

Summary
Survivin, an inhibitor of apoptosis family molecule, has been proposed as a crucial intermediate in the signaling pathways leading to T-cell development, proliferation, and expansion. However, the importance of survivin to T-cell-driven inflammatory responses has not been demonstrated. Here, we show that survivin transgenic mice exhibit an increased antigen-driven Th2 lung inflammation and that constitutive expression of survivin reversed the defective lung inflammation even in the absence of OX40 costimulation. We found that OX40-deficient mice were compromised in generating Th2 cells, airway eosinophilia, and IgE responses. In contrast, OX40-deficient/survivin transgenic mice generated normal Th2 responses and exhibited strong lung inflammation. These results suggested that OX40 costimulation crucially engaged survivin during antigen-mediated Th2 responses. These findings also promote the notion that OX40 costimulation regulated allergic responses or lung inflammation by targeting survivin thereby enhancing T-cell proliferation and resulting in more differentiated Th2 cells in the allergic inflammatory response.
Keywords: costimulation, lung inflammation, murine model, survivin, Th2 cells
Introduction
Two major types of signals are required for optimal T-cell activation, with one derived from the T-cell receptor (TCR) and the other from a number of costimulatory receptors. Emerging evidence has suggested that the protein survivin, a member of the inhibitor of apoptosis (IAP) family (also called the baculoviral inhibitor of apoptosis repeat-containing 5 or BIRC5), may be an important intermediate contributing to the development of efficient T-cell responses [1, 2]. Homozygous survivin knockout mice died prematurely at embryonic day 3.5 with defects in cell proliferation, spindle formation, and apoptosis [3–5], demonstrating a requirement for survivin during cell development. Knockdown of survivin induced mitotic defects (e.g. centrosomal abberations, multipolar spindles, and missegregating chromosomes) and apoptosis in thymocytes and peripheral T cells [6, 7], suggesting an essential role for survivin in T-cell development, maturation, and homeostasis. Survivin also synergized with aurora B kinase to regulate T-cell costimulation-mediated cell cycle progression and proliferation [8]. In further support of an important role in T-cell activation, peripheral T cells isolated from T-cell-specific survivin-deficient mice exhibited an impaired homeostatic and mitogen-induced proliferation [6, 7]. Overexpression of survivin has also been shown to promote T-cell persistence and tumor regression in a murine cancer model [9].
Although these data strongly support a critical role for survivin in T cells, there have been few studies to date showing the importance of this molecule in the T-cell-driven inflammatory responses in vivo. We previously found that the TNFR costimulatory family member OX40 (CD134) promoted survivin expression in T cells [9]. OX40 has been shown to control clonal expansion, cytokine production, and memory development of T cells in several experimental settings involving Th1, Th2, and Th17 responses [10, 11]. In this study, we assessed the impact of survivin transgenic (Tg) expression in a Th2-driven response in vivo, using both wild-type (Wt) and OX40-deficient (OX40−/−) mice, and determining their susceptibility to the development of lung inflammation. Our results demonstrated that survivin can support optimal Th2 responses in vivo and compensate for a costimulatory defect brought about by the lack of OX40 expression, thereby promoting effector T-cell expansion that produced high levels of IL-4 and IL-5, recruited large numbers of eosinophils to the airways, and induced goblet cell hyperplasia as well as mucus overproduction. These data show that survivin can be a critical driver of the development of large numbers of Th2 cells and subsequent allergic asthmatic reactions.
Results
Tg survivin is expressed in thymocytes, bone marrow (BM) cells, immature B cells and T cells
In normal immune responses, survivin is only expressed in activated immune cells such as T cells [1]. To obtain constitutive survivin expression in B cells, a construct containing the murine survivin gene under the control of the human CD19 promoter region was used to generate survivin Tg mice. The construct was injected into the fertilized oocytes of C57BL/6 mice (Fig. 1A). Four in 34 founder lines were identified that expressed the transgene. Tg survivin was expressed in the BM cells, thymocytes, peripheral lymph nodes (LNs), and spleen, but not in the peripheral blood (Fig. 1B). B220+ B-cell populations (IgM−IgD−, IgM+IgD−, and IgM+IgD+) from the BM cells were sorted and further analyzed for the transgene expression (Fig. 1C). IgM−IgD− (pre-B cells) and IgM+IgD− (immature B cells), but not IgM+IgD+(mature B cells) BM cells were observed to have a specific expression of survivin, which correlated with a previous study showing the expression of a transgene by using the human CD19 promoter-based construct [12]. In addition, in vitro BM-derived mast cells and dendritic cells (DCs) did not express survivin (Fig. 1D). To further confirm the observation that mature B cells did not express Tg survivin, germinal center (GC) B cells and plasmablasts were isolated from the spleens of immunized mice. GC B cells and plasmablasts did not express survivin by western blot analysis (Fig. 1E), suggesting that Tg survivin was expressed in the early pro-B-cell stage when the CD19 promoter began to express and was maintained to the late stage of B-cell differentiation before maturation.
Figure 1. Expression of survivin in Tg mice.

(A) A schematic diagram of the construct to express survivin. (B) Thymus, BM cells, LN, spleen and peripheral blood from survivin Tg mice were analyzed for the expression of survivin and β-actin by western blots. (C) BM cells from survivin Tg mice were examined for the expression of IgM and IgD by flow cytometry, after gating on B220+ cells. (D) B-cell populations (IgM−IgD−, IgM+IgD− and IgM+IgD+) of B220+ BM cells, in vitro BM-derived mast cells and DCs were determined for the expression of survivin and β-actin by western blots. (E) Total T cells, B cells, CD4+ and CD8+ T cells from the spleens and LNs of Tg mice, and GC B cells and plasmablasts from the spleens of immunized Tg mice were analyzed for the expression of survivin and β-actin (as a control) by western blots. All data shown are representative of three experiments performed.
In addition to B cells, CD3+TCRβ+ T cells from the spleens and LNs of all four founder lines expressed Tg survivin, and Tg survivin expression was higher in CD4+ T cells than CD8+ T cells (Fig. 1E). Collectively, despite the human CD19 promoter, immature B cells in the BM, thymus, and spleen, and mature T cells in the spleen and LN of survivin Tg mice expressed the Tg survivin.
Survivin Tg mice have no gross defect in thymic selection and bear normal T-cell profiles
Because Tg survivin was highly expressed in the thymus, which could potentially impact T-cell thymic selection, we analyzed T-cell populations in the thymus. Thymi from survivin Tg or normal Wt mice were analyzed for CD4 and CD8 expression by flow cytometry. There were no differences in the percentages and absolute numbers of CD4+ CD8− (CD4 single positve - SP), CD4− CD8+ (CD8 SP), and CD4+ CD8+ (CD4 CD8 double positve - DP) T cells between the two groups (Figure 2 A and B). Thus, the survivin Tg mice have no gross defect in thymic selection during T-cell development.
Figure 2. T-cell profiles of survivin Tg mice.

(A–D) The thymi, pooled LNs, and spleens of C57BL/6 or survivin Tg mice were analyzed for T-cell numbers and surface protein expression. (A) Flow cytometric analysis for CD4+ and CD8+ T cells, after gating on live cells. One representative of three independent experiments is shown. (B) Total numbers of CD4+ or CD8+ SP and CD4+CD8+ DP T cells in the thymus. (C) Total numbers of CD4+ T cells in the spleens and LNs. (B, C) Data are shown as mean ± SEM of three mice per group and are representative of/pooled from three experiments performed (p > 0.05; unpaired t test with two-tailed p value). (D) Surface expression of CD62L and CD44 on CD4+ or CD8+ T cells in naive mice, after gating on CD4+ or CD8+ cells. One representative of three independent experiments is shown. (E–J) Naive CD4+ T cells (Th0) were isolated from the LNs and spleens of C57BL/6 or survivin Tg mice, and stimulated under (E–H) nonpolarizing condition as plate-coated anti-CD3 plus soluble ant-CD28 Abs, or (I–J) under Th1 and Th2 polarizing conditions. (E) Surface expression of CD25 and CD69, gated on naive or post-activation (72 h) of CD4+ population, after gating on CD4+ cells. One representative of three independent experiments is shown. (F) Cytokines at 40 h were measured in triplicate cultures by ELISA (p > 0.05; unpaired t test with two-tailed p value). (G) Proliferation at day 3 was measured in triplicate cultures by the incorporation of 3H-thymidine (1 μCi/well) during the last 12 h of culture (*p < 0.05; unpaired t test with two-tailed p value). (H) T-cell survival in vitro was determined by Trypan blue exclusion. Recovery is shown as a percentage of the input cell number. Data are shown as the mean ± SEM percentage change from three separate experiments (p > 0.05; unpaired t test with two-tailed p value). (I) IL-4 and IFN-γ production under Th1 or Th2 polarizing conditions. One representative of three independent experiments is shown. (J)Survivin expression under differentiated conditions was determined by western blots. β-actin served as a loading control. Data shown are representative of three experiments performed.
In addition to the thymus, Tg survivin was also expressed in T cells from the spleen and LN, which could potentially influence T-cell numbers and function. To determine this, we further analyzed T-cell populations in the spleen and LN. Spleens and LNs from survivin Tg or normal Wt mice were analyzed for CD4 and CD8 expression by flow cytometry. Similar to the observation in the thymus, there were no differences in the percentages and absolute numbers of CD4 SP, CD8 SP, and CD4 CD8 DP T cells between these two groups (Figure 2 A, C). Additionally, the surface expression of CD62L and CD44 on CD4+ and CD8+ T cells from spleens and LNs of survivin Tg or Wt mice was analyzed by flow cytometry, with similar populations of memory-phenotype T cells observed in both groups (Figure 2D). To investigate the potential impact of Tg survivin on T-cell function, we further examined activation markers, cytokine secretion, proliferation, and survival following TCR stimulation and costimulatory receptor ligation. Naive CD4+ T cells were isolated from the spleens and LNs of survivin Tg or Wt mice and stimulated with plate-coated anti-CD3 and soluble anti-CD28 antibodies (Abs). Naive CD4+ T cells from both strains of mice showed no surface expession of CD25 and CD69 before stimulation, but upregulated expression of both molecules following stimulation (Figure 2E). After stimulation, the activated CD4+ T cells from survivin Tg mice secreted equivalent amounts of IL-2 and IFN-γ as Wt mice (Figure 2F), but proliferated more strongly (Figure 2G), and showed enhanced clonal expansion profiles compared with that of activated CD4+ T cells from Wt mice (Figure 2H). Furthermore, under Th1 and Th2 polarizing conditions, the percentages of CD4+ T cells that secreted IFN-γ and IL-4 were similar between strains (Figure 2I). Although they overexpressed exogenous survivin, CD4+ T cells from survivin Tg mice also expressed endogenous survivin similiar to CD4+ T cells from Wt mice after activation (Figure 2J).
Taken together, survivin Tg mice have no gross defect in thymic selection and bear normal numbers of peripheral T cells. In addition, the T cells in survivin Tg mice were not constitutively activated, but displayed an enhanced proliferation in response to anti-CD3/CD28 stimulation.
Tg expression of survivin enhanced the number of differentiated Th2 cells
To assess the role of survivin played during in vivo T-cell activation, we used a murine allergic airway inflammatory model where Th2 cells developed and drove eosinophilic responses reminiscent of human asthma. We observed an enhanced inflammation in survivin Tg mice compared with that in Wt C57BL/6 mice. In this initial study, survivin Tg mice and Wt mice were sensitized with ovalbuman (OVA) in alum to generate an allergic Th2 response, and subsequently challenged by inhalation of OVA three weeks later. To assess whether survivin influences airway inflammation, bronchoalveolar lavages (BALs) were carried out on sacrificed mice 24 hours after the last OVA challenge and the influx of total leukocytes was determined. In survivin Tg mice, the total numbers of BAL leukocytes (Fig. 3A), eosinophils (Fig. 3B), Th2 cytokine production (Fig. 3C) and CD4+ T cells in the lung (Fig. 3D) increased significantly after OVA inhalation challenge, compared with the numbers of leukocytic infiltrates and cytokine production in Wt mice (p < 0.05 and p < 0.01, separately; unpaired t test with two-tailed p value). Comparing Wt and survivin Tg mice, there was no difference in lung inflammation (Supporting Information Fig. 1) or in the frequencies of IL-4 and IFN-γ producing CD4+ T cells (Fig. 3E). However, higher absolute numbers of IL-4 producing CD4+ T cells were observed in survivin Tg mice than in Wt mice (Fig. 3F). These results suggested that Tg expression of survivin might enhance the proliferation of differentiated T cells in the allergic inflammatory response and demonstrated that the survivin transgene was active and could modulate the immunity.
Figure 3. Allergic lung eosinophilia and Th2 cytokine production are significantly enhanced in survivin Tg mice.

Groups of C57BL/6 and survivin Tg mice were immunized i.p. with OVA in alum. Mice were challenged by inhalation of OVA or PBS control 25 days later. (A) Total leukocyte numbers were counted in BAL recovered from mice (n=5/group) 24 h after the last OVA challenge. (B) Total numbers of eosinophils were calculated from differentially stained BAL cytospins. (C) One day after the last OVA challenge, BAL fluid from mice was assessed for Th2 cytokines (IL-4, IL-5, IL-9, and IL-13) by ELISA. (D) Pooled lung cells from each group were processed for flow cytometry and analyzed for CD3+CD4+ and CD3+CD8+ cells. (A–D) Data are shown as the mean ± SEM from five mice per group. (E) Cytokine-producing CD4+ T cells were assessed by intracellular staining and flow cytometry of lung cells after 5 h of in vitro stimulation with PMA and ionomycin in the presence of brefeldin A. (F) The absolute numbers of IL-4 producing CD4+ T cells are shown. All results are shown as the mean number or percentage of cells ± SEM from three separate experiments with five mice per group in each experiment. Significance was tested by unpaired t test with two-tailed p value with ** p < 0.01 and *p < 0.05.
Survivin restored the numbers of leukocytes and eosinophils in the airways and AHR of OX40−/− mice
Because we previously found that survivin expression in T cells was regulated by signals from the costimulatory receptor OX40 [1, 8], we sought to determine how important survivin was to an inflammatory response in the absence of OX40 costimulation.
In the absence of OX40-OX40L interactions, allergic inflammatory responses in the lungs of mice were severely impaired in several different model systems [13–17]. To determine the role for survivin in the in vivo immune response, we generated a new Tg mouse model, the OX40−/−/survivin Tg mouse, and tested whether survivin could reverse the defective allergic inflammatory response in the absence of OX40 costimulation.
Wt C56BL/6, OX40−/− and OX40−/−/survivin Tg mice were directly immunized to test the allergic inflammatory response. To assess whether survivin influenced airway inflammation, BALs were carried out on mice as described above and the influx of total leukocytes was determined. In Wt mice, the total numbers of BAL leukocytes (Fig. 4A) and eosinophils (Fig. 4B) increased dramatically after OVA inhalation compared with the low numbers of leukocytic infiltrates in OX40−/− mice. In contrast, the numbers of leukocytes and eosinophils in the airways of OX40−/−/survivin Tg mice were significantly increased by approximately 10-fold compared with those of OX40−/− mice (p < 0.01). Importantly, no significant difference in leukocyte numbers was seen between Wt and OX40−/−/survivin Tg mice (p > 0.05). In this murine asthmatic model, eosinophils occupy a large fraction of the leukocytic infiltrates in the lungs; however, OX40−/− animals showed dramatically reduced numbers of eosinophils in the BAL, but no change in the numbers of infiltrating neutrophils and monocytes.
Figure 4. Survivin reverses the defect in eosinophilia and AHR in the lungs of OX40−/− mice.
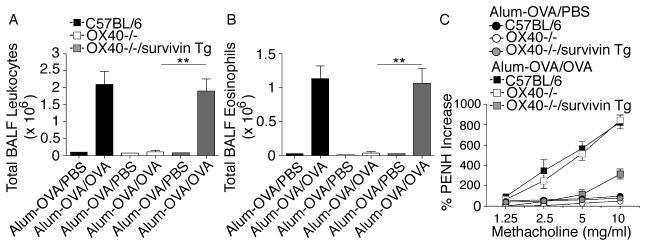
Groups of C57BL/6, OX40−/−, and OX40−/−/survivin Tg mice were immunized i.p. with OVA in alum. Mice were challenged by inhalation of OVA or PBS control 25 days later. (A) Total leukocyte numbers were counted in BAL recovered from mice (n=6/group) 24 hr after the last OVA or PBS challenge. (B) Total numbers of eosinophils were calculated from differentially stained BAL cytospins. (A, B) Data are shown as the mean number of cells ± SEM from three separate experiments with six mice per group in each experiment. Significance was tested by one-way ANOVA with Bonferroni multiple comparison test with **p < 0.01. (C) Individual mice were assessed 1–3 hours after the last aerosol challenge for AHR by barometric plethysmography. Results are shown as the mean percent change in PenH levels above baseline (PenHPBS for Alum-OVA/PBS: C57BL/6, 0.81 ± 0.05; OX40−/−, 0.92 ± 0.03; OX40−/−/survivin Tg, 0.83 ± 0.04; PenHPBS for Alum-OVA/OVA: C57BL/6, 0.80 ± 0.04; OX40−/−, 0.82 ± 0.03; OX40−/−/survivin Tg, 0.81 ± 0.03) after exposure to different concentrations of methacholine. Values were calculated from six mice in each group per experiment. Similar results were seen in three experiments.
Airway hyperreactivity (AHR) is a hallmark clinical symptom of the pulmonary allergic response and has been defined as an exaggerated airflow obstruction in response to bronchoconstrictors. In addition, AHR and mucous overproduction were consistently linked to asthma onset and its morbidity. Both defects have been observed in OX40−/− mice in various murine models of asthma [16, 17]. To determine whether survivin could overcome the airway dysfunction of OX40−/− mice, we measured the airway reactivity to methacholine. Wt mice developed AHR in response to methacholine challenge. In contrast, OX40−/− mice showed an attenuation in methacholine sensitivity as characterized by a reduced AHR. OX40−/−/survivin Tg mice, however, demonstrated a similar pulmonary resistance as Wt mice (Fig. 4C).
Survivin reversed the defect in mucus production in the lungs of OX40−/− mice
To determine whether survivin could overcome the airway dysfunction of OX40−/− mice, we further analyzed lung mucus profiles with Periodic acid-Schiff (PAS) staining. A large number of PAS+ cells were presented in the lungs of Wt mice. In contrast, OX40−/− mice only showed a few PAS+ cells. In OX40−/−/survivin Tg mice, however, we observed similar frequencies of PAS+ cells as in Wt mice (Fig. 5A).
Figure 5. Survivin reverses the defect in mucus production in the lungs of OX40−/− mice.

Groups of C57BL/6, OX40−/− and OX40−/−/survivin Tg mice were immunized and challenged as described in Fig. 4. (A) 24 hr after the final OVA challenge, lung tissue sections were stained with periodic acid-Schiff (PAS) to highlight the mucus-secreting goblet cells (original magnificaiton, × 40) in bronchioles. Lung sections were graded by % PAS cells for mucus production. (B) 24 h after the final OVA challenge, lung tissue sections were stained with H&E for the quantitation of inflammatory infiltrates by light microscopy (original magnification × 20). Lung sections were graded on a scale from 0–5 for inflammation severity. Results are shown as the mean score ± SEM from three separate experiments with six mice per group in each experiment. Significance was tested by one-way ANOVA with Bonferroni multiple comparison test with ***p < 0.001. (C) Pooled lung cells from each group were processed for flow cytometry and analyzed for CD3+CD4+ and CD3+CD8+ cells. (D) The absolute numbers of cytokine-producing CD4+ T cells are shown. (E) The percentage of cytokine-producing CD4+ T cells was assessed by intracellular staining and flow cytometry of lung cells. (C–E) Data are shown as the mean number or percentage of cells ± SEM from three separate experiments with six mice per group in each experiment. Significance was tested by one-way ANOVA with Bonferroni multiple comparison test with *p < 0.05.
The increased airway inflammation observed in Wt and OX40−/−/survivin Tg, but not in OX40−/− mice, was further supported by the histological analysis of lung sections. Wt and OX40−/−/survivin Tg mice developed marked perivascular and peribronchiolar cellular infiltrates which were composed predominantly of eosinophils and mononuclear cells. In addition, there was also pronounced goblet cell hypertrophy of the bronchiolar epithelium. In sharp contrast, lung sections from OX40−/− mice had far fewer cellular infiltrates close to the bronchioles and blood vessels and had histologically normal bronchiolar epithelium (Fig. 5B). Although survivin Tg expression significantly increased numbers of CD4+ T cells, including IL-4-producing CD4+ T cells in the lung, in the absence of OX40 (Fig. 5C and D), the frequencies of IL-4 and IFN-γ producing CD4+ T cells in OX40−/−/survivin Tg mice were not different from those found in Wt and OX40−/− mice (Fig. 5E). Taken together, these results suggested that survivin Tg expression could overcome the defect in the lung allergic response in the absence of OX40.
Survivin reversed defective IgE and Th2 cytokine production driven by allergen in OX40−/− mice
Eosinophils, AHR, and mucus overproduction are induced by Th2 cytokines, such as IL-4, IL-5, IL-9 and IL-13. To determine if survivin impacted Th2 cytokine production in the lungs of OX40−/− mice, 24 hours after the last OVA challenge, serum was assayed for OVA-specific IgE and BAL was assayed for Th2 cytokines. In OX40−/− animals, the levels of IgE, IL-4, IL-5, IL-9 and IL-13 were significantly reduced compared with those in Wt mice (p < 0.05) (Fig. 6). In contrast, the magnitude of the IgE response and production of other cytokines in OX40−/−/survivin Tg mice were similar to those seen in Wt mice (P > 0.05). Collectively, these results showed that Tg expression of survivin reversed the histological and functional defects of OX40−/− mice in generating asthmatic lung inflammation driven by Th2 cells.
Figure 6. Survivin reverses defective antigen-induced IgE and Th2 cytokine production driven by allergen in OX40−/− mice.

Groups of mice were immunized and challenged as described in Fig. 4. (A–E) One day after the last OVA challenge, BAL fluid from mice was assessed for (A) IgE and Th2 cytokines (B) IL-4, (C) IL-5, (D) IL-9 and (E) IL-13 by ELISA. Results are shown as the mean ± SEM from three separate experiments with six mice per group in each experiment. Significance was tested by one-way ANOVA with Bonferroni multiple comparison test with ** p < 0.01 and *p < 0.05.
Discussion
In this study, we determined the impact of Tg survivin expression on a Th2-driven response in vivo. Our experiments clearly demonstrated that Tg expression of survivin enhanced the number of differentiated Th2 cells in the allergic inflammatory response. Furthermore, Tg expression of survivin compensated for the OX40 deficiency in driving Th2 development and allergic inflammation in the lung, including induction of high levels of IL-4, and IL-5, large numbers of eosinophils, hyperplasia of goblet cell and overproduction of mucus. Our results suggested that OX40 signaling might regulate survivin to enhance T-cell proliferation and result in more differentiated Th2 cells that can mediate the allergic responses or lung inflammation.
Despite the human CD19 promoter, Tg survivin was lymphocyte specific, but was not exclusively expressed in B cells. CD19 is a B-cell-specific member of the immunoglobulin superfamily expressed that is from early pre-B-cell development until plasma cell differentiation. Previous studies showed that the promoter of the CD19 gene was a target for the B-cell-specific transcription factor – BSAP (B-cell-specific activator protein) [18], and suggested that Tg mice using the human CD19 promoter have B-cell-specific expression of the transgene [19, 20]. Several groups that generated Tg mice with constructs containing the human CD19 promoter also found a high expression of Tg genes in B cells [21–25]. Our initial project was to generate B-cell-specific survivin Tg mice by subcloning a Flag-tagged murine survivin gene into the pBSIIKS construct containing the human CD19 promoter region (Fig. 1A). After microinjection into the fertilized oocytes, four survivin Tg founder mice were obtained. Unexpectedly, in addition to Tg survivin expression in pre-B to immature B cells, CD3+TCRβ+ T cells from the spleens and LNs of all four founder lines expressed the transgene, with a higher expression level seen in CD4+ T cells than in CD8+ T cells (Fig. 1E). These results differed from previous observations of B cell-specific Tg mice using the human CD19 promoter. Although Tg expression of MHC II (I-Eαd), Bruton’s tyrosine kinase (Btk), or CD70 under the human CD19 promoter were shown in B220+ cells [22, 23, 25], B220 expression was not restricted to B cells and could also be expressed on activated T cells, DCs and other APCs. Therefore, gating on B220+ cells in these cited studies did not exclude the potential Tg expression in other cell types, such as T cells. In addition, our data showed that mature B and plasma cells in the peripheral immune system did not express the Tg survivin driven by the human CD19 promoter, which was in agreement with a previous study [22].
Survivin was undetectable in Wt naive CD4+ T cells, but upregulated after activation under either nonpolarizing conditions, or Th1 and Th2 polarizing conditions (Fig. 2J). Our previous study showed that survivin controlled in vivo antigen-driven clonal expansion and proliferation, resulting in an accumulation of large numbers of effector cells [1]. The current study supported this observation and demonstrated that expression of Tg survivin promoted T-cell proliferation (Figs. 2G and 2H). Under Th1 and Th2 polarizing conditions, there was no difference in the percentages of CD4+ T cells that produced IFN-γ (23.8% vs 21.9%) or IL-4 (23.5% vs 20.2%) from survivin Tg and Wt mice (Fig. 2I). Although survivin has the potential to affect Th17 differentiation, under Th17 polarizing conditions we observed similiar numbers of CD4+ T cells that produced Th17 from both survivin Tg and Wt mice (data not shown). These results indicated that survivin has the ability to enhance the proliferation of differentiated T cells that resulted in increased numbers of differentiated T cells after activation. Indeed, during the allergic inflammatory response, compared with Wt mice, survivin Tg mice had a higher number of differentiated Th2 cells (Fig. 3F). Furthermore, survivin Tg expression significantly increased the number of IL-4-producing CD4+ T cells in the lungs in the absence of OX40 (Fig. 5D). Thus, Tg expression of survivin reversed the defective allergic responses or lung inflammation of OX40−/− mice through enhanced proliferation of differentiated Th2 cells.
In this current study, using barometric whole-body plethysmography and increases in enhanced pause (PenH) as an index of airway obstruction, we measured AHR based on the responses to inhaled methacholine after OVA sensitization and airway challenge in conscious and unrestrained mice. Although PenH values have been shown to correlate with measures of pulmonary resistance in small animal models of asthma, PenH is a controversial measure of AHR as changes in PenH and respiratory resistances often do not correlate. Therefore, the conclusions of this study regarding AHR need to be further validated by using direct measurements of airways resistance rather than PenH.
Survivin is highly expressed in most cancers and has been associated with chemotherapy resistance, increased tumor recurrence, and shorter patient survival [19, 26]. Thus, the idea of targeting survivin has attracted attention as a potential cancer treatment strategy [27]. None of the offspring of any of the survivin Tg founders exhibited developmental defects or any increased susceptibility to malignancies. In addition, the survivin Tg mice used in this study survived normally, and we did not observe any prominent tumor growth or spontaneous inflammatory phenotypes in these mice, suggesting that in this case survivin activity was still highly regulated by exogenous stimuli. As such, our results likely reflected a specific activity in compensating for the defective survivin expression that accompanies T-cell activation when costimulation is absent or less than optimal.
The data demonstrated that survivin can compensate for OX40-deficiency in Th2 responses under Th2 dominant conditions, and indicated that survivin regulates the activity of Th2 cells that induce the airway asthmatic inflammation. Because T-cell-specific deletion of survivin (survivin−/−) resulted in T-cell development/maturation defects, survivin−/− mice were compromised in generating airway eosinophilia and AHR in the lungs (Supporting Information Fig. 2). To support our conclusion, the use of other inducible deletions (i.e., CreERT2 or OX40-Cre mice) will be more informative. Future studies will include other costimulatory molecules, such as CD28, that also drive Th2 responses and regulate survivin expression [8]. This will determine whether survivin can also compensate for other costimulatory deficiencies. In addition, the potential role of survivin during costimulation-driven Th1 and Th17 responses still needs to be defined. Furthermore, survivin Tg mice in this study were generated by using the human CD19 promoter, which is not T-cell-specific. Tg expression of survivin in pre-B to immature B cells might impact T-cell activity within the murine asthmatic model. Using T-cell-specific survivin Tg mice will exclude this potential effect. Nevertheless, these results extend our and others’ previous findings showing the importance of survivin to T-cell responsiveness [1, 6–9], and highlight the critical role of survivin in T-cell–driven inflammation in vivo.
Materials and methods
Mice
OX40−/− mice were bred on a C57BL/6 background [28]. The pCD19-TIAP Tg vector encoding mouse survivin (TIAP) under control of the human CD19 promoter [18] was generated as described for pCD19-FLIP [29]. Survivin Tg mice were originally generated on a C57BL/6 background and were backcrossed for greater than five generations onto the OX40−/− background to generate OX40−/−/Survivin Tg mice. All experiments were in compliance with the regulations of the Pennsylvania State University College of Medicine Animal Care committee in accordance with guidelines of the Association for the Assessment and Accreditation of Laboratory Animal Care.
Cell culture
Naive CD4+ T cells were stimulated with 1 μg/ml plate-bound anti-CD3 mAb in the presence of 1 μg/ml anti-CD28 mAb in a 48-well plate [8]. Where indicated, cells were cultured under either Th1-polarizing conditions (10 ng/ml IL-2, 1 ng/ml IL-12, and 10 μg/ml anti–IL-4 mAb), or Th2-polarizing conditions (10 ng/ml IL-2 and IL-4 and 10 μg/ml anti–IFN-γ mAb). The cells were cultured for 2 days for western blotting, or cultured for 4 days then rested in fresh medium containing 10ng/ml of IL-2/ml (Shenandoah) for 1.5 days. The cells were stimulated with phorbol myristate acetate (PMA) and ionomycin (Calbiochem) for intracellular cytokine staining.
Induction of BM-derived mast cells and DCs
BM-derived mast cells and DCs were generated as described [30, 31]. Briefly, red blood cells were removed from mouse femoral BM cells by using ACK lysing buffer (Lonza), and isolated nucleated cells were cultured in the presence of 10 ng/ml rmIL-3 (PeproTech) for 4 weeks for mast cells or 20 ng/ml of GM-CSF plus 20 ng/ml rmIL-4 (PeproTech) for 7 days for DCs.
Immunization
Mice were injected intraperitoneally with 100 μg 4-Hydroxy-3-nitrophenylacetyl-Chicken Gamma Globulin (NP-CGG, Biosearch Technologies) suspended in alum (Pierce). Spleens were harvested 10 days after immunization and dissociated by using a cell strainer, and subjected to red blood cell lysis. Single cell suspension was stained with PE-B220 (BioLegend) and FITC-PNA (Vector Laboratories), or PE-B220, allophycocyanin-CD138 and FITC-CD93 (all from BioLegend). GC B-cell populations (B220+/PNA+) and plasmablasts (B220+/CD138+/CD93−) were sorted to determine expression of survivin and β-actin (both from Santa Cruz Biotech) by western blots.
Induction of allergic airway inflammation
Mice were sensitized and challenged as described previously to induce a Th2 response [17]. Briefly, mice were sensitized by intraperitoneal injection of 20 μg OVA protein (chicken egg albumin; Sigma), adsorbed to 2 mg aluminum hydroxide (Alum; Pierce) in phosphate buffered saline (PBS) on day 0. Nonsensitized (control) mice received 2 mg Alum in PBS alone. On day 25, mice were challenged via the airways with OVA (5 mg/ml in 15 ml of PBS) or PBS control for 30 min, once a day for four consecutive days by ultrasonic nebulization. Mice were sacrificed 18–24 hr after the last aerosol challenge and assessed for inflammation of the lung.
Measurement of airway hyperreactivity (AHR)
AHR was determined 1–3 hr after the last aerosolized OVA exposure by whole-body plethysmography (Buxco Technologies) in response to inhaled methacholine (MCh; 1.25–10 mg/ml; Santa Cruz Biotech) as described previously [16]. Briefly, AHR was determined using the parameter PenH that is calculated based on the mean pressure generated in plethysmograph chambers during inspiration and expiration, combined with the time of each phase. Values were represented as the mean percent change in PenH at each methacholine concentration compared with baseline readings, calculated from groups of four mice.
Bronchoalveolar lavage (BAL) measurements
Lung inflammation was assessed by BAL as described previously [17]. Briefly, lungs were lavaged with PBS containing 2% BSA. Cells in the recovered BAL fluid (BALF) fractions were centrifuged, counted, and then pelleted onto glass slides by cytocentrifugation. Cell type counts were performed with Hema 3 manual staining system (Fisher). Cytokines were assessed in BAL by using commercially available ELISA kits (eBiosciences). All samples were collected 24 hr after the final antigen challenge.
Characterization of lung histology and cellular infiltrates
Lung sections were characterized as described previously [17]. Briefly, lungs were removed and placed directly into Formical-4™ (Decal Cehmical Corp), paraffin embedded, and sections were stained with hematoxylin and eosin (HE) for quantitation of inflammatory infiltrates or with periodic acid-Schiff (PAS) for quantitation of bronchial mucus gland hypertrophy.
Statistics
Unpaired t test or one-way analysis of variance (ANOVA) was used for the statistical analysis between groups and significance was set at 5%. All statistics were calculated using GraphPad Prism (San Diego, CA).
Supplementary Material
Acknowledgments
The authors thank Dr. F. Beerman (ISREC, Epalinges, Switzerland) and the late Prof. J.T. Tschopp (Biochemistry, Univ. of Lausanne, Switzerland) for their help with the generation of survivin Tg mice, and Dr. Tak W. Mak (Ontario Cancer Institute) for providing Lck-survivinflox/flox mice. The authors also thank Drs. Aron Lukacher and Laurie Ludgate, and Mr. Gregory Berry (Penn State University) for their critical reviews of our manuscript. This project was funded, in part, under grants with the National Institute of Health Grant K18CA151798 and the Pennsylvania Department of Health to J.S., the National Institute of Health Grant R01AI079056 to D.F., and the Dutch Cancer Society (UU2009-4311, UU2011-5143) and the Netherlands Organization for Scientific Research (NWO Vici 918.12.610) to S.L.
Footnotes
Conflict of interest
The authors declare no financial or commercial conflict of interest.
References
- 1.Song J, So T, Cheng M, Tang X, Croft M. Sustained survivin expression from OX40 costimulatory signals drives T cell clonal expansion. Immunity. 2005;22:621–631. doi: 10.1016/j.immuni.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 2.Shinomiya N, Koike Y, Koyama H, Takayama E, Habu Y, Fukasawa M, Tanuma S, Seki S. Analysis of the susceptibility of CD57 T cells to CD3-mediated apoptosis. Clin Exp Immunol. 2005;139:268–278. doi: 10.1111/j.1365-2249.2004.02687.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Uren AG, Wong L, Pakusch M, Fowler KJ, Burrows FJ, Vaux DL, Choo KH. Survivin and the inner centromere protein INCENP show similar cell-cycle localization and gene knockout phenotype. Curr Biol. 2000;10:1319–1328. doi: 10.1016/s0960-9822(00)00769-7. [DOI] [PubMed] [Google Scholar]
- 4.Grossman D, Kim PJ, Blanc-Brude OP, Brash DE, Tognin S, Marchisio PC, Altieri DC. Transgenic expression of survivin in keratinocytes counteracts UVB-induced apoptosis and cooperates with loss of p53. J Clin Invest. 2001;108:991–999. doi: 10.1172/JCI13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–3257. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 6.Okada H, Bakal C, Shahinian A, Elia A, Wakeham A, Suh WK, Duncan GS, et al. Survivin loss in thymocytes triggers p53-mediated growth arrest and p53-independent cell death. J Exp Med. 2004;199:399–410. doi: 10.1084/jem.20032092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xing Z, Conway EM, Kang C, Winoto A. Essential role of survivin, an inhibitor of apoptosis protein, in T cell development, maturation, and homeostasis. J Exp Med. 2004;199:69–80. doi: 10.1084/jem.20031588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song J, Salek-Ardakani S, So T, Croft M. The kinases aurora B and mTOR regulate the G1-S cell cycle progression of T lymphocytes. Nat Immunol. 2007;8:64–73. doi: 10.1038/ni1413. [DOI] [PubMed] [Google Scholar]
- 9.Zhao B, Song A, Haque R, Lei F, Weiler L, Xiong X, Wu Y, et al. Cooperation between molecular targets of costimulation in promoting T cell persistence and tumor regression. J Immunol. 2009;182:6744–6752. doi: 10.4049/jimmunol.0804387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Croft M. Control of immunity by the TNFR-related molecule OX40 (CD134) Annu Rev Immunol. 2010;28:57–78. doi: 10.1146/annurev-immunol-030409-101243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vincenti F, Luggen M. T cell costimulation: a rational target in the therapeutic armamentarium for autoimmune diseases and transplantation. Annu Rev Med. 2007;58:347–358. doi: 10.1146/annurev.med.58.080205.154004. [DOI] [PubMed] [Google Scholar]
- 12.Shankar M, Nixon JC, Maier S, Workman J, Farris AD, Webb CF. Anti-nuclear antibody production and autoimmunity in transgenic mice that overexpress the transcription factor Bright. J Immunol. 2007;178:2996–3006. doi: 10.4049/jimmunol.178.5.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Duan W, So T, Croft M. Antagonism of airway tolerance by endotoxin/lipopolysaccharide through promoting OX40L and suppressing antigen-specific Foxp3+ T regulatory cells. J Immunol. 2008;181:8650–8659. doi: 10.4049/jimmunol.181.12.8650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seshasayee D, Lee WP, Zhou M, Shu J, Suto E, Zhang J, Diehl L, et al. In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J Clin Invest. 2007;117:3868–3878. doi: 10.1172/JCI33559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damayanti T, Kikuchi T, Zaini J, Daito H, Kanehira M, Kohu K, Ishii N, et al. Serial OX40 engagement on CD4+ T cells and natural killer T cells causes allergic airway inflammation. Am J Respir Crit Care Med. 2010;181:688–698. doi: 10.1164/rccm.200910-1598OC. [DOI] [PubMed] [Google Scholar]
- 16.Jember AG, Zuberi R, Liu FT, Croft M. Development of allergic inflammation in a murine model of asthma is dependent on the costimulatory receptor OX40. J Exp Med. 2001;193:387–392. doi: 10.1084/jem.193.3.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salek-Ardakani S, Song J, Halteman BS, Jember AG, Akiba H, Yagita H, Croft M. OX40 (CD134) controls memory T helper 2 cells that drive lung inflammation. J Exp Med. 2003;198:315–324. doi: 10.1084/jem.20021937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kozmik Z, Wang S, Dorfler P, Adams B, Busslinger M. The promoter of the CD19 gene is a target for the B-cell-specific transcription factor BSAP. Mol Cell Biol. 1992;12:2662–2672. doi: 10.1128/mcb.12.6.2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Yang L, Yang J, Kuropatwinski K, Wang W, Liu XQ, Hauser J, Brattain MG. Transforming growth factor beta induces apoptosis through repressing the phosphoinositide 3-kinase/AKT/survivin pathway in colon cancer cells. Cancer Res. 2008;68:3152–3160. doi: 10.1158/0008-5472.CAN-07-5348. [DOI] [PubMed] [Google Scholar]
- 20.Zhou LJ, Smith HM, Waldschmidt TJ, Schwarting R, Daley J, Tedder TF. Tissue-specific expression of the human CD19 gene in transgenic mice inhibits antigen-independent B-lymphocyte development. Mol Cell Biol. 1994;14:3884–3894. doi: 10.1128/mcb.14.6.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maas A, Dingjan GM, Grosveld F, Hendriks RW. Early arrest in B cell development in transgenic mice that express the E41K Bruton’s tyrosine kinase mutant under the control of the CD19 promoter region. J Immunol. 1999;162:6526–6533. [PubMed] [Google Scholar]
- 22.Nixon JC, Ferrell S, Miner C, Oldham AL, Hochgeschwender U, Webb CF. Transgenic mice expressing dominant-negative bright exhibit defects in B1 B cells. J Immunol. 2008;181:6913–6922. doi: 10.4049/jimmunol.181.10.6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arens R, Tesselaar K, Baars PA, van Schijndel GM, Hendriks J, Pals ST, Krimpenfort P, et al. Constitutive CD27/CD70 interaction induces expansion of effector-type T cells and results in IFNgamma-mediated B cell depletion. Immunity. 2001;15:801–812. doi: 10.1016/s1074-7613(01)00236-9. [DOI] [PubMed] [Google Scholar]
- 24.Gisler R, Jacobsen SE, Sigvardsson M. Cloning of human early B-cell factor and identification of target genes suggest a conserved role in B-cell development in man and mouse. Blood. 2000;96:1457–1464. [PubMed] [Google Scholar]
- 25.Kleindienst P, Chretien I, Winkler T, Brocker T. Functional comparison of thymic B cells and dendritic cells in vivo. Blood. 2000;95:2610–2616. [PubMed] [Google Scholar]
- 26.Rahman KM, Banerjee S, Ali S, Ahmad A, Wang Z, Kong D, Sakr WA. 3,3′-Diindolylmethane enhances taxotere-induced apoptosis in hormone-refractory prostate cancer cells through survivin down-regulation. Cancer Res. 2009;69:4468–4475. doi: 10.1158/0008-5472.CAN-08-4423. [DOI] [PubMed] [Google Scholar]
- 27.Altieri DC. Survivin, cancer networks and pathway-directed drug discovery. Nat Rev Cancer. 2008;8:61–70. doi: 10.1038/nrc2293. [DOI] [PubMed] [Google Scholar]
- 28.Song J, So T, Croft M. Activation of NF-kappaB1 by OX40 contributes to antigen-driven T cell expansion and survival. J Immunol. 2008;180:7240–7248. doi: 10.4049/jimmunol.180.11.7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Faideau B, Briand JP, Lotton C, Tardivel I, Halbout P, Jami J, Elliott JF, et al. Expression of preproinsulin-2 gene shapes the immune response to preproinsulin in normal mice. J Immunol. 2004;172:25–33. doi: 10.4049/jimmunol.172.1.25. [DOI] [PubMed] [Google Scholar]
- 30.Fathman JW, Gurish MF, Hemmers S, Bonham K, Friend DS, Grusby MJ, Glimcher LH, Mowen KA. NIP45 controls the magnitude of the type 2 T helper cell response. Proc Natl Acad Sci U S A. 2010;107:3663–3668. doi: 10.1073/pnas.0914700107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat Med. 2005;11:748–756. doi: 10.1038/nm1257. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.