi.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 107.El Bayoumy K, et al. Molecular targets of the chemopreventive agent 1,4-phenylenebis (methylene)-selenocyanate in human non-small cell lung cancer. Carcinogenesis. 2006;27:1369–1376. doi: 10.1093/carcin/bgi328. [DOI] [PubMed] [Google Scholar]
- 108.Das A, Desai D, Pittman B, Amin S, El Bayoumy K. Comparison of the chemopreventive efficacies of 1,4-phenylenebis(methylene)selenocyanate and selenium-enriched yeast on 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone induced lung tumorigenesis in A/J mouse. Nutr Cancer. 2003;46:179–185. doi: 10.1207/S15327914NC4602_11. [DOI] [PubMed] [Google Scholar]
- 109.Franklin MR, Moos PJ, El Sayed WM, Aboul-Fadl T, Roberts JC. Pre- and post-initiation chemoprevention activity of 2-alkyl/aryl selenazolidine-4(R)-carboxylic acids against tobacco-derived nitrosamine (NNK)-induced lung tumors in the A/J mouse. Chem Biol Interact. 2007;168:211–220. doi: 10.1016/j.cbi.2007.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Li L, et al. Chemopreventive activity of selenocysteine prodrugs against tobacco-derived nitrosamine (NNK) induced lung tumors in the A/J mouse. J Biochem Mol Toxicol. 2005;19:396–405. doi: 10.1002/jbt.20105. [DOI] [PubMed] [Google Scholar]
- 111.Lee HY, et al. Chemopreventive effects of deguelin, a novel Akt inhibitor, on tobacco-induced lung tumorigenesis. J Natl Cancer Inst. 2005;97:1695–1699. doi: 10.1093/jnci/dji377. [DOI] [PubMed] [Google Scholar]
- 112.Kim WY, et al. A novel derivative of the natural agent deguelin for cancer chemoprevention and therapy. Cancer Prev Res. 2008;1:577–587. doi: 10.1158/1940-6207.CAPR-08-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lantry LE, et al. 5-aza-2′-deoxycytidine is chemopreventive in a 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone-induced primary mouse lung tumor model. Carcinogenesis. 1999;20:343–346. doi: 10.1093/carcin/20.2.343. [DOI] [PubMed] [Google Scholar]
- 114.Belinsky SA, et al. Inhibition of DNA methylation and histone deacetylation prevents murine lung cancer. Cancer Res. 2003;63:7089–7093. [PubMed] [Google Scholar]
- 115.Soria JC, et al. Chemoprevention of lung cancer. Lancet Oncol. 2003;4:659–669. doi: 10.1016/s1470-2045(03)01244-0. [DOI] [PubMed] [Google Scholar]
- 116.Cohen V, Khuri FR. Chemoprevention of lung cancer. Curr Opin Pulm Med. 2004;10:279–283. doi: 10.1097/01.mcp.0000129754.97392.d5. [DOI] [PubMed] [Google Scholar]
- 117.Winterhalder RC, Hirsch FR, Kotantoulas GK, Franklin WA, Bunn PA., Jr Chemoprevention of lung cancer--from biology to clinical reality. Ann Oncol. 2004;15:185–196. doi: 10.1093/annonc/mdh051. [DOI] [PubMed] [Google Scholar]
- 118.Hirsch FR, Lippman SM. Advances in the biology of lung cancer chemoprevention. J Clin Oncol. 2005;23:3186–3197. doi: 10.1200/JCO.2005.14.209. [DOI] [PubMed] [Google Scholar]
- 119.Keith RL, Miller YE. Lung cancer: genetics of risk and advances in chemoprevention. Curr Opin Pulm Med. 2005;11:265–271. doi: 10.1097/01.mcp.0000166493.77412.2d. [DOI] [PubMed] [Google Scholar]
- 120.Gray J, et al. Lung cancer chemoprevention: ACCP evidence-based clinical practice guidelines (2nd Edition) CHEST. 2007;132:56S–68S. doi: 10.1378/chest.07-1348. [DOI] [PubMed] [Google Scholar]
- 121.NIH/NCI. Phase II Randomized Chemoprevention Study of Sulindac in Current or Former Smokers With Bronchial Dysplasia. 2009 http://www.cancer.gov/search/viewclinicaltrials.aspx?cdrid=496457&version=healthprofessional&protocolsearchid=5772556. Current clinical trials for chemoprevention of lung carcinogenesis are summarized here.
- 122.Szabo E. Selecting targets for cancer prevention: where do we go from here? Nat Rev Cancer. 2006;6:867–874. doi: 10.1038/nrc2008. This paper presents a good review of target selection in cancer prevention. [DOI] [PubMed] [Google Scholar]
- 123.Church TR, et al. A prospectively measured serum biomarker for a tobacco-specific carcinogen and lung cancer in smokers. Cancer Epidemiol Biomarkers & Prev. 2009;18:260–266. doi: 10.1158/1055-9965.EPI-08-0718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Yuan Y-M, et al. A urinary biomarker of a tobacco-specific carcinogen is prospectively related to lung cancer in smokers in Singapore and Shanghai, China. Cancer Res. 2009 in press. [Google Scholar]
- 125.Gerner EW, Meyskens FL., Jr Combination chemoprevention for colon cancer targeting polyamine synthesis and inflammation. Clin Cancer Res. 2009;15:758–761. doi: 10.1158/1078-0432.CCR-08-2235. This paper demonstrates the clinical efficacy of a mixture of agents in chemoprevention of colon cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.U.S. Department of Health and Human Services. The Health Consequences of Smoking: A Report of the Surgeon General. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; Atlanta, GA: 2004. [Google Scholar]
- 127.Luo S, Crainiceanu CM, Louis TA, Chatterjee N. Analysis of smoking cessation patterns using a stochastic mixed-effects model with a latent cured state. J Am Stat Assoc. 2008;103:1002–1013. doi: 10.1198/016214507000001030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hoving EF, Mudde AN, de Vries H. Smoking and the O pattern; predictors of transitions through the stages of change. Health Educ Res. 2006;21:305–314. doi: 10.1093/her/cyl033. [DOI] [PubMed] [Google Scholar]
- 129.Joseph AM, Hennrikus D, Thoele MJ, Krueger R, Hatsukami D. Community tobacco control leaders’ perceptions of harm reduction. Tob Control. 2004;13:108–113. doi: 10.1136/tc.2003.004242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kass NE. An ethics framework for public health. Am J Public Health. 2001;91:1776–1782. doi: 10.2105/ajph.91.11.1776. This paper provides a framework for ethics analysis of public health programs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Childress JF, et al. Public health ethics: mapping the terrain. J Law Med Ethics. 2002;30:170–178. doi: 10.1111/j.1748-720x.2002.tb00384.x. [DOI] [PubMed] [Google Scholar]
- 132.Chung FL. Chemoprevention of lung cancer by isothiocyanates and their conjugates in A/J mouse. Exp Lung Res. 2001;27:319–330. doi: 10.1080/01902140118500. [DOI] [PubMed] [Google Scholar]
- 133.Wattenberg LW. Chemoprevention of pulmonary carcinogenesis by myo-inositol. Anticancer Res. 1999;19:3659–3661. [PubMed] [Google Scholar]
- 134.Chung FL, et al. Inhibition of lung carcinogenesis by black tea in Fischer rats treated with a tobacco-specific carcinogen: caffeine as an important constituent. Cancer Research. 1998;58:4096–4101. [PubMed] [Google Scholar]
- 135.Yan Y, et al. Chemopreventive effect of aerosolized polyphenon E on lung tumorigenesis in A/J mice. Neoplasia. 2007;9:401–405. doi: 10.1593/neo.07160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tyagi A, et al. Growth inhibition and regression of lung tumors by silibinin: modulation of angiogenesis by macrophage-associated cytokines and nuclear factor-κβ and signal transducers and activators of transcription 3. Cancer Prev Res (Phila Pa) 2009;2:74–83. doi: 10.1158/1940-6207.CAPR-08-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Witschi H, Espiritu I, Ly M, Uyeminami D. The chemopreventive effects of orally administered dexamethasone in Strain A/J mice following cessation of smoke exposure. Inhal Toxicol. 2005;17:119–122. doi: 10.1080/08958370590899712. [DOI] [PubMed] [Google Scholar]
- 138.Estensen RD, et al. Effect of chemopreventive agents on separate stages of progression of benzo[α]pyrene induced lung tumors in A/J mice. Carcinogenesis. 2004;25:197–201. doi: 10.1093/carcin/bgg196. [DOI] [PubMed] [Google Scholar]
- 139.Liby K, et al. The rexinoid LG100268 and the synthetic triterpenoid CDDO-methyl amide are more potent than erlotinib for prevention of mouse lung carcinogenesis. Mol Cancer Ther. 2008;7:1251–1257. doi: 10.1158/1535-7163.MCT-08-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Pepin P, Bouchard L, Nicole P, Castonguay A. Effects of sulindac and oltipraz on the tumorigenicity of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in A/J mouse lung. Carcinogenesis. 1992;13:341–348. doi: 10.1093/carcin/13.3.341. [DOI] [PubMed] [Google Scholar]
- 141.Castonguay A, Rioux N. Inhibition of lung tumorigenesis by sulindac: comparision of two protocols. Carcinogenesis. 1997;18:491–496. doi: 10.1093/carcin/18.3.491. [DOI] [PubMed] [Google Scholar]
- 142.Moody TW, et al. Indomethacin reduces lung adenoma number in A/J mice. Anticancer Res. 2001;21:1749–1755. [PubMed] [Google Scholar]
- 143.Zerbe LK, et al. Inhibition by erlotinib of primary lung adenocarcinoma at an early stage in male mice. Cancer Chemother Pharmacol. 2008;62:605–620. doi: 10.1007/s00280-007-0644-z. [DOI] [PubMed] [Google Scholar]
- 144.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mauderly JL, et al. Chronic inhalation exposure to mainstream cigarette smoke increases lung and nasal tumor incidence in rats. Toxicol Sci. 2004;81:280–292. doi: 10.1093/toxsci/kfh203. [DOI] [PubMed] [Google Scholar]
- 146.Hutt JA, et al. Life-span inhalation exposure to mainstream cigarette smoke induces lung cancer in B6C3F1 mice through genetic and epigenetic pathways. Carcinogenesis. 2005;26:1999–2099. doi: 10.1093/carcin/bgi150. [DOI] [PubMed] [Google Scholar]
- 147.Hecht SS, et al. Rapid single-dose model for lung tumor induction in A/J mice by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone and the effect of diet. Carcinogenesis. 1989;10:1901–1904. doi: 10.1093/carcin/10.10.1901. [DOI] [PubMed] [Google Scholar]
- 148.Morse MA, et al. Structure-activity relationships for inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone lung tumorigenesis by arylalkyl isothiocyanates in A/J mice. Cancer Res. 1991;51:1846–1850. [PubMed] [Google Scholar]
- 149.Belinsky SA, Devereux TR, Foley JF, Maronpot RR, Anderson MW. Role of the alveolar type II cell in the development and progression of pulmonary tumors induced by 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in the A/J mouse. Cancer Res. 1992;52:3164–3173. [PubMed] [Google Scholar]