

Abstract
A mucosal vaccine against Helicobacter pylori infection could help prevent gastric cancers and peptic ulcers. While previous attempts to develop such a vaccine have largely failed because of the requirement for safe and effective adjuvants or large amounts of well defined antigens, we have taken a unique approach to combining our strong mucosal CTA1-DD adjuvant with selected peptides from urease B (UreB). The protective efficacy of the selected peptides together with cholera toxin (CT) was first confirmed. However, CT is a strong adjuvant that unfortunately is precluded from clinical use because of its toxicity. To circumvent this problem we have developed a derivative of CT, the CTA1-DD adjuvant, that has been found safe in non-human primates and equally effective compared to CT when used intranasally. We genetically fused the selected peptides into the CTA1-DD plasmid and found after intranasal immunizations of Balb/c mice using purified CTA1-DD with 3 copies of an H. pylori urease T cell epitope (CTA1-UreB3T-DD) that significant protection was stimulated against a live challenge infection. Protection was, however, weaker than with the gold standard, bacterial lysate+CT, but considering that we only used a single epitope in nanomolar amounts the results convey optimism. Protection was associated with enhanced Th1 and Th17 immunity, but immunizations in IL-17A-deficient mice revealed that IL-17 may not be essential for protection. Taken together, we have provided evidence for the rational design of an effective mucosal subcomponent vaccine against H. pylori infection based on well selected protective epitopes from relevant antigens incorporated into the CTA1-DD adjuvant platform.
Introduction
Helicobacter pylori is a gram negative microaerophilic bacterium which infects the gastric mucosa of approximately half of the world's population and is a risk factor for both peptic ulcer disease and gastric cancers [1], [2]. The bacteria live in the mucus layer overlying gastric epithelial cells, an environment from which it is able to provoke host inflammatory and immune responses. These host responses are unable to eradicate the infection, however, so that without treatment, the infection can persist for decades or even the life of the host. Although pharmacologic agents can cure the infection, multi-drug regimens which can have significant side effects are required. Using combinations of antibiotics and agents such as proton pump inhibitors it is possible to achieve eradication rates as high as 80–90%, but failures can lead to antibiotic resistance and re-infection is not uncommon [3], [4]. An alternative and more attractive approach is vaccination which not only leads to more vigorous immune responses than infection but it is also likely to provide herd immunity, dramatically reducing spread of infection. Several candidate vaccines and mucosal vaccines, in particular, have been shown in animal models to reduce or eliminate bacterial load and disease in the stomach [5], [6].
Although an abundance of purified/recombinant H. pylori antigens and vaccine adjuvants have been successfully used in animal models of H. pylori vaccination, bacterial lysates and whole cell vaccines combined with the holotoxins cholera toxin (CT) or the closely related E. coli heat labile toxin (LT) as mucosal adjuvants have been the gold standard in animal models of H. pylori vaccination [5]. Most vaccine regimens require an adjuvant and work best intranasally (i.n) [7] or sublingually [8]. Numerous studies in animal models have also demonstrated that antibodies are not required for (but may participate in or even impair) protective immunity, but, in contrast, specific CD4 T cell responses are required for H. pylori vaccine efficacy [9], [10], [11], [12], [13]. Among subunit and vector vaccines, H. pylori urease has been a leading candidate [14], [15], [16] and both CD4 T cell and B cell peptide epitopes have been defined [17], [18].
Cholera toxin or LT have been the most effective and widely used adjuvants for H. pylori mucosal vaccines in animal models. These bacterial toxins are well tolerated when used at adjuvant effective doses in mice and other small animal models of H. pylori infection. CT and LT are too toxic for humans, however, and in a human clinical H. pylori vaccine trial, the use of holotoxin LT resulted in significant diarrhea in 2/3 of the vaccine recipients [19]. Mutations targeting the active sites of these molecules can reduce the toxicity while retaining adjuvant function and these mutant toxins have been used with some success as mucosal adjuvants for H. pylori vaccines [20], [21]. Our approach has been to create chimeric CT-derived molecules which retain the full enzymatic activity of the holotoxin, but which specifically target immune cells instead of all nucleated GM1-receptor carrying cells, including nerve cells [22]. In this approach we have linked the enzymatically active CTA1 fragment of CT to two copies of the D-fragment of Staphylococcus aureus protein A, a strong immunoglobulin binding domain, to create an adjuvant that we have named CTA1-DD [23]. We have shown that this molecule is non-toxic when delivered i.n to mice and non-human primates [24], [25] and significantly reduces the bacterial burden when used as a mucosal adjuvant for an H. pylori lysate vaccine in mice [26], [27]. We recently demonstrated a related approach in which a peptide from influenza virus, the M2e peptide, was inserted into CTA1-DD (CTA1-M2e-DD) and found to effectively protect against infection in mice [28], [29]. We now report that an MHC class II restricted H. pylori peptide inserted into CTA1-DD also can successfully immunize and protect Balb/c mice against H. pylori infection [18].
Materials and Methods
H. pylori growth, mice, immunization, and challenge
H. pylori for infection and preparation of bacterial lysate was grown as previously published [30]. Balb/c female mice aged 6–8 weeks were obtained from Charles River Laboratories (Portage, MI) or from Harlan Laboratories (the Netherlands) and were housed in microisolater cages under specific pathogen free (spf) conditions with free access to autoclaved food and water. IL-17 KO mice [31] on a Balb/c background, were bred in ventilated cages and kept together with the Balb/c wild-type mice under spf conditions at the University of Gothenburg Animal Facility (Gothenburg, Sweden). All mice infected with H. pylori were housed in A-BSL-2 rooms. Mice were immunized intranasally (i.n) while fully awake by administering 100 µg of H. pylori lysate plus 5 µg of CT adjuvant, 100 µg of H. pylori peptides plus 5 µg of CT adjuvant or 5 µg of CTA1-peptide-DD in 20 µl of saline, 10 µl at a time slowly to each nare. Immunizations were done as a single immunization or as series of 4 immunizations, once a week, as indicated in the figure legends. Immunized mice were challenged twice over three days with ca 107 cfu of H. pylori strain SS-1 [32] in 0.5 ml of brucella broth at 4 weeks after completion of immunizations using flexible polyethylene tubing on the end of a tuberculin syringe. At 3 or 7 weeks after challenge mice were killed and stomachs were removed aseptically. These time intervals post-challenge were chosen based upon our previously published work showing a stable level of gastric H. pylori colonizing bacteria in non-immunized challenged mice and a consistent drop in the bacterial load from intranasally immunized mice over this time period [33], [34]. These published results were in C57BL/6 mice, but we have observed essentially identical kinetics for the drop in H. pylori colonizing bacteria also in immunized BALB/c mice, the mouse strain used in the present research (JGN, unpublished results). Noteworthy, protection induced by CTA1-UreB3T-DD immunizations was similar irrespective if we assessed it at 3 or 7 weeks after immunizations (see Results). A strip of stomach including both antrum and body was removed from the greater curvature and used for quantitative culture [33]. Additional sections of stomach were fixed in buffered formalin, embedded in paraffin, and sections were stained with H&E for scoring of gastric inflammation. Each experiment included challenge of both immunized and the internal control of non-immunized (administered saline) mice and most experiments also included the positive control of mice immunized with H. pylori lysate plus CT.
Ethics statement:All mouse protocols were approved by the Case Western Reserve University IACUC protocol number 09-0111 and the Ethical committee, the Board of Agriculture in Gothenburg, Sweden, ethical protocol number 60/11.
H. pylori peptides
For immunizations, a linear B cell epitope peptide and two CD4 T cell epitope peptides were produced by Genscript (Piscataway, NJ) at >95% purity. The 19 amino acid ureB linear B cell epitope ureB321–339 (CHHLDKSIKEDVQFADSRI) represents a site which a urease neutralizing monoclonal antibody recognizes [17] and a fusion protein between this epitope and CTB as been shown to reduce H. pylori colonization when used as an oral vaccine [35]. The two CD4 T cell epitopes, 15 amino acid ureB237–251 (VADKYDVQVAIHTDT) (I-Ad restricted) and 16 amino acid ureB546–561 (FVDGKEVTSKPANKVS) (I-Ed restricted) [18] have been shown to act synergistically to induce T cell proliferation after subcutaneous immunization with CFA/IFA in BALB/c mice [18]. For peptide immunization of Balb/c mice, 100 µg of peptide was mixed with 5 µg of holotoxin CT (List Biological Laboratories, Campell, CA) in a total of 20 µl of PBS and 10 µl was administered i.n to each nare. When a combination of two different peptides were used for immunization, 50 µg of each peptide (100 µg total) were admixed with CT adjuvant, as described above.
Genetic constructs incorporating H. pylori peptides into CTA1-DD fusion proteins
CTA1-DD fusion protein alone or CTA1-DD constructions containing a single copy or 3 copies of UreB237–251 peptide, CTA1-UreB3T-DD, or three copies of the influenza virus antigen M2e (amino acid sequence VADKYDVQVAIHTDT), were produced in Escherichia coli as previously described [23]. Another fusion protein containing single copies of the CD4 T cell epitope peptide UreB237–251 plus the B cell epitope peptide UreB321—339 incorporated into the CTA1-DD backbone was produced in a similar manner. ADP-ribosyltransferase enzymatic activity was tested using the NAD:agmatine assay as described previously [36]. Protein analysis was performed with SDS-PAGE, and concentrations were determined using the Bio-Rad DC protein assay, according to the manufacturer's instructions. Endotoxin levels were assessed by the Limulus amebocyte lysate test (LAL Endochrome; Charles River Endostestafe). The endotoxin levels were <1 EU/mg protein in all preparations used.
Determinations of antibodies, cytokines and cell proliferation
Serum IgG anti-H. pylori antibodies were analyzed by ELISA as previously described using H. pylori lysate (10 µg/ml) as the coating antigen [33]. At the time of harvest, spleens were removed and cultured for 48–72 hr together with recall antigen in RPMI media or Iscove's media (IMDM, HyClone), supplemented with 10% fetal bovine serum, 5×10−5 M beta mercaptoethanol and antibiotics at 2×106 cells/ml. For antigen recall responses we used H. pylori lysate (10 µg/ml), recombinant UreB (rUreB) (10 µg/ml) (kind gift from C. Flach and J. Holmgren, University of Gothenburg, Sweden), CTA1-UreB3T-DD (1 µM), CTA1-DD (1 µM) or purified peptides, UreB T 237–251 and UreB B 321–339, Innovagen AB (Lund, Sweden), at 5 µg/ml or as specified in the figure legends and text describing each experiment. To assess UreB-specific CD4 T cell responses we enriched splenocytes from immunized mice with AutoMacs (Miltenyi Biotech, Köln, Germany) (purity >96%) and cultured these cells at 100.000 cells/well together with irradiated splenocytes from nu/nu Balb/c mice and recall antigen (UreB and CTA1-Ureb3T-DD) as described above. CD4 T cell antigen-recall responses were analyzed in culture supernates for IFNγ, IL-17A, IL-17F and other cytokines as indicated using specific Duoset kits (R&D Systems, Minneapolis, MN) according to manufacture's instructions. Specific cell proliferation to recall antigen was assessed after 72 h of culture, by the addition of 1 µCi/well 3H-thymidine (PerkinElmer, USA) for the last 6 hours of culture. The 3H-thymidine uptake was determined using a scintillation counter (Beckman, LKB, Bromma, Sweden). Data were expressed as mean c.p.m ± S.E.M. of 5 mice per group in each experiment.
Statistical analysis
The parametric ANOVA analysis with post-hoc Bonferroni tests for multiple groups was used for analysis of statistical significance in all experimental groups. Statistical significance is also given as p-values: ***p<0.001, **p<0.01 and *p<0.05.
Results
We first confirmed the capacity of two different H. pylori urease B subunit MHC class II restricted peptides, UreB237–251 and UreB546–561, to prime for T cell and antibody responses following i.n immunizations of Balb/c mice using CT adjuvant and challenging with live bacteria. Panel A of Figure 1 shows that after H. pylori challenge of mice previously immunized with a mixture of the two peptides, a significantly elevated IFNγ response was observed after in vitro recall stimulation with H. pylori lysate (P = 0.014). There was also a trend towards elevated responses after immunization with the individual peptides. However, these results did not reach statistical significance. In a similar manner, Figure 1B shows that elevated levels of serum IgG antibody versus H. pylori lysate was observed when mice were immunized with peptide UreB237–251 (P = 0.026), but not with UreB546–561 or a mixture of the two peptides. Most importantly, the capacity of the T cell epitopes to prime for anti-H. pylori immune responses was also shown to have functional activity since mice immunized i.n with a mixture of the two peptides plus CT adjuvant exhibited a 5.5 fold reduction in gastric H. pylori (p = 0.050) (Figure 1C). This reduction in bacterial load was not as great as the 27-fold reduction (p = 0.022) observed after mice were immunized with lysate plus CT, however. There was a positive correlation between protective efficacy and increased priming of IFNγ producing spleen T cells.
Figure 1. Groups of 6 Balb/c mice were left unimmunized (PBS) or were immunized intranasally 4 times with 100 µg of H. pylori lysate+5 µg of CT adjuvant, 100 µg of Hp UreB237–251+5 µg of CT adjuvant, 100 µg of Hp UreB546–561+5 µg of CT adjuvant or a mixture of the two peptides (50 µg each)+5 µg of CT adjuvant, as described in the Methods section.
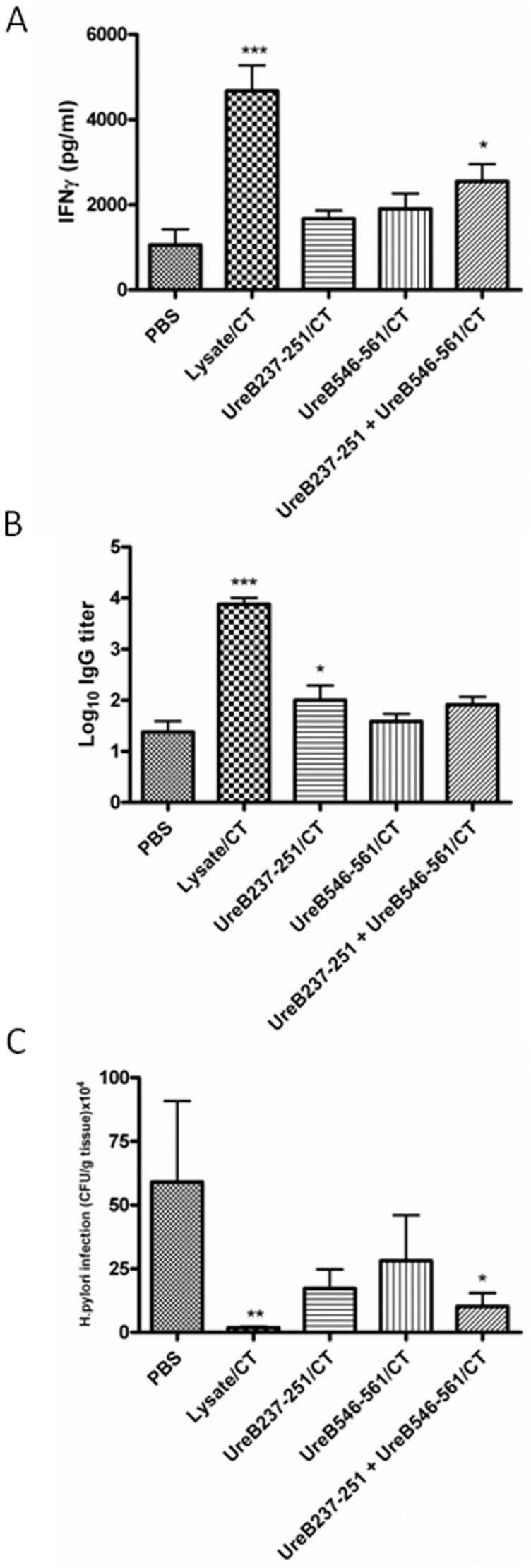
Four weeks after completion of immunizations all mice were challenged twice over three days with ca 2×107 cfu of H. pylori SS-1 and 3 weeks later mice were killed and recall splenic IFNγ responses in supernatants from lysate stimulated cultures with unstimulated background controls subtracted (A), serum IgG anti-H. pylori antibody responses (B) and gastric H. pylori cfu/g of stomach tissue (C) were determined. These are representive data from at least 3 identical experiments giving similar results. ANOVA was used for statistical analysis: * p<0.05, ** p<0.01 and ***p<0.001.
Based upon the capacity of these H. pylori peptides to induce anti-H. pylori immunity following immunization, we elected to generate a genetic fusion protein construct incorporating 3 copies of UreB237–251 within CTA1-DD, CTA1-UreB3T-DD, as shown in Figure 2A and described under Methods. The resulting fusion protein was expressed and purified as described [23] and Figure 2B shows a western blot of the purified fusion protein probed with an anti-DD monoclonal antibody [23]. We also demonstrated that the fusion protein possessed full ADP-ribosylation activity (a measure of preserved enzymatic function of the CTA1 moiety) comparable to that of native CT (Figure 2C). Next we evaluated this fusion protein as an i.n H. pylori vaccine and compared it with i.n H. pylori lysate plus CT, UreB237–251 plus CT, or a mixture of UreB237–251 and a B cell epitope peptide, UreB321–339, plus CT. As shown in Figure 3, lysate significantly primed IFNγ producing CD4 T cells as did the mixture of UreB237–251 and UreB321–339 (p<0.05)(Fig. 3A). However, similar to the results in Figure 1 only lysate and UreB237–251 plus CT adjuvant yielded enhanced antibody responses (p<0.001 and p<0.05, respectively) (Fig. 3B). When the bacterial load was examined after challenge of mice immunized i.n with CTA1-UreB3T-DD significant reduction in cfu was achieved (p = 0.02, 4-fold reduction), although this effect was not as great as that achieved by lysate plus CT (p = 0.004, 90-fold reduction) (Fig. 3C). Noteworthy, the dose of peptide incorporated in the fusion protein was in the 1 nanomolar range, whereas the dose of UreB237–251 (p = 0.10, 2-fold reduction) or a mixture of UreB237–251 and UreB321–339 plus CT adjuvant (p = 0.06, 3-fold reduction) was 60-fold higher, but still CTA1-UreB3T-DD resulted in lower cfu and stronger protection (Fig. 3C). Since we and others have observed enhanced gastritis in protectively immunized mice [33], [34], we also examined gastric inflammation in the mice from this experiment. As expected, mice immunized with H. pylori lysate plus CT exhibited enhanced gastritis after challenge (gastritis score 2.33±1.03 compared with naive challenged mice score of 1.0±0.82, p = 0.002 by ANOVA). Mice immunized with CTA1-UreB3T-DD did not exhibit enhanced gastritis (gastritis score 0.83±0.75, p = 0.68), perhaps reflecting the lower degree of protection when compared to mice immunized with bacterial lysate plus CT.
Figure 2. Schematic drawing of the CTA1-DD plasmid vector with incorporated 1–3 UreB 237–251 epitopes in single or tandem repeats.

(A). Protein analysis was performed with SDS-PAGE on purified material from the E.coli expression system and concentrations were determined using the Bio-Rad DC protein assay, according to the manufacturer's instructions. Western blot analysis using HRP-labelled anti-DD antibodies (B). The ADP-ribosylating ability of CTA1-UreB3T-DD, but not the mutant inactive CTA1R7K-UreB3T-DD protein, was comparable to that of CT at a molar level, as shown in the NAD-agmatine assay diagram (C). These are representive data from at least 5 identical experiments giving similar results.
Figure 3. Groups of 6 or 7 Balb/c mice were left unimmunized (PBS) or were immunized intranasally 4 times with H. pylori lysate+5 µg of CT adjuvant, a fusion protein of CTA1-UreB3T-DD containing three copies of UreB237–251, UreB237–251 peptide alone+5 µg of CT adjuvant, or a mixture of UreB237–251 plus the UreB B321–339 peptide+5 µg of CT adjuvant, as described in Fig. 1.
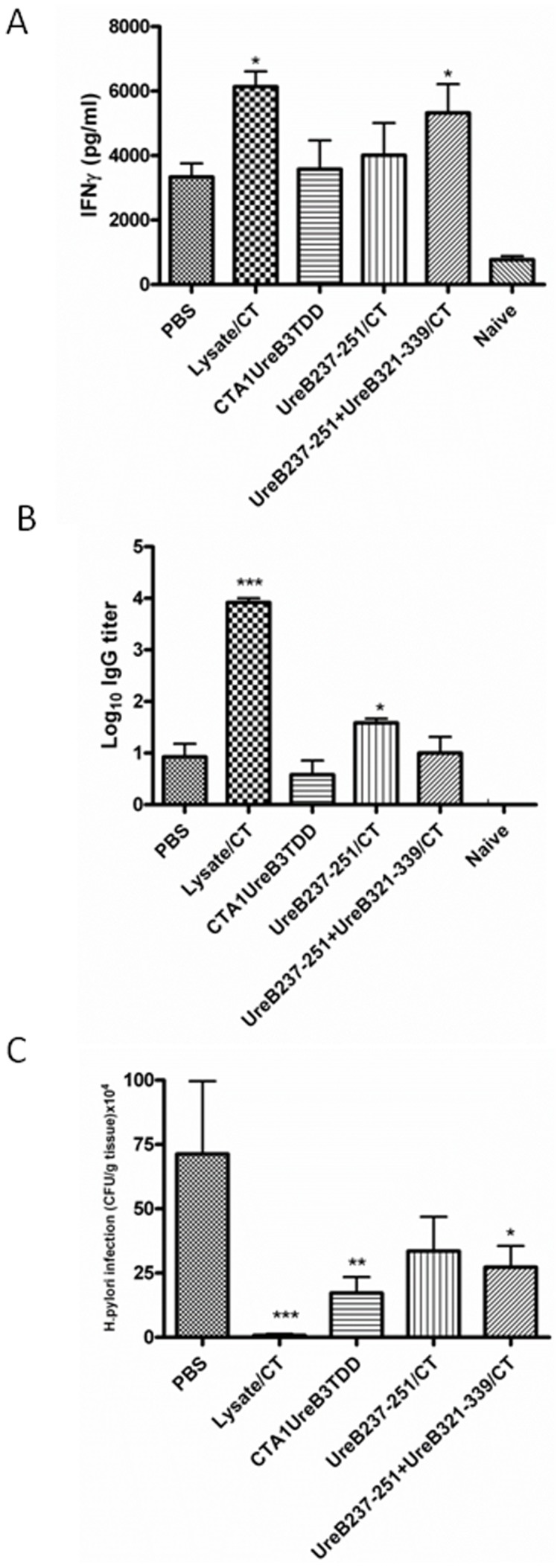
Four weeks after completion of immunization a separate group of 3 naïve mice were left unchallenged (Naïve) and all other mice were challenged twice over three days with ca 2×107 cfu of H. pylori SS-1 and 3 weeks later mice were killed and recall splenic IFNγ responses in supernatants from lysate stimulated cultures with unstimulated background controls subtracted (A), serum IgG anti-H. pylori antibody responses (B) and gastric H. pylori cfu/g of stomach tissue (C) were determined. For IFNγ determinations (A) only one experiment with CTA1-UreB3T-DD was included, but apart from that these are representive data from at least 3 identical experiments giving similar results. ANOVA was used for statistical analysis :* p<0.05, ** p<0.01 and ***p<0.001.
To verify that our CTA1-UreB3T-DD construct stimulated UreB-specific CD4 T cells we sorted CD4 T cells from the spleens of CTA1-UreB3T-DD or CTA1-DD-immunized or unimmunized mice by AutoMacs (>96% purity) and cultured these cells together with irradiated antigen-presenting cells (nu/nu splenocytes) and CTA1-UreB3T-DD or rUreB (Fig. 4A). We found that rUreB stimulated significant CD4 T cell proliferation in mice immunized i.n with the CTA1-UreB3T-DD construct, while CD4 T cells from CTA1-DD or unimmunized mice did not respond to recall rUreB (Fig. 4A). Moreover, the response to in vitro stimulation by CTA1-UreB3T-DD was stronger in mice immunized with the whole molecule than it was in mice immunized with CTA1-DD, without the UreB237–251 peptide. This indicated that UreB237–251, indeed, stimulated an UreB-specific CD4 T cell response (Fig. 4A). In fact, in 4 of 5 experiments, which included the CTA1-UreB3T-DD fusion protein, i.n immunization induced CD4 T cell responses that were associated with a 3–8 fold reduction in bacterial load as opposed to H. pylori challenge of unimmunized controls (not shown). The results from one of these experiments are shown in Figure 4. In this experiment immunization with CTA1-UreB3T-DD resulted in elevated levels of spleen cell recall proliferation, and in splenic IL-5, IFNγ and IL-17 cytokine production, as well as a 5-fold reduction in bacterial cfu after challenge. (Fig. 4B–C). As expected, i.n immunizations with CTA1-DD, lacking the UreBT-epitope, failed to stimulate anti-bacterial protection (Fig. 4C). Noteworthy, the recall responses to CTA1-UreB3T-DD, as opposed to those after CTA1-DD alone, which did not contain the UreB237–251 peptide, confirmed a significant CD4 T cell response against the UreB237–251-epitope (Fig. 4A–B). By contrast, immunizations with a mutant, enzymatically inactive, CTA1R7K-UreB3T-DD construct failed to stimulate protection or CD4 T cell responses, in agreement with our results from previous studies using inactive mutants (not shown) [29].
Figure 4. Mice (n = 5) were immunized once i.n with CTA1-UreB3T-DD or CTA1-DD alone and 10 days after the immunization spleen CD4 T cells were enriched using AutoMACS and cultured together with irradiated splenocytes from naïve nu/nu mice (T cell deficient) in the absence (\) or presence of CTA1-UreB3T-DD or rUreB at 1 µM and 10 µg/ml, respectively (A).

Next, groups of 8 or 10 Balb/c mice were left unimmunized (PBS) or were immunized intranasally 4 times with 5 µg/dose of CTA1-UreB3T-DD (B–C) or 5 µg/dose CTA1-DD (C). Four weeks after completion of immunization a separate group of 3 naïve mice were left unchallenged and all other mice were challenged twice over three days with ca 2×107 cfu of H. pylori SS-1 and 7 weeks later mice were killed and recall splenic T cell proliferation (B) and splenic whole cell recall responses of IL-5, IFNγ and IL-17A were determined in unstimulated (\) or stimulated (CTA1-UreB3T-DD (1 µM) or CTA1-DD (1 µM)) culture supernatants (B) and gastric bacterial colonization was assessed with H. pylori in cfu/g of stomach tissue (C). A is representative of 2 separate experiments while B–C are representive data from at least 5 identical experiments giving similar results. ANOVA was used for statistical analysis : * p<0.05, ** p<0.01 and ***p<0.001.
These results, along with those in Figure 3, showing that a mixture of UreB237–251 and UreB321–339 (B cell epitope) plus CT-adjuvant yielded an enhanced IFNγ response and a nearly significant reduction in bacterial load, encouraged us to generate a CTA1-DD fusion protein, which contained a single copy each of both the T and B epitopes. However, in two experiments this CTA1-UreBBT-DD fusion protein failed to achieve any reduction in gastric H. pylori load, even though the non-fusion protein mixture of the two peptides plus CT did result in a slightly elevated IFNγ recall response and a reduction in bacterial load comparable to lysate in one of these experiments, as shown in Figure 5. Thus, we concluded that the fusion protein with 3 copies of the T cell epitope UreB237–251 was a more effective vaccine than a similar fusion protein with only a single copy of UreB237–251 (which also included a single copy of the B cell epitope UreB321–339) and significantly more effective than soluble peptide plus CT.
Figure 5. Groups of 6 or 7 Balb/c mice were left unimmunized (PBS) or were immunized intranasally 4 times with H. Pylori lysate+5 µg of CT adjuvant, CTA1-UreB T237–251/B 321–339-DD (a fusion protein containing 1 copy of the T epitope (UreB 237–251)+1 copy of the B epitope (B321–339)), UreB 237–251 alone+5 µg of CT adjuvant, or a mixture of UreB 237–251 and UreB B321–339+5 µg of CT adjuvant.
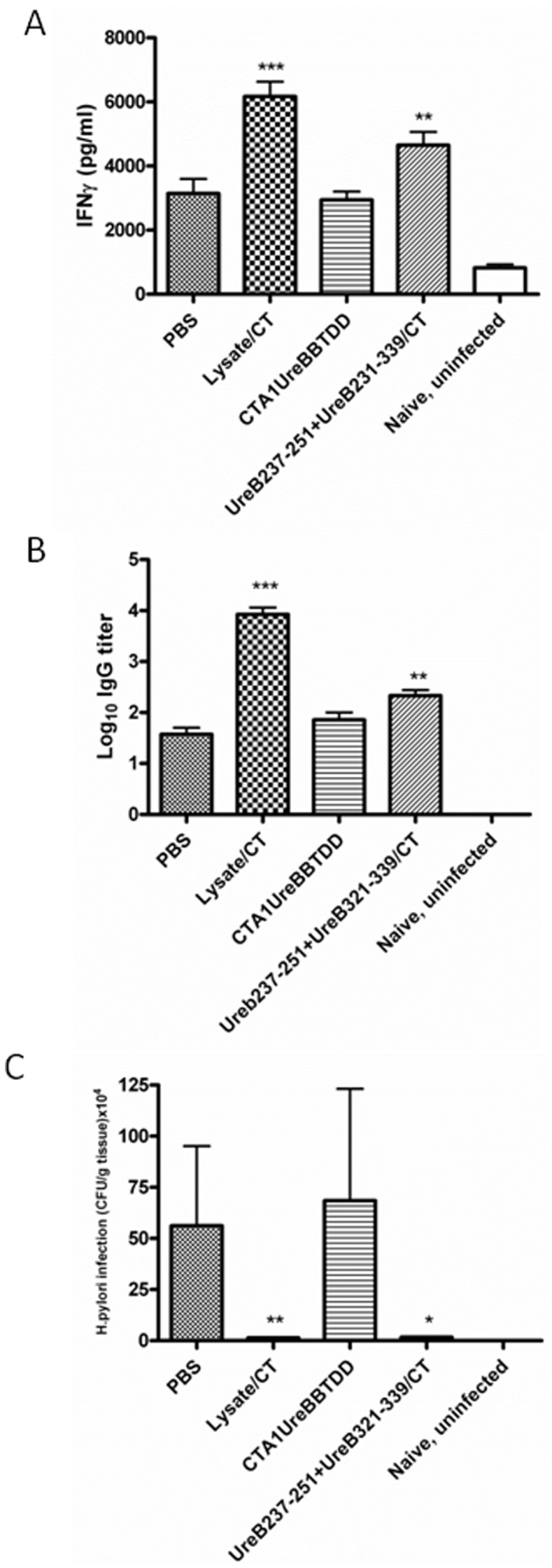
Four weeks after completion of immunization a separate group of 3 naïve mice (Naïve) were left unchallenged and all other mice were challenged twice over three days with ca 2×107 cfu of H. pylori SS-1 and 7 weeks later mice were killed and recall splenic IFNγ responses in supernatants from lysate-stimulated cultures with unstimulated background controls subtracted (A), serum IgG anti-H. pylori antibody responses (B) and gastric H. pylori cfu/g of stomach tissue (C) were determined. These are representive data from 2 identical experiments giving similar results. ANOVA was used for statistical analysis : *p<0.05, **p<0.01 and ***p<0.001.
Because recent publications have investigated a possible role for IL-17 in vaccine-mediated immunity and protection against H. pylori infection [37], [38], [39] and our results with CTA1-UreB3T-DD showed enhanced priming of IL-17A producing T cells following i.n immunizations (Fig. 4B) we investigated the protective effect of CTA1-UreB3T-DD in IL-17A-deficient and wild-type Balb/c mice [31]. Strikingly, CTA1-UreB3T-DD was equally effective in IL-17A-deficient as compared to WT mice with regard to protection and also effectively primed CD4 T cells in IL-17A-deficient mice (Fig. 6A–C). Hence, when immunized IL-17A-deficient mice were challenged with live H. pylori they showed a 5-fold reduction in bacterial load compared with mice immunized with CTA1-DD, without the UreB peptide, or given PBS only (Figure 6C). Again supernatants stimulated with CTA1-UreB3T-DD and CTA1-DD, respectively, differed with regard to the cytokine responses, indicating that CD4 T cells recognizing the UreB237–251 peptide were stimulated by CTA1-UreB3T-DD (Fig. 6B). As expected, CD4 T cells from mice immunized i.n with CTA1-UreB3T-DD had much stronger recall responses to the fusion protein than against bacterial lysate, whereas lysate immunized mice had fewer CD4 T cells that could respond to the fusion protein (Fig. 6A). The results also showed that, as expected, no IL-17A was produced in CD4 T cell recall responses in IL-17A deficient mice, while supernatants exhibited significantly elevated levels of IL-5, IFNγ and IL-17F when taken from cultures of immunized mice (Figure 6B).
Figure 6. Groups of 8 or 10 wild-type (Balb/c) or IL-17A-deficient mice (H-2d background) under SPF conditions were left unimmunized (PBS) or were immunized intranasally 4 times with 5 µg/dose of CTA1-UreB3T-DD or H. Pylori lysate+5 µg of CT adjuvant (IL-17KO only).

Four weeks after completion of immunization a separate group of 3 naïve mice were left unchallenged and all other mice were challenged twice over three days with ca 2×107 cfu of H. pylori SS-1 and 7 weeks later mice were killed and recall splenic T cell proliferation (A) and IL-5, IFNγ, IL-17F or IL-17A in culture supernatants were assessed after no stimulation (\) or stimulation in vitro with lysate, CTA1DD (1 µM) or CTA1UreB3TDD (1 µM) following i.n immunizations of IL-17KO mice, as indicated (B) and gastric H. pylori bacterial colonization as cfu/g of stomach tissue (C) was determined in both wild-type (WT) and IL-17KO mice. This is one representive experiment of three with IL-17 KO mice and one experiment comparing also with WT mice (C). ANOVA was used for statistical analysis: * p<0.05, **p<0.01 and ***p<0.001.
Discussion and Conclusions
The unexpectedly slow progress in anti-Helicobacter vaccine research can be ascribed to a poor understanding of the immunopathology and immune evasion mechanisms employed by H. pylori, subsequent waning pharmaceutical/vaccine company enthusiasm, as well as the lack of safe and effective mucosal vaccine formulations and delivery systems/adjuvants [6], [40]. Therefore, unravelling the mechanisms of Helicobacter-specific protective immunity using the mouse model is still highly relevant and much needed, even from an industrial perspective [41]. The poor outcome of clinical vaccine trials in the past has certainly also contributed to a declining interest in H. pylori-vaccine development. Apart from whole killed bacteria, also urease or combinations of CagA, VacA and neutrophil-activating protein (NAP) have been tested in human clinical trials with quite poor outcome [42]. Although parenteral vaccination has been reported to induce memory T cells specific for H. pylori, most experimental studies have clearly documented that mucosal immunizations are more effective at stimulating Helicobacter-specific protective immunity in the gastric mucosa. Particularly interesting are vaccines that effectively have stimulated H. pylori-specific CD4 T cells and the successful immunizations using subcomponent vaccines in mouse models [18].
While whole bacterial cell lysate approaches to vaccine-development has been attempted for a long time these are unrealistic for commercial production and more and more subcomponent vaccines have been described. Antioxidants have attracted interest as subcomponent vaccines and catalase, thiolperoxidase or superoxide dismutase, all proteins involved in bacterial colonization and survival, are three candidates to potentially be included in a vaccine [43]. Also, Urease B (UreB) has been found to be a strong vaccine candidate and it has provided excellent results in experimental models. Indeed, recent attempts to immunize with whole recombinant UreB and a truncated form of neuraminyllactose-binding hemagglutinin (HpaA), a H. pylori-specific lipoprotein, induced strong immune protection comparable to that of whole cell lysate [44]. Attempts to develop UreB peptide-based immunization protocols have also been reported in mice and immunodominant epitopes in H. pylori-infected subjects have recently been reported [18], [45], [46].
We have developed a peptide-based CTA1-DD adjuvanted H. pylori vaccine for mucosal administrations. We hypothesized that by exploring the CTA1-DD adjuvant as a carrier and immunomodulator of UreB-specific MHC class II-restricted peptides, we could achieve strong protection against infection, and with minimal amounts of peptide [18], [28], [29]. For comparison we initially also used rather large doses of peptide alone together with CT holotoxin, as the adjuvant, to generate a significant level of protection against a live H. pylori challenge infection. We found that protection in the range of 3–8-fold reduction of bacterial colonization was effectively achieved with a fusion protein that carried the UreB 327–251 peptide, in 3 tandem repeats, while a single epitope or just a single linear B cell epitope of UreB 321–339, was without protective effect following 4 i.n immunizations. Noteworthy, the assessment of protection following immunizations and challenge was 3 or 7 weeks, a time difference that could have contributed to the variablity in protective capacity of CTA1-UreB3T-DD (see Materials & methods). However, previous studies have clearly shown only very small differences in bacterial load in immunized and protected mice at 3 and 7 weeks [33], [34]. Interestingly, also i.n immunizations with the constructs gave comparable protection in IL-17A-deficient mice, indicating that IL-17A was not absolutely required to elicit a protective immune response. This result confirms our previous published report which utilized an H. pylori whole cell lysate vaccine [37], but it is in contrast to some other reports which used anti-IL17 monoclonal antibodies to neutralize IL-17A during the effector phase after H. pylori challenge [38], [39]. In our present results protection in IL-17 null mice correlated with IFNγ levels, while in wild-type (WT) mice also IL-17A and IL-5 levels were strongly up-regulated in CD4 T cells mediating protective immunity, as compared to CD4 T cell responses in unprotected mice. Thus the role of IL-17A or IL-17F in protective immunity remains unclear, but could be related to the well known role of IL-17A/F in promoting the induction of neutrophil chemokines, which could be substituted for by other neutrophil chemotactic factors, as discussed in our previous publication [37]. Nevertheless, our study clearly suggests that in the absence of IL-17A and Th17 cells, Th1 cells may compensate and provide significant protection following vaccination. This is in agreement with recent results exploring vaccine-induced protection in IL23 p19-deficient mice and the fact that IL-12 i.p treatment of H. pylori-infected mice promoted eradication of infection even without prior vaccination [47]. In total, our study clearly demonstrates that it is feasible to design and develop novel mucosal vaccines against H. pylori infections by exploring the CTA1-DD adjuvant platform and selected CD4 T cell epitopes. In conclusion, we have provided evidence for the rational design of an effective mucosal subcomponent vaccine against H. pylori infection based on well selected protective epitopes from relevant antigens incorporated into the CTA1-DD adjuvant platform.
Funding Statement
This work was supported by US National Institutes of Health grant AI 083694 to JGN and NYL and Swedish Research Council, the Swedish Cancer Foundation grant 4992-B10 06-03XCC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. NIH Consensus Conference (1994) Helicobacter pylori in peptic ulcer disease. Journal of the American Medical Association 272: 65–69.8007082 [Google Scholar]
- 2.World Health Organization (1994) Infection with Helicobacter pylori Schistosomes, Liver Flukes and Helicobacter pylori. Lyon: International Agency for Research on Cancer. pp. 177–241. [Google Scholar]
- 3. Malfertheiner P, Megraud F, O'Morain CA, Atherton J, Axon AT, et al. (2012) Management of Helicobacter pylori infection–the Maastricht IV/Florence Consensus Report. Gut 61: 646–664. [DOI] [PubMed] [Google Scholar]
- 4. Tepes B, O'Connor A, Gisbert JP, O'Morain C (2012) Treatment of Helicobacter pylori infection 2012. Helicobacter 17 Suppl 1: 36–42. [DOI] [PubMed] [Google Scholar]
- 5.Blanchard TG, Nedrud JG (2010) Helicobacter pylori Vaccines. In: Sutton P, Mitchell H, editors. Helicobacter Pylori in the 21st Century. Wallingford, Oxfordshire United Kingdom: CAB International. pp. 167–189. [Google Scholar]
- 6. Czinn SJ, Blanchard T (2011) Vaccinating against Helicobacter pylori infection. Nat Rev Gastroenterol Hepatol 8: 133–140. [DOI] [PubMed] [Google Scholar]
- 7. Garhart CA, Heinzel FP, Czinn SJ, Nedrud JG (2003) Vaccine-Induced Reduction of Helicobacter pylori Colonization in Mice Is Interleukin-12 Dependent but Gamma Interferon and Inducible Nitric Oxide Synthase Independent. Infect Immun 71: 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Raghavan S, Ostberg AK, Flach CF, Ekman A, Blomquist M, et al. (2010) Sublingual immunization protects against Helicobacter pylori infection and induces T and B cell responses in the stomach. Infect Immun 78: 4251–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ermak TH, Giannasca PJ, Nichols R, Myers GA, Nedrud JG, et al. (1998) MHC-class II but not MHC-class I or B cell responses are required for vaccine-induced protection against murine Helicobacter pylori infection. Abstracts to Third International Workshop on Pathogenesis and Host Response in Helicozbcter Infections, Helsingor, Denmark.
- 10. Blanchard TG, Czinn SJ, Redline RW, Sigmund N, Harriman G, et al. (1999) Antibody-independent protective mucosal immunity to gastric Helicobacter infection in mice. Cellular Immunology 191: 74–80. [DOI] [PubMed] [Google Scholar]
- 11. Pappo J, Torrey D, Castriotta L, Savinainen A, Kabok Z, et al. (1999) Helicobacter pylori Infection in Immunized Mice Lacking Major Histocompatibility Complex Class I and Class II Functions. Infect Immun 67: 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sutton P, Wilson J, Kosaka T, Wolowczuk I, Lee A (2000) Therapeutic immunization against Helicobacter pylori infection in the absence of antibodies. Immunol Cell Biol 78: 28–30. [DOI] [PubMed] [Google Scholar]
- 13. Akhiani AA, Schon K, Franzen LE, Pappo J, Lycke N (2004) Helicobacter pylori-specific antibodies impair the development of gastritis, facilitate bacterial colonization, and counteract resistance against infection. J Immunol 172: 5024–5033. [DOI] [PubMed] [Google Scholar]
- 14. Corthesy-Theulaz I, Porta N, Glauser M, Saraga E, Vaney AC, et al. (1995) Oral immunization with Helicobacter pylori urease B subunit as a treatment against Helicobacter infection in mice. Gastroenterology 109: 115–121. [DOI] [PubMed] [Google Scholar]
- 15. Gomez-Duarte OG, Lucas B, Yan ZX, Panthel K, Haas R, et al. (1998) Protection of mice against gastric colonization by Helicobacter pylori by single oral dose immunization with attenuated Salmonella typhimurium producing urease subunits A and B. Vaccine 16: 460–471. [DOI] [PubMed] [Google Scholar]
- 16. Smythies LE, Novak MJ, Waites KB, Lindsey JR, Morrow CD, et al. (2005) Poliovirus replicons encoding the B subunit of Helicobacter pylori urease protect mice against H. pylori infection. Vaccine 23: 901–909. [DOI] [PubMed] [Google Scholar]
- 17. Hirota K, Nagata K, Norose Y, Futagami S, Nakagawa Y, et al. (2001) Identification of an antigenic epitope in Helicobacter pylori urease that induces neutralizing antibody production. Infect Immun 69: 6597–6603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shi Y, Wu C, Zhou WY, Mao XH, Guo G, et al. (2007) Identification of H-2d restricted Th epitopes in Urease B subunit of Helicobacter pylori . Vaccine 25: 2583–2590. [DOI] [PubMed] [Google Scholar]
- 19. Michetti P, Kreiss C, Kotloff KL, Porta N, Blanco JL, et al. (1999) Oral immunization with urease and Escherichia coli heat-labile enterotoxin is safe and immunogenic in Helicobacter pylori-infected adults. Gastroenterology 116: 804–812. [DOI] [PubMed] [Google Scholar]
- 20. Marchetti M, Rossi M, Giannelli V, Giuliani MM, Pizza M, et al. (1998) Protection against Helicobacter pylori infection in mice by intragastric vaccination with H. pylori antigens is achieved using a non-toxic mutant of E. coli heat-labile enterotoxin (LT) as adjuvant. Vaccine 16: 33–37. [DOI] [PubMed] [Google Scholar]
- 21. Summerton NA, Welch RW, Bondoc L, Yang HH, Pleune B, et al. (2010) Toward the development of a stable, freeze-dried formulation of Helicobacter pylori killed whole cell vaccine adjuvanted with a novel mutant of Escherichia coli heat-labile toxin. Vaccine 28: 1404–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ågren LC, Ekman L, Löwenadler B, Nedrud JG, Lycke NY (1999) Adjuvanticity of the cholera toxin A1-based gene fusion protein, CTA1-DD, is critically dependent on the ADP-ribosylatransferase and Ig-binding activity. Journal of Immunology 162: 2432–2440. [PubMed] [Google Scholar]
- 23. Ågren LC, Ekman L, Löwenadler B, Lycke NY (1997) Genetically engineered nontoxic vaccine adjuvant that combines B cell targeting with immunomodulation by cholera toxin A1 subunit. Journal of Immunology 158: 3936–3946. [PubMed] [Google Scholar]
- 24. Eriksson AM, Schon KM, Lycke NY (2004) The Cholera Toxin-Derived CTA1-DD Vaccine Adjuvant Administered Intranasally Does Not Cause Inflammation or Accumulate in the Nervous Tissues. J Immunol 173: 3310–3319. [DOI] [PubMed] [Google Scholar]
- 25. Sundling C, Schon K, Morner A, Forsell MN, Wyatt RT, et al. (2008) CTA1-DD adjuvant promotes strong immunity against human immunodeficiency virus type 1 envelope glycoproteins following mucosal immunization. J Gen Virol 89: 2954–2964. [DOI] [PubMed] [Google Scholar]
- 26. Akhiani AA, Stensson A, Schon K, Lycke N (2006) The nontoxic CTA1-DD adjuvant enhances protective immunity against Helicobacter pylori infection following mucosal immunization. Scand J Immunol 63: 97–105. [DOI] [PubMed] [Google Scholar]
- 27. Lycke N, Bemark M (2010) Mucosal adjuvants and long-term memory development with special focus on CTA1-DD and other ADP-ribosylating toxins. Mucosal Immunol [DOI] [PubMed] [Google Scholar]
- 28. Eliasson DG, El Bakkouri K, Schon K, Ramne A, Festjens E, et al. (2008) CTA1-M2e-DD: a novel mucosal adjuvant targeted influenza vaccine. Vaccine 26: 1243–1252. [DOI] [PubMed] [Google Scholar]
- 29. Eliasson DG, Helgeby A, Schon K, Nygren C, El-Bakkouri K, et al. (2011) A novel non-toxic combined CTA1-DD and ISCOMS adjuvant vector for effective mucosal immunization against influenza virus. Vaccine 29: 3951–3961. [DOI] [PubMed] [Google Scholar]
- 30. Blanchard TG, Nedrud JG (2012) Laboratory Maintenance of Helicobacter Species. Current Protocols in Microbiology 24: 8B.1.1–8B.1.19. [DOI] [PubMed] [Google Scholar]
- 31. Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, et al. (2002) Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17: 375–387. [DOI] [PubMed] [Google Scholar]
- 32. Lee A, O'Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, et al. (1997) A standardised mouse model of Helicobacter pylori infection. Introducing the Sydney strain. Gastroenterology 112: 1386–1397. [DOI] [PubMed] [Google Scholar]
- 33. Garhart CA, Redline RW, Nedrud JG, Czinn SJ (2002) Clearance of Helicobacter pylori infection and resolution of post immunization gastritis in a kinetic study of prophylactically immunized mice. Infection and Immunity 70: 3529–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. DeLyria ES, Redline RW, Blanchard TG (2009) Vaccination of mice against H pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterology 136: 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao W, Wu W, Xu X (2007) Oral vaccination with liposome-encapsulated recombinant fusion peptide of urease B epitope and cholera toxin B subunit affords prophylactic and therapeutic effects against H. pylori infection in BALB/c mice. Vaccine 25: 7664–7673. [DOI] [PubMed] [Google Scholar]
- 36. Spangler BD (1992) Structure and function of cholera toxin and the related Escherichia coli heat-labile enterotoxin. Microbiological Reviews 56: 622–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Delyria ES, Nedrud JG, Ernst PB, Alam MS, Redline RW, et al. (2011) Vaccine-Induced Immunity Against Helicobacter pylori in the Absence of IL-17A. Helicobacter 16: 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Flach C-F, Ostberg AK, Nilsson A-T, Malefyt RDW, Raghavan S (2011) Proinflammatory Cytokine Gene Expression in the Stomach Correlates with Vaccine-Induced Protection against Helicobacter pylori Infection in Mice: an Important Role for Interleukin-17 during the Effector Phase. Infect Immun 79: 879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Velin D, Favre L, Bernasconi E, Bachmann D, Pythoud C, et al. (2009) Interleukin 17 is a Critical Mediator of Vaccine-Induced reduction of Helicobacter infection in the mouse model. Gastroenterology 136: 2237–2246 e2231. [DOI] [PubMed] [Google Scholar]
- 40. Sutton P, Chionh YT (2013) Why can't we make an effective vaccine against Helicobacter pylori ? Expert Rev Vaccines 12: 433–441. [DOI] [PubMed] [Google Scholar]
- 41. Ruggiero P, Peppoloni S, Rappuoli R, Del Giudice G (2003) The quest for a vaccine against Helicobacter pylori/how to move from mouse to man? Microbes Infect 5: 749–756. [DOI] [PubMed] [Google Scholar]
- 42. Malfertheiner P, Schultze V, Rosenkranz B, Kaufmann SH, Ulrichs T, et al. (2008) Safety and immunogenicity of an intramuscular Helicobacter pylori vaccine in noninfected volunteers: a phase I study. Gastroenterology 135: 787–795. [DOI] [PubMed] [Google Scholar]
- 43. Every AL (2013) Key host-pathogen interactions for designing novel interventions against Helicobacter pylori . Trends Microbiol [DOI] [PubMed] [Google Scholar]
- 44. Flach CF, Svensson N, Blomquist M, Ekman A, Raghavan S, et al. (2011) A truncated form of HpaA is a promising antigen for use in a vaccine against Helicobacter pylori . Vaccine 29: 1235–1241. [DOI] [PubMed] [Google Scholar]
- 45. Yang WC, Chen L, Li HB, Li B, Hu J, et al. (2013) Identification of two novel immunodominant UreB CD4(+) T cell epitopes in Helicobacter pylori infected subjects. Vaccine 31: 1204–1209. [DOI] [PubMed] [Google Scholar]
- 46. Zhou WY, Shi Y, Wu C, Zhang WJ, Mao XH, et al. (2009) Therapeutic efficacy of a multi-epitope vaccine against Helicobacter pylori infection in BALB/c mice model. Vaccine 27: 5013–5019. [DOI] [PubMed] [Google Scholar]
- 47. Ding H, Nedrud JG, Blanchard TG, Zagorski BM, Li G, et al. (2013) Th1-Mediated Immunity against Helicobacter pylori Can Compensate for Lack of Th17 Cells and Can Protect Mice in the Absence of Immunization. PLoS One 8: e69384. [DOI] [PMC free article] [PubMed] [Google Scholar]