

Abstract
Regulators of G protein signaling (RGS) proteins are key players in regulating signaling via G protein-coupled receptors. RGS proteins directly bind to the Gα-subunits of activated heterotrimeric G-proteins, and accelerate the rate of GTP hydrolysis, thereby rapidly deactivating G-proteins. Using atomistic simulations and NMR spectroscopy, we have studied in molecular detail the mechanism of action of CCG-50014, a potent small molecule inhibitor of RGS4 which covalently binds to cysteine residues on RGS4. We apply temperature-accelerated molecular dynamics (TAMD) to carry out enhanced conformational sampling of apo RGS4 structures, and consistently find that the α5-α6 helix pair of RGS4 can spontaneously span open-like conformations, allowing binding of CCG-50014 to the buried side-chain of Cys95. Both NMR experiments and MD simulations reveal chemical shift perturbations in residues in the vicinity of inhibitor binding site as well as in the RGS4-Gα binding interface. Consistent with a loss of G-protein binding, GAP activity, and allosteric mechanism of action of CCG-50014, our simulations of the RGS4-Gα complex in the presence of inhibitor suggest a relatively unstable protein-protein interaction. These results have potential implications for understanding how the conformational dynamics among RGS proteins may play a key role in the sensitivity of inhibitors.
G protein-coupled receptors (GPCRs) are membrane proteins of profound clinical relevance because they mediate cellular responses to a variety of hormones, neurotransmitters, and extracellular ligands.1 GPCR signaling pathways are negatively modulated by a class of intracellular proteins known as the regulators of G protein signaling (RGS).2,3 RGS proteins directly associate with the GTP-bound Gα subunits of activated G-proteins and accelerate the rate of GTP-hydrolysis, leading to the rapid deactivation of Gα and termination of GPCR signaling. The crystal structure of RGS4, an R4 subfamily member,2 bound to Giα1 (PDB code 1AGR) provides molecular details on this protein-protein interaction. Specifically, a conserved ~120 residue core domain of RGS4 (also known as the “RGS-box”)2,4 with a typical helical fold5 (Figure 1a) directly contacts three switch regions of Gα and stabilizes them in a transition state configuration for catalysis (Figure 1b).6 Given the direct role of RGS proteins in regulating GPCR signaling, they have emerged as potential drug targets,7–10 and significant efforts are underway to design potent small molecule inhibitors targeting these proteins.11–16
Figure 1.
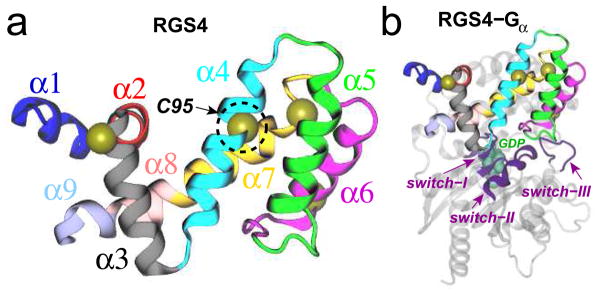
Cartoon representations of (a) the box domain of RGS4; and (b) the RGS4-Giα1 complex. Each of the nine helices of RGS4 is distinctly colored and labeled. The Cα atoms of four cysteine residues in RGS4 are shown as yellow spheres, while the buried residue Cys95 (C95) is labeled and highlighted in a dotted circle. In panel b, three switch regions of Giα1 are shown as magenta cartoons and labeled, and the GDP molecule is rendered as green space-filling spheres.
Protein-protein interactions (PPIs) similar to that between RGS and Gα represent critical therapeutic targets for their direct role in abnormal signaling mechanisms in disease,17 although targeting PPIs is a significant challenge.12,18–20 These challenges arise from difficulty in overcoming the binding energy associated with PPIs by a small molecule and targeting the large, relatively featureless interaction interfaces that commonly lack well-defined pockets into which small molecules can be targeted. Alternatively, targeting allosteric sites may provide greater specificity, and importantly remove the need to compete with the protein binding partner. The RGS4 allosteric site (also known as the “B-site”),7 located away from the Gα binding site (termed as the “A-site”), is a promising alternative because physiological regulators of RGS4 directly interact with the B-site.21,22 However, there is currently no structural data on RGS4 bound to an inhibitor molecule, making it difficult to pursue structure-based drug design strategies targeting the RGS4 B-site to modulate function.
CCG-50014 (Figure S1) is a potent (IC50 = 30 nM) and selective inhibitor of RGS4 that was discovered in a high-throughput biochemical screen.23 Like all previously reported RGS inhibitors, it acts by forming a covalent adduct to cysteine residues in RGS proteins.15,16,23 We have previously proposed15 a docked model of CCG-50014 binding to RGS8 (a homologue of RGS4), which suggested that the inhibitor molecule docks near the Cys107 residue of RGS8 (Cys95 in RGS4; Figure 1). In this model, the reactive group of CCG-50014 is located ~8–13 Å away from two cysteine residues in RGS8, where formation of a covalent adduct is unlikely. Therefore, we hypothesized that a conformational change in the helix bundle occurs to allow binding of the inhibitor molecule to the buried side-chain of Cys95/Cys107 in RGS4/RGS8. In this work, we expose the existence of a previously uncharacterized open-like conformation of RGS4 using atomistic simulations and NMR experiments. In characterizing the molecular mechanism of CCG-50014, we show that the compound acts as a chemical probe of this novel conformational state, which may provide an invaluable model for structure-based design of novel inhibitors.
1 RESULTS AND DISCUSSION
Using two independent starting structures of RGS4 (PDB codes 1AGR and 1EZT),6,24 we first conducted molecular dynamics (MD) simulations (Table S1) of apo wild-type RGS4, and a mutant form of RGS4, where all cysteine residues except Cys95 were mutated to Ala. The single-cysteine mutant was created to specifically understand the effect of CCG-50014 binding to Cys95 on RGS4, and was also used in the NMR experiments (vide infra). These conventional MD simulations reveal no significant differences in the overall structure of RGS4 (Figure 2a and S2a), given the relatively low root-mean-squared-deviation (RMSD; Cα) from starting structures (red and green traces in Figure 2c and S2c). To probe the solvent-accessibility of the residue Cys95, we measured the buried surface area (BSA) between the α5-α6 helix pair and the rest of RGS4 (red and green traces in Figure 2d and S2d) along with the solvent-accessible-surface-area (SASA) of residue Cys95 (Figure S3). Throughout each 20-ns MD simulation, we observe no change in the aforementioned BSA, and the SASA of residue Cys95 remains negligible (~2–3 Å2). Therefore, the underlying dynamics of RGS4 that facilitate access to Cys95 are not yet clear. However, this is not surprising because long time-scale conformational flexibility in proteins is seldom revealed using short unbiased MD simulations due to the underlying free-energy barriers. Therefore, we used an enhanced sampling algorithm for proteins25 based upon temperature-accelerated molecular dynamics (TAMD)26,27 to carry out conformational sampling of RGS4 (see Supporting Information for details). In this algorithm, enhanced conformational sampling of proteins is achieved by exploring the dynamics of relatively rigid subdomains (spatially contiguous groups of residues) by accelerating via high fictitious temperature the Cartesian coordinates of the centers-of-mass (COM) of subdomains as collective variables (CVs).28–32 We conducted four independent TAMD simulations (Table S1) of RGS4 at fictitious thermal energies of β̄−1= 2 and 3 kcal/mol, where β̄ =1/kBT̄, kB is Boltzmann’s constant, and T̄ is the fictitious temperature. The RMSD and BSA data (magenta traces in Figure 2c and 2d) from a typical TAMD run at β̄−1= 3 kcal/mol reveal the existence of relatively large-scale conformational flexibility in the α5-α6 helix pair, where these helices span open-like conformational states away from the rest of RGS4 (Figure 2b). However, similar data from a TAMD run at a lower β̄−1= 2 kcal/mol (blue traces in Figure 2c and 2d) suggest that the conformational change in the α5-α6 helix pair cannot be observed on similar time-scale with thermal energies lower or comparable to 2 kcal/mol, which also explains why this conformational change was not observed in conventional MD simulations (vide supra). We note that this conformational change is reproducibly observed in multiple independent TAMD simulations of RGS4 (Figures S2 and S4).
Figure 2.

MD and TAMD simulation data for RGS4 runs with initial coordinates from PDB code 1AGR. (a and b) Overlay of cartoon representations of apo-RGS4 (red, beginning; blue, end of simulations). All helices of RGS4, except the α5-α6 pair, are shown in white cartoons. (c) The Cα -RMSD traces with reference to starting conformations. (d) Buried surface area (BSA) between the α5-α6 helix pair and the rest of RGS4.
It is well established that large-amplitude collective motions in proteins are governed by their intrinsically accessible low-frequency normal modes,33–37 and conformational sampling via TAMD in Cartesian CVs is also governed by these modes.30 Therefore, we carried out a Cα-based elastic network normal mode analysis (NMA) of RGS4 with both initial structures (PDB codes 1AGR and 1EZT), and computed the individual and cumulative projections or overlaps (θm) for 20 low-frequency (non-zero) normal modes of apo RGS4 onto the functional displacement vector (d⃗) between initial and final conformations generated by a typical TAMD simulation of each structure (Figure 3). These data suggest that low-frequency modes 1, 3 and 5 consistently contribute to this conformational change (see overlaps, θm, for these modes in Figure 3), while either mode 7 (Figure 3b) or modes 8, 9, and 10 (Figure 3a) can contribute significantly to this conformational change. Also, the first 10 low-frequency modes can describe the majority of the conformational change in the α5-α6 helix pair, as indicated by the cumulative overlap traces (red dotted lines in Figure 3). These results reinforce the point that the conformational flexibility observed in the TAMD simulations is intrinsic to the structure of RGS4 due to the presence of such low-frequency (high-amplitude) modes. This also means that our NMA analysis supports the definition of subdomain COM as our collective variables in TAMD since both calculations show significant overlap in conformational changes.
Figure 3.
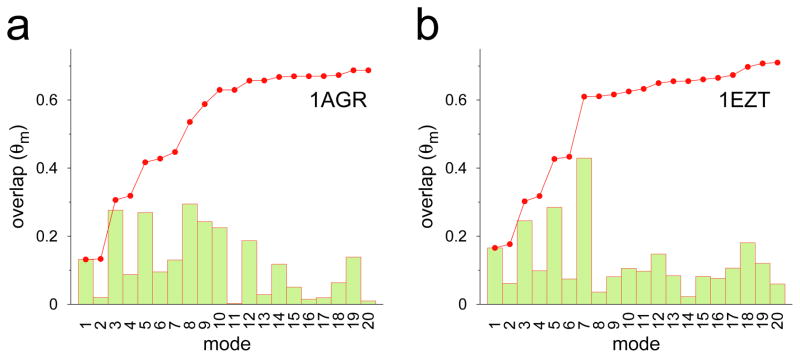
Individual (bars) and cumulative (red dotted line) projections of 20 low-frequency normal modes of RGS4 onto TAMD generated conformations; projection of the nth mode θmn is the normalized scalar product between the eigen vector (I⃗n) of the corresponding mode and functional displacement d⃗ i.e θmn = I⃗n·d⃗/(|I⃗n|×|d⃗|), while the cumulative overlap is . The functional displacement vector d⃗ is defined as the vector difference between starting and ending conformations from a typical TAMD simulation.
A key implication of the conformational change in the α5-α6 helix bundle is that the side-chain of residue Cys95 is now accessible, and the inhibitor molecule could be successfully accommodated in the TAMD-generated conformations of RGS4 (Figure S5). To understand specifically the effect of CCG-50014 binding to Cys95, we carried out seven independent 40-ns long MD equilibration trajectories (Table S1) of the single-cysteine mutant of RGS4 with a colvalently-linked inhibitor. In 6 out of 7 simulations, we observe that intercalation of CCG-50014 into the helix bundle restricts closure of the α5-α6 helix pair (Figure 4a, S6a–b, and S7a–c), as indicated by a nearly constant BSA between these two helices and the rest of RGS4 (magenta traces in Figure 4d, S6a–b, and S7a–c). This is primarily because the covalently-linked (to Cys95) inhibitor molecule experiences only small conformational rearrangements due to minor rotations of aromatic groups (Figure S8, and top three panels in Figure S9). However, in one simulation, we observe that CCG-50014 rotates (around the disulfide bond) by ~90° with respect to its initially docked conformation (lowest panel in Figure S9), which also allows partial closing of the α5-α6 helix pair (see snapshots, and the BSA trace in Figure S7d). To further understand the role of the inhibitor in keeping the α5-α6 helix pair in a relatively open conformation, we also carried out a 40-ns long MD equilibration of the TAMD-generated open-conformation of RGS4 in the absence of CCG-50014. The data from this simulation showed a spontaneous recovery of the α5-α6 helix pair to a near crystallographic conformation (snapshot in Figure 4b, and green traces for RMSD and BSA in Figure 4c–d) unlike the inhibitor-bound simulation (magenta traces for RMSD and BSA in Figure 4c–d). This suggests that apo-RGS4 likely exists predominantly in a closed-conformation, but the open-conformation that exposes Cys95 is favored when RGS4 is bound to CCG-50014.
Figure 4.

Data from MD equilibrations of TAMD-generated conformations of RGS4 (PDB code 1AGR) with (panel a) and without (panel b) CCG-50014 (red, beginning; blue, end of simulations). (c) RMSD with reference to the crystallographic conformation of RGS4 (PDB code 1AGR). (d) Same BSA as in Figure 2d are shown.
To further dissect the effect of inhibitor binding on the structure of RGS4, we computed the root-mean-squared-fluctuation (RMSF) per residue from three independent MD simulations of mutant-RGS4 with and without CCG-50014 (Figure S10). Based upon these data, we show a change in RMSF per residue on binding of the inhibitor (Figure 5). On inhibitor binding, simulations from both RGS4 structures (PDB codes 1AGR and 1EZT) reveal a consistent and significant increase in the fluctuations of residues located in the α1-helix (51–60), as well as residues (~110 to 135) in the helices α5, α6, and the loop region connecting these two helices (see Figure 1a for designation of helices). Additionally, simulations of the NMR structure (PDB code 1EZT) suggest increased flexibility in the C-terminus of the α3-helix, and helices α7 and α8 (bottom panel in Figure 5) on binding of CCG-50014. These results have potential implications for the interaction of inhibitor-bound RGS4 with the activated Gα -subunit because the residues in the helices α3, α7, α8 of RGS4 directly contact switch-I and switch-II regions on Gα, while residues in the loop region connecting helices α5 and α6 directly contact the switch-III region on Gα (see Figure 1b for RGS4-Gα complex). The increase in the fluctuations of residues located away from the inhibitor binding site suggest an allosteric effect of CCG-50014 binding, which may contribute to an unstable RGS4-Gα interface.
Figure 5.

Change in RMSF per residue (ΔRMSF) on binding of CCG-50014 to two different structures of RGS4. Data represent difference in RMSF from at least three independent MD simulations, each for apo and inhibitor-bound structures, of RGS4. See also Fig. S10 in the supplemental material.
Using NMR methods for RGS4,24,38 we further carried out a detailed structural analysis of the effects of CCG-50014 on RGS4 (see Supporting Information for methodological details). Specifically, we recorded 1H-15N HSQC spectra (Figure S11) for two different constructs of RGS4 (cysteine-less RGS4, cysless-RGS4; and single-cysteine RGS4, Cys95-RGS4) in the presence of CCG-50014 at a 1:1 molar ratio. The absence of peak shifts, new peaks, or significant change in signal intensity in the HSQC spectrum of cysless-RGS4 in the presence of CCG-50014 (top panel in Figure S11) suggest that the compound is not interacting with the protein. This is consistent with the mechanism of (cysteine) thiol modification necessary for activity of CCG-50014.15,16 However, CCG-50014 modification of Cys95-RGS4 produces significant perturbations in the HSQC spectrum in the form of attenuation of existing peaks and the appearance of new peaks, as observed in the spectral overlay of CCG-50014-treated protein with the DMSO-treated reference sample (bottom panel in Figure S11). The appearance of new peaks with lower dispersion in the presence of CCG-50014 demonstrates that the perturbed regions of the structure undergo greater conformational averaging than in apo-RGS4. Consistent with extensive conformational changes in the presence of CCG-50014, the normalized intensity ratios of the HSQC peaks before and after the CCG-50014 modification (Figure 6) indicate that signal intensity decreased for most of the peaks between residues 61 and 182 (Figure 6b) with the exception of those in the unstructured portion of the C-terminus of RGS4 starting with Gly184.
Figure 6.

Effect of CCG-50014 on the 1H-15N HSQC spectra of RGS4. (a and b) Normalized intensity ratios of the HSQC peaks after and before CCG-50014 modification as a function of residue number for the Cysteine less (Cysless) and single Cysteine (Cys95) RGS4. (c) Percentage attenuation in peak intensity for each residue after treatment with CCG-50014. Solid and dotted horizontal lines indicate values at mean and 1 standard deviation (SD). See Figure 7 for residue mapping.
In the presence of the covalent inhibitor, where koff ≪ the chemical shift timescale, structural perturbations or inhibitor interactions result in partial or full attenuation of peaks as they are split between two (or possibly more) chemical states.39,40 Therefore, we computed % attenuation in peak intensity per residue on CCG-50014 binding (Figure 6c), and mapped residues experiencing above-average structural perturbations on the RGS4 structure (Figure 7a). These data suggest significant perturbations in many residues (91, 101, 110, 142, and 153) in the vicinity of Cys95 (Figure S12), which is likely a result of direct interaction with the aromatic rings of the inhibitor, as seen in MD simulations (Figures S7 and S8). It is worth noting that the most perturbed peak in the HSQC spectrum comes from Phe91 (Figure 6c), a residue located exactly below Cys95, and in direct contact with one of the phenyl rings of CCG-50014 in our simulations (Figures S7 and S8). Consistent with RMSF results (Figure 5), additional significant perturbations are also seen in residues (61, 63, 64, 66, 77, 167, and 178) in the N- and C-terminal helices of RGS4, as well as residues (124 and 126) in the the loop region connecting helices α5 and α6. Based upon our MD trajectories, we also carried out a theoretical chemical shift perturbation (CSP) analysis (see methods in Supporting Information) using software packages SHIFTX241 (Figure S13) and SPARTA+42 (Figure S14). The residues that are predicted by these software as perturbed more than average are compared to the NMR data in Table S3. A subset of experimentally-observed residue perturbations is compared to those perturbed in simulations (Figure 7b). Overall, we find that ~66% of perturbed residues seen in NMR experiments are also consistently observed in MD simulations. Remaining residues are not captured in theoretical CSP analysis, but some of those residues indicate increased flexibility in the RMSF data (Figure 5). Some disagreement between the experimental observations and predictions from simulations may be due to a variety of factors such as difficulty in unambiguously resolving overlapping peak positions in the NMR spectra, and the absence of certain rotameric conformational states of residues that occur on long time-scale.43 Consistent with a loss of G-protein binding and GAP activity,15 residues in the G-protein binding interface, such as Thr124 and Glu126, are perturbed both in experiments and simulations in the presence of CCG-50014.
Figure 7.
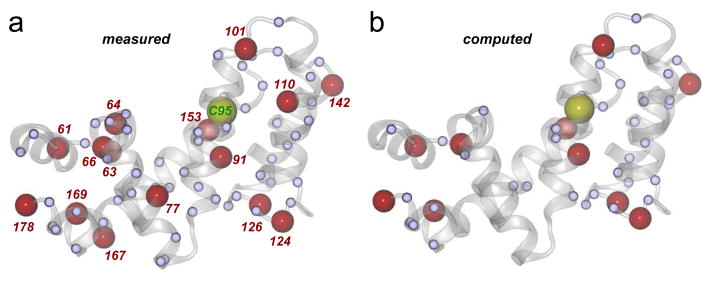
Perturbed residues are mapped on the crystal structure of RGS4 (PDB code 1AGR): (a) measured (from NMR experiments); and (b) computed (from MD simulations). Red labeled spheres indicate residues with % peak attenuation larger than 1 SD, while small blue spheres are residues with % peak attenuation between mean and 1 SD in NMR experiments. Residue Cys95, which is covalently modified by CCG-50014, is shown as a labeled yellow sphere. Protein backbone is rendered as a transparent white cartoon.
Finally, we carried out MD simulations of the RGS4-Gα complex with and without inhibitor to better understand the role of CCG-50014 in inhibiting this protein-protein interaction. For each simulation, we assess the stability of interaction between RGS4 and Gα by measuring (a) BSA between the entire RGS4 and Gα structures in the complex; and (b) total non-bonded interaction energy and its components between key residues (see Figure 8a) in the RGS4-Gα interface. These metrics, computed from a 20-ns MD equilibration of the apo crystal structure of RGS4-Gα complex (PDB code 1AGR), indicate that the interface between RGS4 and Gα remains preserved (Figure 8b, red trace for BSA in Figure 8d, and the interaction energy traces in Figure 8e). The interaction energy traces (Figure 8e) suggest that electrostatic interactions significantly contribute to the total non-bonded energy. Because the conformation of RGS4 used in the inhibitor-bound complex simulation was generated via TAMD, we enforced restraints (on all Cα atoms) for the first 20 ns of MD simulation to maintain the integrity of RGS4-Gα interface, and thereafter continued a 25-ns long free (unrestrained) MD equilibration. We observe that although RGS4-Gα interface remains largely intact during the 20-ns long restrained simulation (green trace in the gray background in Figure 8d, and snapshots in Figure S15), it gradually becomes unstable on removing the restraints (green trace in the white background in Figure 8d, and Figure 8c) due likely to a loss of dominant electrostatic interactions in this interface (Figure 8f). These data further suggest that binding of CCG-50014 to Cys95 on RGS4 results in allosteric perturbations to residues in the RGS4-Gα interface thereby inhibiting this protein-protein interaction.
Figure 8.

Snapshots of RGS4-Gα complex in (a) crystallographic conformation (PDB code 1AGR); and conformations at the end of MD equilibrations without (b) and with (c) inhibitor. The Cα -atoms of key interacting residues in the RGS4-Gα interface are labeled (panel a), and rendered as space-filling spheres in each snapshot. (d) Traces of BSA between RGS4 and Gα in the complex (green, with inhibitor; and red, without inhibitor). (e–f) Traces for non-bonded interaction energy and its components computed between residues of RGS4 and Gα (see panel a) from MD simulations without (panel e) and with (panel f) inhibitor. (Gray background in panels d and f) The first 20 ns of restrained MD simulation with inhibitor. See Figure S15 for additional snapshots from the inhibitor-bound simulation. The red trace in panel d is deliberately shifted by 20 ns to highlight the fact that simulation was not restrained.
2 CONCLUSIONS
In summary, we have studied the effect of CCG-50014 (a covalent inhibitor) binding to RGS4 as well as on the interaction of RGS4 with the Gα-subunit. Enhanced conformational sampling of RGS4 using TAMD consistently reveals the existence of open-like conformations of the α5-α6 helix pair, which facilitates binding of CCG-50014 to the buried side-chain of Cys95. We can further consistently identify a large number of key perturbed residues from NMR experiments and MD simulations, especially in the vicinity of the inhibitor binding site and in the RGS4-Gα interface. The steric effects of CCG-50014 interacting with specific residues in α4-α7, particularly with Phe91, contribute to the stabilization of the RGS4 open-conformation by restricting the closure of the α5-α6 helix pair. This allosteric effect translates into structural perturbations downstream in the Gα binding interface of RGS4. Finally, results from a simulation of the RGS4-Gα complex in the presence of CCG-50014 suggest a relatively unstable interface between RGS4 and Gα, unlike the simulation of the apo complex where a stable interface is observed.
3 METHODS
We generated all simulation trajectories using NAMD44 and the CHARMM force-field with CMAP correction.45,46 VMDv1.9 was used for system creation and protein rendering.47 We carried out all MD and TAMD simulations of RGS4 structures in explicit TIP3P water with two different initial structures (PDB codes 1AGR and 1EZT). Details of all simulations appear in Table S1. We computed the eigen-values and eigen-vectors corresponding to the first 20 non-zero normal modes of RGS4 for each of the two structures (PDB codes 1AGR and 1EZT) using their Cα-based elastic network models with the help of AD-ENM server (http://enm.lobos.nih.gov/).48,49 Overlap analyses were carried out using tcl-scripts under VMD. Data from overlap analyses are shown in Fig. 3. Details on protein expression, purification, and NMR HSQC experiments are also described in the supplemental material.
Supplementary Material
Acknowledgments
This work was supported by the NIH (grants GM037554 to CLB and DA-023252 to RRN) and the NSF funded Center for Theoretical Biological Physics (grant PHY0216576 to CLB). AJS was supported by the pharmacological sciences NIGMS training grant (GM007767), and the contents of this work are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS. We also thank J. Yesselman for help in parameterizing CCG-50014 using MATCH,50 and T. Cierpicki for providing both time and expertise in running the NMR experiments.
Footnotes
Supporting Information Available
Methodological details of simulation set-up and execution as well as NMR experiments along with additional results appear in the supplemental material. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Venkatakrishnan A, Deupi X, Lebon G, Tate CG, Schertler GF, Babu MM. Molecular signatures of G-protein-coupled receptors. Nature. 2013;494:185–194. doi: 10.1038/nature11896. [DOI] [PubMed] [Google Scholar]
- 2.Neubig R, Siderovski D. Regulators of G-protein signalling as new central nervous system drug targets. Nat Rev Drug Discov. 2002;1:187–197. doi: 10.1038/nrd747. [DOI] [PubMed] [Google Scholar]
- 3.Hollinger S, Hepler J. Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev. 2002;54:527–559. doi: 10.1124/pr.54.3.527. [DOI] [PubMed] [Google Scholar]
- 4.Tesmer JJ. Structure and function of regulator of G protein signaling homology domains. Prog Mol Biol Transl Sci. 2009;86:75–113. doi: 10.1016/S1877-1173(09)86004-3. [DOI] [PubMed] [Google Scholar]
- 5.Soundararajan M, Willard F, Kimple A, Turnbull A, Ball L, Schoch G, Gileadi C, Fedorov O, Dowler E, Higman V, Hutsell S, Sundström M, Doyle D, Siderovski D. Structural diversity in the RGS domain and its interaction with heterotrimeric G protein α-subunits. Proc Natl Acad Sci. 2008;105:6457–6462. doi: 10.1073/pnas.0801508105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tesmer J, Berman D, Gilman A, Sprang S. Structure of RGS4 bound to AlF4−-activated Giα1: Stabilization of the transition state for GTP hydrolysis. Cell. 1997;89:251–261. doi: 10.1016/s0092-8674(00)80204-4. [DOI] [PubMed] [Google Scholar]
- 7.Zhong H, Neubig RR. Regulator of G protein signaling proteins: novel multi-functional drug targets. J Pharmacol Exp Ther. 2001;297:837–845. [PubMed] [Google Scholar]
- 8.Neubig R. Regulators of G protein signaling (RGS proteins): novel central nervous system drug targets. J Pept Res. 2002;60:312–316. doi: 10.1034/j.1399-3011.2002.21064.x. [DOI] [PubMed] [Google Scholar]
- 9.Sjögren B, Neubig R. Thinking outside of the “RGS box”: new approaches to therapeutic targeting of regulators of G protein signaling. Mol Pharmacol. 2010;78:550–557. doi: 10.1124/mol.110.065219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roman DL, Traynor JR. Regulators of G protein signaling (RGS) proteins as drug targets: Modulating G-protein-coupled-receptor (GPCR) signal transduction. J Med Chem. 2011;54:7433–7440. doi: 10.1021/jm101572n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimple AJ, Willard FS, Giguère PM, Johnston CA, Mocanu V, Siderovski DP. The RGS protein inhibitor CCG-4986 is a covalent modifier of the RGS4 Gα-interaction face. Biochimica et Biophysica Acta. 2007;1774:1213–1220. doi: 10.1016/j.bbapap.2007.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blazer L, Neubig R. Small molecule protein-protein interaction inhibitors as CNS therapeutic agents: current progress and future hurdles. Neuropsychopharmacology. 2009;34:126–141. doi: 10.1038/npp.2008.151. [DOI] [PubMed] [Google Scholar]
- 13.Blazer L, Roman D, Chung A, Larsen M, Greedy B, Husbands S, Neubig R. Reversible, allosteric small-molecule inhibitors of regulator of G protein signaling proteins. Mol Pharmacol. 2010;78:524–533. doi: 10.1124/mol.110.065128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Roman D, Blazer L, Monroy C, Neubig R. Allosteric inhibition of the regulator of G protein signaling-Gα protein-protein interaction by CCG-4986. Mol Pharmacol. 2010;78:360–365. doi: 10.1124/mol.109.063388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Blazer L, Zhang H, Casey E, Husbands S, Neubig R. A nanomolar potency small molecule inhibitor of regulator of G protein signaling (RGS) proteins. Biochemistry. 2011;50:3181. doi: 10.1021/bi1019622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turner E, Blazer L, Neubig R, Husbands S. Small molecule inhibitors of regulators of G Protein signaling (RGS) proteins. ACS Med Chem Lett. 2012;3:146–150. doi: 10.1021/ml200263y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ryan DP, Matthews JM. Protein-protein interactions in human disease. Curr Op Struct Biol. 2005;15:441–446. doi: 10.1016/j.sbi.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 18.Arkin MR, Wells JA. Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 19.Roman DL, Talbot JN, Roof RA, Sunahara RK, Traynor JR, Neubig R. Identification of small-molecule inhibitors of RGS4 using a high-throughput flow cytometry protein interaction assay. Mol Pharmacol. 2007;71:169–175. doi: 10.1124/mol.106.028670. [DOI] [PubMed] [Google Scholar]
- 20.Blazer LL, Roman DL, Muxlow MR, Neubig RR. Use of Flow Cytometric Methods to Quantify Protein-Protein Interactions. Chapter 13. Wiley; New York: 2010. pp. 11–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Popov SG, Krishna UM, Falck J, Wilkie TM. Ca2+/Calmodulin reverses phosphatidylinositol 3, 4, 5-trisphosphate-dependent inhibition of regulators of G protein-signaling GTPase-activating protein activity. J Biol Chem. 2000;275:18962–18968. doi: 10.1074/jbc.M001128200. [DOI] [PubMed] [Google Scholar]
- 22.Ishii M, Fujita S, Yamada M, Hosaka Y, Kurachi Y. Phosphatidylinositol 3, 4, 5-trisphosphate and Ca2+/calmodulin competitively bind to the regulators of G-protein-signalling (RGS) domain of RGS4 and reciprocally regulate its action. Biochem J. 2005;385:65–73. doi: 10.1042/BJ20040404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Roman DL, Ota S, Neubig RR. Polyplexed flow cytometry protein interaction assay: a novel high-throughput screening paradigm for RGS protein inhibitors. J Biomol Screening. 2009;14:610–619. doi: 10.1177/1087057109336590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moy FJ, Chanda PK, Cockett MI, Edris W, Jones PG, Mason K, Semus S, Powers R. NMR structure of free RGS4 reveals an induced conformational change upon binding Gα. Biochemistry. 2000;39:7063–7073. doi: 10.1021/bi992760w. [DOI] [PubMed] [Google Scholar]
- 25.Abrams CF, Vanden-Eijnden E. Large-scale conformational sampling of proteins using temperature-accelerated molecular dynamics. Proc Natl Acad Sci USA. 2010;107:4961–4966. doi: 10.1073/pnas.0914540107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maragliano L, Vanden-Eijnden E. A temperature accelerated method for sampling free energy and determining reaction pathways in rare events simulations. Chem Phys Lett. 2006;426:168–175. [Google Scholar]
- 27.Maragliano L, Vanden-Eijnden E. Single-sweep methods for free energy calculations. J Chem Phys. 2008;128:184110. doi: 10.1063/1.2907241. [DOI] [PubMed] [Google Scholar]
- 28.Vashisth H, Maragliano L, Abrams CF. “DFG-flip” in the insulin receptor kinase is facilitated by a helical intermediate state of the activation loop. Biophys J. 2012;102:1979–1987. doi: 10.1016/j.bpj.2012.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vashisth H, Skiniotis G, Brooks CL., III Using enhanced sampling and structural restraints to refine atomic structures into low-resolution electron microscopy maps. Structure. 2012;20:1453–1462. doi: 10.1016/j.str.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vashisth H, Brooks CL., III Conformational sampling of maltose-transporter components in Cartesian collective variables is governed by the low-frequency normal modes. J Phys Chem Lett. 2012;3:3379–3384. doi: 10.1021/jz301650q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vashisth H, Abrams CF. All-atom structural models of insulin binding to the insulin receptor in the presence of a tandem hormone-binding element. Proteins. 2013;81:1017–1030. doi: 10.1002/prot.24255. [DOI] [PubMed] [Google Scholar]
- 32.Vashisth H, Skiniotis G, Brooks CL., III Enhanced sampling and overfitting analyses in structural refinement of nucleic acids into electron microscopy maps. J Phys Chem B. 2013;117:3738–3746. doi: 10.1021/jp3126297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hayward S, Go N. Collective variable description of native protein dynamics. Ann Rev Phys Chem. 1995;46:223–250. doi: 10.1146/annurev.pc.46.100195.001255. [DOI] [PubMed] [Google Scholar]
- 34.Berendsen HJC, Hayward S. Collective protein dynamics in relation to function. Curr Op Struct Biol. 2000;10:165–169. doi: 10.1016/s0959-440x(00)00061-0. [DOI] [PubMed] [Google Scholar]
- 35.Tama F, Sanejouand YH. Conformational change of proteins arising from normal mode calculations. Prot Eng. 2001;14:1–6. doi: 10.1093/protein/14.1.1. [DOI] [PubMed] [Google Scholar]
- 36.Yang L, Song G, Jernigan RL. How well can we understand large-scale protein motions using normal modes of elastic network models? Biophys J. 2007;93:920–929. doi: 10.1529/biophysj.106.095927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang L, Song G, Carriquiry A, Jernigan RL. Close correspondence between the motions from principal component analysis of multiple HIV-1 protease structures and elastic network modes. Structure. 2008;16:321–330. doi: 10.1016/j.str.2007.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Storaska AJ, Neubig RR. NMR methods for detection of small molecule binding to RGS4. Methods Enzymol. 2012;522:133–152. doi: 10.1016/B978-0-12-407865-9.00008-X. [DOI] [PubMed] [Google Scholar]
- 39.Girotto S, Sturlese M, Bellanda M, Tessari I, Cappellini R, Bisaglia M, Bubacco L, Mammi S. Dopamine-derived quinones affect the structure of the redox sensor DJ-1 through modifications at Cys-106 and Cys-53. J Biol Chem. 2012;287:18738–18749. doi: 10.1074/jbc.M111.311589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsuo H, Walters KJ, Teruya K, Tanaka T, Gassner GT, Lippard SJ, Kyogoku Y, Wagner G. Identification by NMR spectroscopy of residues at contact surfaces in large, slowly exchanging macromolecular complexes. J Am Chem Soc. 1999;121:9903–9904. [Google Scholar]
- 41.Han B, Liu Y, Ginzinger S, Wishart D. SHIFTX2: significantly improved protein chemical shift prediction. J Biomol NMR. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shen Y, Bax A. SPARTA+: a modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J Biomol NMR. 2010;48:13–22. doi: 10.1007/s10858-010-9433-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Robustelli P, Stafford KA, Palmer AG., III Interpreting protein structural dynamics from NMR chemical shifts. J Am Chem Soc. 2012;134:6365–6374. doi: 10.1021/ja300265w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kalé L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.MacKerell AD, Jr, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 46.MacKerell AD, Jr, Feig M, Brooks CL., III Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J Comput Chem. 2004;25:1400–1415. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 47.Humphrey W, Dalke A, Schulten K. VMD - Visual Molecular Dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 48.Zheng WJ, Brooks BR, Doniach S, Thirumalai D. Network of dynamically important residues in the open/closed transition in polymerases is strongly conserved. Structure. 2005;13:565–577. doi: 10.1016/j.str.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 49.Zheng WJ, Brooks BR, Thirumalai D. Low-frequency normal modes that describe allosteric transitions in biological nanomachines are robust to sequence variations. Proc Natl Acad Sci USA. 2006;103:7664–7669. doi: 10.1073/pnas.0510426103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yesselman JD, Price DJ, Knight JL, Brooks CL., III MATCH: An atom-type toolset for molecular mechanics force fields. J Comput Chem. 2012;33:189–202. doi: 10.1002/jcc.21963. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.