

Abstract
The past twenty years have revealed the existence of numerous ion channel mutations resulting in human pathology. Ion channels provide the basis of diverse cellular functions, ranging from hormone secretion, excitation-contraction coupling, cell signaling, immune response, and trans-epithelial transport. Therefore, the regulation of biophysical properties of channels is vital in human physiology. Only within the last decade has the role of non-ion channel components come to light in regard to ion channel spatial, temporal, and biophysical regulation in physiology. A growing number of auxiliary components have been determined to play elemental roles in excitable cell physiology, with dysfunction resulting in disorders and related manifestations. This review focuses on the broad implications of such dysfunction, focusing on disease-causing mutations that alter interactions between ion channels and auxiliary ion channel components in a diverse set of human excitable cell disease.
Keywords: Ion channels, auxiliary subunits, ion channel regulation, cardiac arrhythmia, long-QT syndromes, Liddle syndrome, catecholaminergic polymorphic ventricular tachycardia, ankyrin-B syndrome, Brugada syndrome, permanent neonatal diabetes mellitus
1. Introduction
Most ion channels are recognized as central players of larger multi-subunit complexes. Ion channels associate with a number of scaffolding, anchoring, regulatory, and signaling proteins. Co-targeting of these auxiliary subunits to the channel complex has evolved to allow for rapid and localized regulation of ion channels in response to specific stimuli. Electrophysiological properties of cells, once thought to be exclusively attributed to ion channels, have expanded to include these auxiliary subunits. While mutations in ion channel genes can cause dysfunction, a growing number of ion channel-associated proteins have come to the forefront in the study of human excitable cell pathology. This new paradigm has emerged based on growing evidence demonstrating variants in genes involved in the expression, localization, and regulation of ion channels also result in excitable cell disease. These findings have expanded the view of excitable cell disease, uncovered new mechanisms for ion channel regulation, and provided novel targets for pharmacological treatment of aberrant ion channel activity in excitable cells.
2. Liddle Syndrome: Dysregulation of Nedd4–2-mediated regulation of ENaC and hypertension
The last, and rate-limiting, step in Na+ reabsorption by the kidney occurs in the distal convoluted tubules, connecting tubules, and collecting ducts where the epithelial sodium channel (ENaC) regulates Na+ absorption. 1,2 ENaC selectively transports Na+ down an electrochemical gradient established by the Na+K+ ATPase at the basolateral membrane of polarized epithelial cells. 3 Consequently, the directional movement of Na+ produces an osmotic gradient that drives the movement of water in the same direction. 4,5,6
ENaC is selectively expressed at the apical membrane in a variety of epithelia, such as the kidney, lung, and colon. 7 Since entry of Na+ through ENaC is the last and rate-limiting step for Na+ absorption, 3,7,8 regulation of this channel is vital to the regulation of extracellular fluid volume, blood volume, and blood pressure. The activity of ENaC is tightly controlled by a number of hormones (aldosterone, vasopressin, and insulin) and intracellular mediators (Na+, Ca2+, pH, cAMP, protein kinases A and C, and extracellular proteases). 3,9 The necessity of this tight regulation is illustrated by the number of human diseases that have been linked to dysfunction in ENaC, including Liddle syndrome, 10,11 cystic fibrosis, 12 pseudohypoaldosteronism type I (PHA-I), 13,14 and pulmonary edema. 15,16
Liddle syndrome, first described in 1963, was originally determined to be an autosomal dominant form of endocrine hypertension. 17 Activating mutations of the epithelial sodium channel (ENaC) were initially described, resulting in increased sodium reabsorption/potassium wasting in the distal nephron. As a result, affected patients experience hypertension, hypokalemia, low aldosterone and renin levels, salt sensitivity volume expansion, and metabolic alkalosis. 17 ENaC is composed of three similar subunits, α, β, and γ. While the stoichiometric ratio of these subunits in a channel complex is debated, the current view is a ratio of 1:1:1. 18. Each subunit consists of intracellular N- and C-termini, two transmembrane domains, and an extracellular loop. 19 The C-terminal region of each subunit contains a conserved proline-rich sequence, called the PY motif. 19 The original gene variants (frameshifts and premature stop codons) identified in the etiology of Liddle syndrome were found to result in the truncation of the C-terminus, with specific deletions of the PY motifs (PPxY) of the β (SCNN1B) and γ (SCNN1G) subunits. 10,11
Sodium absorption through ENaC is altered in two non-mutually exclusive ways: altering the open probability (Po) of the channel 20 or varying the membrane density of the channel. 21,22,23,24 There is overwhelming evidence to demonstrate that the most potent down-regulation of ENaC is via ubiquitination of the membrane channel and subsequent endocytosis. Liddle syndrome is linked to a defect in the ubiquitin-mediated down-regulation of β and γ ENaC. Specifically, it has been demonstrated that it is the Nedd4–2-mediated ubiquitination of ENaC that lead to endocytosis of the channel 25 and that dysfunction in the ability of Nedd4–2 to ubiquitinate ENaC results in enhanced Na + absorption and hypertension in Liddle syndrome (Figure 1). 26 Specifically, the E3 ubiquitin ligase Nedd4–2 binds to the PY motif in the C-terminal regions of ENaC subunits and facilitates ubiquitination of lysine residues located in the N-terminal domains of the β and γ ENaC subunits. Nedd4–2 has been shown to effectively suppress ENaC activity by enhancing the endocytosis of the channel from the apical membrane. 27,28,29,30 Liddle syndrome mutations effectively remove the PY motifs, resulting in an inability of Nedd4–2 to mediate the ubiquitination and endocytosis of ENaC. The result is an accumulation of active channels at the cell surface, sustained Na+ (and fluid) absorption in the distal nephron, and hypertension. In summary, the dynamic relationship between ENaC and Nedd4–2 illustrate the complex mechanisms underlying human disease as well as potential new molecular targets to tune membrane excitability.
Figure 1. Regulation of ENaC by Nedd4–2.
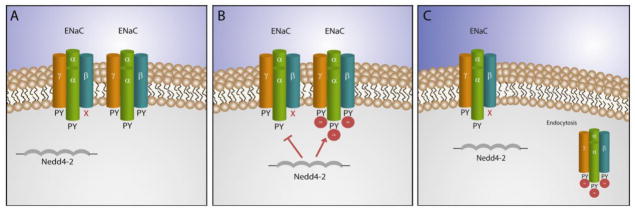
A–B. The ubiquitin ligase Nedd4–2 recognizes PY motifs in wild type ENaC but not mutated forms of the β or γ subunits, which lack the PY motif. C. Ubiquitinated ENaC is endocytosed, leading to a reduction in ENaC at the plasma membrane, reducing activity. On the other hand, mutated forms of ENaC, that are resistant to ubiquitination, remain at the plasma membrane, and increase channel activity.
3. Yotiao and LQT1: PKA-mediated phosphorylation of KCNQ1 and the development of long QT syndrome
Late phase repolarization of the cardiac action potential is largely determined by the slow delayed rectifier current, IKs.31 This cardiac membrane current is controlled by potassium channel comprised of four pore-forming KCNQ1 α-subunits and auxiliary KCNE1 β-subunits.32,33 Individuals with variants in KCNQ1 or KCNE1 may develop type 1 or type 5 long QT syndromes, respectively. Specifically, these patients demonstrate arrhythmias and sudden cardiac death in response to stimulation of the sympathetic nervous system. 34
IKs is regulated by stimulation of the sympathetic nervous system, contributing to shortening of the cardiac action potential despite concurrent increases in heart rate. β-adrenergic receptors (β-ARs) modulate excitation-contraction coupling through signaling cascades that lead to PKA-dependent phosphorylation of several proteins. β-AR agonists, such as norepinephrine, activate stimulatory G-proteins, resulting in the activation of adenylate cyclase and an increase in cAMP production. The increase in cAMP activates PKA 35. Sympathetic regulation of the cardiac action potential is mediated via PKA-mediated phosphorylation of KCNQ1.36,37 Facilitation of PKA-mediated phosphorylation of KCNQ1 requires assembly with the A-kinase anchoring protein (AKAP) Yotiao (Figure 2).38 A decade ago, Marx and colleagues identified Yotiao as a necessary targeting protein for the KCNQ1 channel complex in the cardiomyocyte.38 Studies in brain had previously identified Yotiao as an AKAP.39,40
Figure 2. Regulation of IKs by the AKAP Yotiao.
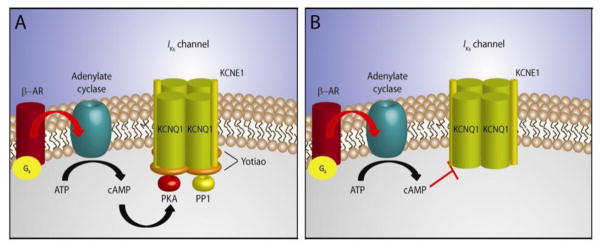
A. The IKs channel consists of KCNQ1 α-subunits and KCNE1 β-subunits. The AKAP Yotiao complexes protein kinase A (PKA) and protein phosphatase 1 (PP1) to the IKs channel. Stimulation of the β-adrenergic receptor (β-AR) leads to activation of adenylate cyclase, the production of cAMP, and activation of protein kinase A. PKA and PP1 regulate IKs via phosphorylation/dephosphorylation activities. B. Mutations which disrupt the association between Yotiao and the IKs channel complex become insensitive to β-adrenergic stimulation.
Mutations in KCNQ1 and Yotiao which disrupt the complex result in type 1 and type 11 long QT syndromes, respectively.41 For example, a mutation in KCNQ1 (G589D), originally identified by Piippo and colleagues,42 was determined to disrupt the interaction between Yotiao and IKS complex, rendering the IKS complex insensitive to β-adrenergic stimulation. 38 As a result, the channel had a significantly increased threshold of activation. 38 Modeling studies suggested that this mutation was likely to produce QTc prolongation and extrasystoles in conjunction with sympathetic stimulation.43 Counterpoint to mutations in KCNQ1, the inherited S1570L mutation in Yotiao also results in a reduced interaction between Yotiao and theIKS complex. 44 Specifically, this mutation resides in a region of Yotiao which coordinates its interaction with KCNQ1. 44 While S1570L does not eliminate the interaction with the IKS complex, it produces a significant reduction in its association with KCNQ1, reduces cAMP-induced KCNQ1 phosphorylation, and eliminates the functional response of the IKS complex to cAMP.44 Overall, this results in delayed repolarization of the action potential.44 Together, these data demonstrate the necessity of macromolecular complexes for normal cardiac physiology.
4. The Ryanodine Receptor and CPVT: defects in a macromolecular regulatory complex
Ryanodine receptors are critical players in cardiac excitation-contraction coupling, where opening of the plasma membrane L-type Ca2+ channel triggers an intensified release of Ca2+ from the sarcoplasmic reticulum, i.e. Ca2+-induced Ca2+ release. Cardiac RyRs are primarily tetramers of RyR2 monomers with large regulatory domains located in the cytosol. 45 RyR2 channels exist as extensive macromolecular complexes, comprised of over a dozen regulatory subunits, including calmodulin (CaM), protein kinase A (PKA), Ca2+/calmodulin-dependent kinase II (CaMKII), protein phosphatases I and 2A (PP1, PP2A), calstabin2 (also known as FK506-binding protein FKBP12.6), phosphodiesterase 4D3 (PDE4D3), triadin, and calsequestrin. 45,46
Human variants in the RyR2 gene have been linked to human catecholaminergic polymorphic ventricular tachycardia (CPVT), 42,47 a disease directly linked with sympathetic-dependent polymorphic arrhythmia with risk of sudden cardiac death. Specifically, CPVT-associated gain-of-function variants sensitize RyR2 to activation by cytosolic Ca2+ 48,49 or delay Ca2+-dependent inactivation. 50 Multiple studies have supported this, demonstrating that cardiomyocytes isolated from transgenic knock-in mice (heterozygous for CPVT-associated mutations) show gain-of-function activity, where catecholamine-induced spontaneous Ca2+ release events were observed. 46,51
In 2003, Marks and colleagues proposed a model to explain the decreased stability of the closed conformation state of mutant RyR2 channels. While another hypothesis suggested that dysfunctional regulation of RyR2 was due to abnormal intramolecular associations within the RyR2 molecule,52 the Marks’ model suggested a role for the channel-stabilizing subunit FKBP12.6. 49 Specifically, their work proposed that binding to FKBP12.6 was decreased in CPVT-linked mutations in RyR2. Further work from the group supported this model, demonstrating that the binding affinity of FKBP12.6 is reduced for CPVT-associated RyR2 mutants.49 Moreover, several studies have demonstrated that the dissociation of FKBP12.6 destabilizes the closed state of the channel, leading to a gain-of-function phenotype. 53 Studies using FKBP12.6 knockout mice further strengthened the link between RyR2-FKBP12.6 interactions and ventricular arrhythmias. 49,54 Following β-adrenergic stimulation, FKBP12.6−/− mice experience ventricular tachycardia, which is very similar to what is observed in transgenic mice harboring CPVT-associated mutations in RyR2. 46,51 Moreover, an experimental drug used to enhance FKBP12.6 binding to RyR2 (JTV519) inhibits arrhythmias in FKBP12.6+/− mice. 55 Despite these findings, there is still controversy involving the role of FKBP12.6 in the pathogenesis of CPVT. 56 Future studies are needed to fully elucidate the molecular mechanisms associated with RyR2-associated CPVT in humans.
5. Ankyrins: Critical components of the excitable cell membrane
Ankyrins have been established as integral components in the targeting, retention, and regulation of a number of ion channels, transporters, pumps, and cytosolic proteins in excitable cells. Three ankyrin gene products, ankyrin-R (ANK1), ankyrin-B (ANK2), and ankyrin-G (ANK3) are expressed in vertebrates, each with unique expression profiles and disease associations. In the past decade, studies in humans and mice have illustrated a critical role for ankyrin function in the heart and pancreatic beta cell. Dysfunction in ankyrin-B has been associated with defective intracellular Ca2+ handling (LQT4 or Ankyrin-B syndrome), as well as permanent neonatal diabetes mellitus, while dysfunction in ankyrin-G binding with Nav1.5 has been linked with Brugada syndrome.
5.1. Ankyrin-B Syndrome: lost anchors lead to excitable cell disease
Nearly 20 years ago, Schott and colleagues described a French cohort displaying an atypical form of long QT syndrome. 57 Affected family members displayed abnormal T-wave morphology, sinus node bradycardia, and atrial fibrillation.57 Notably, certain individuals displayed stress- and exercise-induced sudden cardiac death. 57 Using linkage analysis, Schott identified that this fourth locus for congenital LQT was associated with an 18 cM region corresponding to the ANK2 locus (ankyrin-B). 57 Further analysis revealed a single ANK2 missense variant, A4274G, resulting in E1425G. 58
Since this initial discovery, a number of additional ankyrin-B human gene variants have been identified. 59,60,61,62,63 Patients with ANK2 loss-of-function variants may display sinus node dysfunction, atrial fibrillation, polymorphic ventricular arrhythmias, and/or conduction defects. 58,60,61 Studies in mice that are haploinsufficient in ankyrin-B (ankyrin-B+/−) have been invaluable in the evaluation of molecular mechanisms underlying the role of ankyrin-B in excitable cell physiology. Ankyrin-B+/− mice demonstrate common phenotypes with the original French LQT4 kindred, including sinus bradycardia, conduction defects, and catecholamine-induced polymorphic ventricular arrhythmia with associated syncope/death. 58 Isolated primary cardiac myocytes from ankyrin-B+/− mice were used to conclude that the E1425G variant was a loss-of-function variant of ankyrin-B. Ankyrin-B+/− cardiomyocytes displayed reduced spontaneous contraction rates, aberrant localization of the Na+/Ca2+ exchanger (NCX), and abnormal calcium transients. 58,64,65 Rescue experiments using exogenous ankyrin-B successfully restored wild type myocyte phenotypes. 58,64,65 Conversely, overexpression of exogenous ankyrin-B E1425G in ankyrin-B+/− myocytes was unable to restore abnormal phenotypes. 58
Further evaluation revealed that ankyrin-B+/− myocytes expressed significantly less Na+/Ca2+ exchanger (NCX), Na+/K+ ATPase (NKA), and inositol 1,4,5 trisphosphate receptor (InsP3R). 58 Additionally, it was determined that the NCX and NKA were reduced preferentially at the transverse tubule. 58 Biochemical analyses demonstratedat ankyrin-B directly associates with the NCX, NKA, and InsP3R. 66 Furthermore, it was established that exogenous expression of ankyrin-B cDNA in ankyrin-B+/− cardiomyocytes restored proper expression and localization of the NCX, NKA, and InsP3R. 58,60,61,64,66,67 Eventually it was determined that loss of ankyrin-B-dependent localization of NCX and NKA resulted in increased SR Ca2+ load and elevated SR Ca2+ transients, essentially mimicking the effects of cardiac glycosides. 68,69 These studies have demonstrated that reduced expression/dysfunction of ankyrin-B can be an underlying cause for human arrhythmia susceptibility by altering myocyte electrical activity.
5.2. Ankyrin-B and the KATP channel: a new component in glucose homeostasis
The KATP channel is a hetero-octameric structure of four Kir6x pore-forming subunits and four SUR regulatory subunits. 70 Although KATP channels were originally identified in heart, it is the pancreatic KATP channels that have been most thoroughly investigated. In the pancreatic beta cell, the KATP channel functions as a metabolic sensor, closing the KATP channel in response to increased blood glucose. Closure of the KATP channel results in beta cell depolarization, activation of L-type Ca2+ channels, and stimulation of insulin granule release. 71 Not surprisingly, mutations in KATP channel genes (KCNJ11 and ABCC8) are associated with both diabetes mellitus and hyper-insulinemia syndromes (reviewed in 72). In 2004, a mutation in the Kir6.2 subunit, E322K, was identified in a case of permanent neonatal diabetes mellitus. 73 Several years later, it was determined that this mutation resided in the region of Kir6.2 that associated with ankyrin-B (Figure 3).74 Specifically, it was determined that the E322K variant disrupts the association of Kir6.2 with ankyrin-B in vivo and in vitro and also decreases the sensitivity of the channel to ATP. 74 Based on modeling studies, the disruption of the Kir6.2/ankyrin-B interaction was proposed to result in two seemingly opposing phenotypes: a loss-of-function due to loss of KATP channel density at the membrane and a gain-of-function due to decreased ATP sensitivity.74 It is suggested that the combination of the two phenotypes actually results in a diabetic phenotype less severe than either a gain-of-function mutation or loss-of-function mutation alone. These data suggest that the disruption of the ankyrin-B/Kir6.2 complex via the Kir6.2 E322K mutation results in aberrant localization and regulation of the KATP channel. Further, these data illustrate the complexity of pathways underlying ion channel regulation in health and disease.
Figure 3. Regulation of the pancreatic KATP channel by ankyrin-B.

Ankyrin-B associates with the α-subunit of the KATP channel, Kir6.2, via a C-terminal motif in Kir6.2. Association with ankyrin-B regulates Kir6.2 plasma membrane expression and KATP channel ATP sensitivity.
5.3. Ankyrin-G and Nav1.5: Brugada syndrome
Voltage-gated Na+ (Nav) channels are required for normal electrical activity in a number of cell types, including neurons, skeletal muscle, and cardiomyocytes. 75,76,77,78,79,80,81 Ankyrin-G has been firmly established as a critical player in the trafficking of Nav channels in the central nervous system. 82 In neuronal cells, ankyrin-G is co-expressed with Nav1.2 and Nav1.6 at the axon initial segments, nodes of Ranvier, and the neuromuscular junction. 83,84,85,86 More specifically, it has been demonstrated that ankyrin-G directly associates with Nav channels at these specialized neuronal membrane domains. 85,87 In 2003, two studies published independently identified a unique ankyrin-G binding sequence (ABS) in the cytoplasmic DII/DII loop of Nav1.2. 88,89 The ABS is a highly conserved nine amino acid motif [(V/A)P(I/L)AXXE(S/D)] and found in most Nav channel gene products, including Nav1.1, Nav1.2, Nav1.4, Nav1.5, and Nav1.6. Based on the presence of both ankyrin-G and Nav1.5 at the cardiac intercalated disc, it was proposed that perhaps ankyrin-G filled a similar role in cardiac physiology. 61 Indeed, Bennett and colleagues demonstrated that an ankyrin-G-dependent pathway was necessary for proper targeting of Nav1.5 to the intercalated disc.61 Specifically, their study demonstrated co-localization of ankyrin-G and Nav1.5 to the cardiac intercalated disc, that Nav1.5 and ankyrin-G co-immunoprecipitated from detergent-soluble cardiac lysates, and that deletion of the ABS rendered Nav1.5 incapable of binding ankyrin-G. 61 Using primary cardiomyocytes deficient in ankyrin-G expression, a follow up study by Lowe and colleagues demonstrated that loss of ankyrin-G resulted in decreased Nav1.5 protein expression, decreased Nav1.5 membrane targeting, and reduced cardiomyocyte INa. 79
Brugada syndrome, a cardiac syndrome characterized by ST segment elevation, right bundle branch block, and fatal cardiac arrhythmias90, had long been attributed to loss of function mutations in SCN5A (encoding the Nav1.5 channel).91 Specifically, it was believed that these mutations altered Nav1.5 channel biophysics or drastically altered protein structure. 92 However, in 2004, Priori and colleagues screened a large number of Brugada patients for mutations in the ABS region of SCN5A. 61 One SCN5A missense mutation was identified, E1053K.61 Work by Bennett et al. determined that E1053K prevented associated of Nav1.5 with ankyrin-G. 61 Moreover, Nav1.5 E1053K was not properly targeted to the intercalated disc, implicating an ankyrin-G-based pathway for Nav1.5 targeting in cardiomyocytes.61 In summary, findings from brain, heart, and pancreas identify ankyrin polypeptides as critical nodes for regulation of membrane excitability for normal physiology as well as demonstrate how defects in these pathways underlie human pathophysiology. As ankyrins are expressed far beyond these tissues, future studies will likely yield additional linkages between these genes and other disease etiologies.
6. Caveolin-3 and type 9 long QT syndrome
Caveolae (“little caves”) are invaginations of the plasma membrane critical to membrane endocytosis, cholesterol homeostasis, cell signaling, and tumorigenesis. 93,94 The primary coat proteins of caveolae are encoded by three genes: CAV1 (caveolin-1), CAV2 (caveolin-2) and CAV3 (caveolin-3). 95,96,97,98 While caveolin-1 and caveolin-2 are expressed in most cell types, caveolin-3 is primarily expressed in muscle tissue. 98 Structurally, caveolin proteins are similar, with an N-terminal domain, a scaffolding domain, and hydrophobic domain, and a C-terminal domain. 99 Caveolin-3 has been linked with Duchenne muscular dystrophy, rippling muscle disease, and idiopathic hyperCKemia. 100,101,102,103 At a molecular level, caveolin co-purifies with a number of cardiac membrane proteins, including Nav1.5, HCN4 pacemaker channel, NCX, InsP3R, Kv1.5, PMCA, Cav1.2, and TRP channels.104,105,106,107,108,109,110,111 Caveolin-3 knockout mice display abnormal cardiac T-tubule organization, 112,113 moderate cardiomyopathy, 93 and skeletal muscle myopathy. 112,114 Similar to these findings in caveolin-deficient mice, humans harboring CAV3 variants display parallel phenotypes, 100,101,102,103 suggesting a role for caveolin in human cardiac arrhythmias.
In 2006, Vatta and colleagues screened “genotype negative” LQTS probands for potential variants in CAV3. 115 These individuals displayed a variety of cardiac phenotypes, including non-exertional syncope, sinus bradycardia, and prolonged QTc intervals. 115 Four mutations were identified and termed LQT9.115 Prior to these findings, it had been demonstrated that caveolin-3 associated with Nav channels, 111 leading to further investigation of caveolin-3 and Nav1.5 interactions in the cardiomyocyte. Studies by Vatta and others demonstrated that caveolin-3 and Nav1.5 co-localize and co-immunoprecipitate in vivo. 111 Furthermore, it was determined that the N-terminal domain of caveolin-3 was required for interaction with Nav1.5. 115 Heterologous overexpression of both Nav1.5 and caveolin-3 in HEK cells produced a two- to three-fold increase in late Na+ current, likely contributing to the QT prolongation and cardiac arrhythmias observed in CAV3 mutation carriers. 115 It remains to be determined whether CAV3 mutations result in abnormal subcellular targeting (since CAV3 mutations do not disrupt association with Nav1.5 115), whether CAV3 mutations allosterically affect Nav channels, or whether cardiac dysfunction results from altered localization of other caveolin-3-associated ion channels to the sarcolemma. Of note, two recent studies have found an associated between CAV3 mutations and sudden infant death syndrome. 116,117
7. α-syntrophin and type 12 long QT syndrome
Syntrophins are a family of cytoplasmic adapter proteins that link the actin-based cytoskeleton (via the dystrophin-associated complex) to the extracellular matrix. Syntrophins have an internal PDZ domain that has been demonstrated to interact with the C-terminal region of Nav channels, including Nav1.4 and Nav1.5. 118,119 Interestingly, mdx mice (lacking dystrophin) display loss of cardiomyocyte Nav1.5 expression from the peripheral sarcolemma, 119 presumed to result from dysregulation of syntrophin. In addition to Nav1.5, syntrophins have been linked to a number of ion channels; namely, the plasma membrane calcium ATPase (PMCA) and neuronal nitric oxide synthase (nNOS). 120,121,122 Since nitric oxide (NO) directly affects late INa in LQT3 (where pathogenic mutations in SCN5A accentuate late INa), this connection is relevant to topic. 123 Additionally, variants of CAPON (NOS1AP), a nNOS regulatory protein, have been linked with QT interval regulation in genome-wide association studies. 124,125 Direct sequencing of α-1 syntrophin (SNTA1) in a number of “genotype negative” LQTSB patients (see above) identified a novel missense mutation, A390V, in a patient presenting with syncope and QTc prolongation (now termed LQT12). 123 This mutation was found to decrease association of α-1 syntrophin with PMCA4b and nNOS. Additionally, A390V increased Nav1.5 nitrosylation, as well as increased persistent INa in cardiomyocytes. 123 It is believed that this Nav1.5 dysregulation is the underlying cellular mechanism for LQT12. Considering the number of ion channels known to associate with α-1 syntrophin, it is possible that other forms of cardiac dysfunction may be attributable to dysfunction in α -1 syntrophin.
8. Conclusion
Cardiovascular therapies including anti-platelet drugs, statins, renin-angiotensin system antagonists, beta-blockers, and thrombolytic agents developed in this past century have decreased cardiovascular-based mortality over the past fifty years. However, cardiovascular disease remains the leading cause of death in the United States. Cardiac ion channels and transporters regulate membrane excitability and therefore would appear to represent a critical family of targets for anti-arrhythmia therapy. Unfortunately, ion channel antagonists have been unsuccessful in decreasing mortality in patients at risk for cardiac sudden death- and in select cases have actually increased mortality. 126 It is well established that macromolecular signaling complexes, composed of adaptor proteins, ion channels, and signaling molecules, are essential the creation, maintenance, and regulation of local microenvironments in a number of cell types. The identification of mutations and variants within a larger ion channel complex has provided valuable insights into the molecular basis of human disease. Based on the data discussed in this review, we propose that non-conventional cellular pathways that indirectly regulate cardiac membrane ion channels and transporter activities (ankyrins, spectrins, syntrophins) may serve as exciting new routes to tune cellular excitability in both congenital and acquired forms of cardiovascular disease.
Highlights.
Ion channels and auxiliary subunits are necessary players in excitable cell physiology.
Known mutations in ion channels have long been associated with human disease.
Recent studies have demonstrated a very critical role for regulatory, scaffolding, and signaling molecules in the regulation of ion channel function.
Insight into the roles of auxiliary subunits has provided invaluable information with regard to ion channel regulation and has provided novel targets for future therapies.
Acknowledgments
This work was supported in part by a grant from the Saving Tiny Hearts Society the NIH (HL084583, HL083422), AHA Established Investigator Award and Fondation Leducq Award to the Alliance for Calmodulin Kinase Signaling in Heart Disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Frindt G, Palmer LG. Na channels in the rat connecting tubule. Am J Physiol Renal Physiol. 2004;286(4):F669–74. doi: 10.1152/ajprenal.00381.2003. [DOI] [PubMed] [Google Scholar]
- 2.Duc C, Farman N, Canessa CM, Bonvalet JP, Rossier BC. Cell-specific expression of epithelial sodium channel alpha, beta, and gamma subunits in aldosterone-responsive epithelia from the rat: localization by in situ hybridization and immunocytochemistry. J Cell Biol. 1994;127(6 Pt 2):1907–21. doi: 10.1083/jcb.127.6.1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garty H, Palmer LG. Epithelial sodium channels: function, structure, and regulation. Physiol Rev. 1997;77(2):359–96. doi: 10.1152/physrev.1997.77.2.359. [DOI] [PubMed] [Google Scholar]
- 4.Bankir L, Fernandes S, Bardoux P, Bouby N, Bichet DG. Vasopressin-V2 receptor stimulation reduces sodium excretion in healthy humans. J Am Soc Nephrol. 2005;16(7):1920–8. doi: 10.1681/ASN.2004121079. [DOI] [PubMed] [Google Scholar]
- 5.Borgnia M, Nielsen S, Engel A, Agre P. Cellular and molecular biology of the aquaporin water channels. Annu Rev Biochem. 1999;68:425–58. doi: 10.1146/annurev.biochem.68.1.425. [DOI] [PubMed] [Google Scholar]
- 6.Schrier RW. Body water homeostasis: clinical disorders of urinary dilution and concentration. J Am Soc Nephrol. 2006;17(7):1820–32. doi: 10.1681/ASN.2006030240. [DOI] [PubMed] [Google Scholar]
- 7.Rossier BC. The epithelial sodium channel: activation by membrane-bound serine proteases. Proc Am Thorac Soc. 2004;1(1):4–9. doi: 10.1513/pats.2306007. [DOI] [PubMed] [Google Scholar]
- 8.Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. 2002;82(3):735–67. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- 9.Schild L. The epithelial sodium channel: from molecule to disease. Rev Physiol Biochem Pharmacol. 2004;151:93–107. doi: 10.1007/s10254-004-0023-7. [DOI] [PubMed] [Google Scholar]
- 10.Shimkets RA, Warnock DG, Bositis CM, Nelson-Williams C, Hansson JH, Schambelan M, Gill JR, Jr, Ulick S, Milora RV, Findling JW, et al. Liddle’s syndrome: heritable human hypertension caused by mutations in the beta subunit of the epithelial sodium channel. Cell. 1994;79(3):407–14. doi: 10.1016/0092-8674(94)90250-x. [DOI] [PubMed] [Google Scholar]
- 11.Hansson JH, Nelson-Williams C, Suzuki H, Schild L, Shimkets R, Lu Y, Canessa C, Iwasaki T, Rossier B, Lifton RP. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11(1):76–82. doi: 10.1038/ng0995-76. [DOI] [PubMed] [Google Scholar]
- 12.Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269(5225):847–50. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- 13.Strautnieks SS, Thompson RJ, Gardiner RM, Chung E. A novel splice-site mutation in the gamma subunit of the epithelial sodium channel gene in three pseudohypoaldosteronism type 1 families. Nat Genet. 1996;13(2):248–50. doi: 10.1038/ng0696-248. [DOI] [PubMed] [Google Scholar]
- 14.Chang SS, Grunder S, Hanukoglu A, Rosler A, Mathew PM, Hanukoglu I, Schild L, Lu Y, Shimkets RA, Nelson-Williams C, Rossier BC, Lifton RP. Mutations in subunits of the epithelial sodium channel cause salt wasting with hyperkalaemic acidosis, pseudohypoaldosteronism type 1. Nat Genet. 1996;12(3):248–53. doi: 10.1038/ng0396-248. [DOI] [PubMed] [Google Scholar]
- 15.Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A, Boucher R, Rossier BC. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet. 1996;12(3):325–8. doi: 10.1038/ng0396-325. [DOI] [PubMed] [Google Scholar]
- 16.O’Brodovich HM. The role of active Na+ transport by lung epithelium in the clearance of airspace fluid. New Horiz. 1995;3(2):240–7. [PubMed] [Google Scholar]
- 17.Drager LF, Krieger JE. Genetic aspects of endocrine hypertensive disorders. Arq Bras Endocrinol Metabol. 2004;48(5):659–65. doi: 10.1590/s0004-27302004000500011. [DOI] [PubMed] [Google Scholar]
- 18.Adams ME, Kileny PR, Telian SA, El-Kashlan HK, Heidenreich KD, Mannarelli GR, Arts HA. Electrocochleography as a diagnostic and intraoperative adjunct in superior semicircular canal dehiscence syndrome. Otol Neurotol. 2011;32(9):1506–12. doi: 10.1097/MAO.0b013e3182382a7c. [DOI] [PubMed] [Google Scholar]
- 19.Chen HI, Sudol M. The WW domain of Yes-associated protein binds a proline-rich ligand that differs from the consensus established for Src homology 3-binding modules. Proc Natl Acad Sci U S A. 1995;92(17):7819–23. doi: 10.1073/pnas.92.17.7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benos DJ, Awayda MS, Ismailov, Johnson JP. Structure and function of amiloride-sensitive Na+ channels. J Membr Biol. 1995;143(1):1–18. doi: 10.1007/BF00232519. [DOI] [PubMed] [Google Scholar]
- 21.Butterworth MB, Edinger RS, Frizzell RA, Johnson JP. Regulation of the epithelial sodium channel by membrane trafficking. Am J Physiol Renal Physiol. 2009;296(1):F10–24. doi: 10.1152/ajprenal.90248.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butterworth MB, Edinger RS, Johnson JP, Frizzell RA. Acute ENaC stimulation by cAMP in a kidney cell line is mediated by exocytic insertion from a recycling channel pool. J Gen Physiol. 2005;125(1):81–101. doi: 10.1085/jgp.200409124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butterworth MB, Helman SI, Els WJ. cAMP-sensitive endocytic trafficking in A6 epithelia. Am J Physiol Cell Physiol. 2001;280(4):C752–62. doi: 10.1152/ajpcell.2001.280.4.C752. [DOI] [PubMed] [Google Scholar]
- 24.Rossier BC. Hormonal regulation of the epithelial sodium channel ENaC: N or P(o)? J Gen Physiol. 2002;120(1):67–70. doi: 10.1085/jgp.20028638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimkets RA, Lifton RP, Canessa CM. The activity of the epithelial sodium channel is regulated by clathrin-mediated endocytosis. J Biol Chem. 1997;272(41):25537–41. doi: 10.1074/jbc.272.41.25537. [DOI] [PubMed] [Google Scholar]
- 26.Goulet CC, Volk KA, Adams CM, Prince LS, Stokes JB, Snyder PM. Inhibition of the epithelial Na+ channel by interaction of Nedd4 with a PY motif deleted in Liddle’s syndrome. J Biol Chem. 1998;273(45):30012–7. doi: 10.1074/jbc.273.45.30012. [DOI] [PubMed] [Google Scholar]
- 27.Flores SY, Debonneville C, Staub O. The role of Nedd4/Nedd4-like dependant ubiquitylation in epithelial transport processes. Pflugers Arch. 2003;446(3):334–8. doi: 10.1007/s00424-003-1027-x. [DOI] [PubMed] [Google Scholar]
- 28.Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, Munster C, Chraibi A, Pratt JH, Horisberger JD, Pearce D, Loffing J, Staub O. Phosphorylation of Nedd4–2 by Sgk1 regulates epithelial Na(+) channel cell surface expression. EMBO J. 2001;20(24):7052–9. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Itani OA, Stokes JB, Thomas CP. Nedd4–2 isoforms differentially associate with ENaC and regulate its activity. Am J Physiol Renal Physiol. 2005;289(2):F334–46. doi: 10.1152/ajprenal.00394.2004. [DOI] [PubMed] [Google Scholar]
- 30.Staub O, Abriel H, Plant P, Ishikawa T, Kanelis V, Saleki R, Horisberger JD, Schild L, Rotin D. Regulation of the epithelial Na+ channel by Nedd4 and ubiquitination. Kidney Int. 2000;57(3):809–15. doi: 10.1046/j.1523-1755.2000.00919.x. [DOI] [PubMed] [Google Scholar]
- 31.Jespersen T, Grunnet M, Olesen SP. The KCNQ1 potassium channel: from gene to physiological function. Physiology (Bethesda) 2005;20:408–16. doi: 10.1152/physiol.00031.2005. [DOI] [PubMed] [Google Scholar]
- 32.Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature. 1996;384(6604):78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- 33.Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature. 1996;384(6604):80–3. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- 34.Schwartz PJ, Priori SG, Spazzolini C, Moss AJ, Vincent GM, Napolitano C, Denjoy I, Guicheney P, Breithardt G, Keating MT, Towbin JA, Beggs AH, Brink P, Wilde AA, Toivonen L, Zareba W, Robinson JL, Timothy KW, Corfield V, Wattanasirichaigoon D, Corbett C, Haverkamp W, Schulze-Bahr E, Lehmann MH, Schwartz K, Coumel P, Bloise R. Genotype-phenotype correlation in the long-QT syndrome: gene-specific triggers for life-threatening arrhythmias. Circulation. 2001;103(1):89–95. doi: 10.1161/01.cir.103.1.89. [DOI] [PubMed] [Google Scholar]
- 35.Saucerman JJ, Zhang J, Martin JC, Peng LX, Stenbit AE, Tsien RY, McCulloch AD. Systems analysis of PKA-mediated phosphorylation gradients in live cardiac myocytes. Proc Natl Acad Sci U S A. 2006;103(34):12923–8. doi: 10.1073/pnas.0600137103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kass RS, Wiegers SE. The ionic basis of concentration-related effects of noradrenaline on the action potential of calf cardiac purkinje fibres. J Physiol. 1982;322:541–58. doi: 10.1113/jphysiol.1982.sp014054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walsh KB, Kass RS. Regulation of a heart potassium channel by protein kinase A and C. Science. 1988;242(4875):67–9. doi: 10.1126/science.2845575. [DOI] [PubMed] [Google Scholar]
- 38.Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295(5554):496–9. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 39.Feliciello A, Cardone L, Garbi C, Ginsberg MD, Varrone S, Rubin CS, Avvedimento EV. Gottesman, M. E. Yotiao protein, a ligand for the NMDA receptor, binds and targets cAMP-dependent protein kinase II(1) FEBS Lett. 1999;464(3):174–8. doi: 10.1016/s0014-5793(99)01585-9. [DOI] [PubMed] [Google Scholar]
- 40.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285(5424):93–6. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 41.Ciampa EJ, Welch RC, Vanoye CG, George AL., Jr KCNE4 juxtamembrane region is required for interaction with calmodulin and for functional suppression of KCNQ1. J Biol Chem. 2011;286(6):4141–9. doi: 10.1074/jbc.M110.158865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laitinen PJ, Brown KM, Piippo K, Swan H, Devaney JM, Brahmbhatt B, Donarum EA, Marino M, Tiso N, Viitasalo M, Toivonen L, Stephan DA, Kontula K. Mutations of the cardiac ryanodine receptor (RyR2) gene in familial polymorphic ventricular tachycardia. Circulation. 2001;103(4):485–90. doi: 10.1161/01.cir.103.4.485. [DOI] [PubMed] [Google Scholar]
- 43.Saucerman JJ, Healy SN, Belik ME, Puglisi JL, McCulloch AD. Proarrhythmic consequences of a KCNQ1 AKAP-binding domain mutation: computational models of whole cells and heterogeneous tissue. Circ Res. 2004;95(12):1216–24. doi: 10.1161/01.RES.0000150055.06226.4e. [DOI] [PubMed] [Google Scholar]
- 44.Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007;104(52):20990–5. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wehrens XH, Lehnart SE, Marks AR. Intracellular calcium release and cardiac disease. Annu Rev Physiol. 2005;67:69–98. doi: 10.1146/annurev.physiol.67.040403.114521. [DOI] [PubMed] [Google Scholar]
- 46.Kannankeril PJ, Mitchell BM, Goonasekera SA, Chelu MG, Zhang W, Sood S, Kearney DL, Danila CI, De Biasi M, Wehrens XH, Pautler RG, Roden DM, Taffet GE, Dirksen RT, Anderson ME, Hamilton SL. Mice with the R176Q cardiac ryanodine receptor mutation exhibit catecholamine-induced ventricular tachycardia and cardiomyopathy. Proc Natl Acad Sci U S A. 2006;103(32):12179–84. doi: 10.1073/pnas.0600268103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Priori SG, Napolitano C, Tiso N, Memmi M, Vignati G, Bloise R, Sorrentino V, Danieli GA. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103(2):196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 48.Jiang D, Xiao B, Zhang L, Chen SR. Enhanced basal activity of a cardiac Ca2+ release channel (ryanodine receptor) mutant associated with ventricular tachycardia and sudden death. Circ Res. 2002;91(3):218–25. doi: 10.1161/01.res.0000028455.36940.5e. [DOI] [PubMed] [Google Scholar]
- 49.Wehrens XH, Lehnart SE, Huang F, Vest JA, Reiken SR, Mohler PJ, Sun J, Guatimosim S, Song LS, Rosemblit N, D’Armiento JM, Napolitano C, Memmi M, Priori SG, Lederer WJ, Marks AR. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell. 2003;113(7):829–40. doi: 10.1016/s0092-8674(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 50.Milting H, Lukas N, Klauke B, Korfer R, Perrot A, Osterziel KJ, Vogt J, Peters S, Thieleczek R, Varsanyi M. Composite polymorphisms in the ryanodine receptor 2 gene associated with arrhythmogenic right ventricular cardiomyopathy. Cardiovasc Res. 2006;71(3):496–505. doi: 10.1016/j.cardiores.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 51.Liu N, Colombi B, Memmi M, Zissimopoulos S, Rizzi N, Negri S, Imbriani M, Napolitano C, Lai FA, Priori SG. Arrhythmogenesis in catecholaminergic polymorphic ventricular tachycardia: insights from a RyR2 R4496C knock-in mouse model. Circ Res. 2006;99(3):292–8. doi: 10.1161/01.RES.0000235869.50747.e1. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto T, Ikemoto N. Peptide probe study of the critical regulatory domain of the cardiac ryanodine receptor. Biochem Biophys Res Commun. 2002;291(4):1102–8. doi: 10.1006/bbrc.2002.6569. [DOI] [PubMed] [Google Scholar]
- 53.Chelu MG, Danila CI, Gilman CP, Hamilton SL. Regulation of ryanodine receptors by FK506 binding proteins. Trends Cardiovasc Med. 2004;14(6):227–34. doi: 10.1016/j.tcm.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 54.Wehrens XH, Lehnart SE, Reiken SR, Deng SX, Vest JA, Cervantes D, Coromilas J, Landry DW, Marks AR. Protection from cardiac arrhythmia through ryanodine receptor-stabilizing protein calstabin2. Science. 2004;304(5668):292–6. doi: 10.1126/science.1094301. [DOI] [PubMed] [Google Scholar]
- 55.Bossuyt J, Taylor BE, James-Kracke M, Hale CC. Evidence for cardiac sodium-calcium exchanger association with caveolin-3. FEBS Lett. 2002;511(1–3):113–7. doi: 10.1016/s0014-5793(01)03323-3. [DOI] [PubMed] [Google Scholar]
- 56.George CH, Higgs GV, Lai FA. Ryanodine receptor mutations associated with stress-induced ventricular tachycardia mediate increased calcium release in stimulated cardiomyocytes. Circ Res. 2003;93(6):531–40. doi: 10.1161/01.RES.0000091335.07574.86. [DOI] [PubMed] [Google Scholar]
- 57.Schott JJ, Charpentier F, Peltier S, Foley P, Drouin E, Bouhour JB, Donnelly P, Vergnaud G, Bachner L, Moisan JP, et al. Mapping of a gene for long QT syndrome to chromosome 4q25–27. Am J Hum Genet. 1995;57(5):1114–22. [PMC free article] [PubMed] [Google Scholar]
- 58.Mohler PJ, Schott JJ, Gramolini AO, Dilly KW, Guatimosim S, du Bell WH, Song LS, Haurogne K, Kyndt F, Ali ME, Rogers TB, Lederer WJ, Escande D, Le Marec H, Bennett V. Ankyrin-B mutation causes type 4 long-QT cardiac arrhythmia and sudden cardiac death. Nature. 2003;421(6923):634–9. doi: 10.1038/nature01335. [DOI] [PubMed] [Google Scholar]
- 59.Mank-Seymour AR, Richmond JL, Wood LS, Reynolds JM, Fan YT, Warnes GR, Milos PM, Thompson JF. Association of torsades de pointes with novel and known single nucleotide polymorphisms in long QT syndrome genes. Am Heart J. 2006;152(6):1116–22. doi: 10.1016/j.ahj.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 60.Cunha SR, Bhasin N, Mohler PJ. Targeting and stability of Na/Ca exchanger 1 in cardiomyocytes requires direct interaction with the membrane adaptor ankyrin-B. J Biol Chem. 2007;282(7):4875–83. doi: 10.1074/jbc.M607096200. [DOI] [PubMed] [Google Scholar]
- 61.Mohler PJ, Davis JQ, Davis LH, Hoffman JA, Michaely P, Bennett V. Inositol 1,4,5-trisphosphate receptor localization and stability in neonatal cardiomyocytes requires interaction with ankyrin-B. J Biol Chem. 2004;279(13):12980–7. doi: 10.1074/jbc.M313979200. [DOI] [PubMed] [Google Scholar]
- 62.Sherman J, Tester DJ, Ackerman MJ. Targeted mutational analysis of ankyrin-B in 541 consecutive, unrelated patients referred for long QT syndrome genetic testing and 200 healthy subjects. Heart Rhythm. 2005;2(11):1218–23. doi: 10.1016/j.hrthm.2005.07.026. [DOI] [PubMed] [Google Scholar]
- 63.Zhou X, Shimizu M, Konno T, Ino H, Fujino N, Uchiyama K, Mabuchi T, Kaneda T, Fujita T, Masuda E, Kato H, Funada A, Mabuchi H. Analysis of ankyrin-B gene mutations in patients with long QT syndrome. Nan Fang Yi Ke Da Xue Xue Bao. 2006;26(7):901–3. 909. [PubMed] [Google Scholar]
- 64.Mohler PJ, Gramolini AO, Bennett V. The ankyrin-B C-terminal domain determines activity of ankyrin-B/G chimeras in rescue of abnormal inositol 1,4,5-trisphosphate and ryanodine receptor distribution in ankyrin-B (−/−) neonatal cardiomyocytes. J Biol Chem. 2002;277(12):10599–607. doi: 10.1074/jbc.M110958200. [DOI] [PubMed] [Google Scholar]
- 65.Tuvia S, Buhusi M, Davis L, Reedy M, Bennett V. Ankyrin-B is required for intracellular sorting of structurally diverse Ca2+ homeostasis proteins. J Cell Biol. 1999;147(5):995–1008. doi: 10.1083/jcb.147.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mohler PJ, Davis JQ, Bennett V. Ankyrin-B coordinates the Na/K ATPase, Na/Ca exchanger, and InsP3 receptor in a cardiac T-tubule/SR microdomain. PLoS Biol. 2005;3(12):e423. doi: 10.1371/journal.pbio.0030423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mohler PJ, Splawski I, Napolitano C, Bottelli G, Sharpe L, Timothy K, Priori SG, Keating MT, Bennett V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc Natl Acad Sci U S A. 2004;101(24):9137–42. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Kluwer Academic; Boston, MA: 1991. [Google Scholar]
- 69.Reuter H, Henderson SA, Han T, Ross RS, Goldhaber JI, Philipson KD. The Na+−Ca2+ exchanger is essential for the action of cardiac glycosides. Circ Res. 2002;90(3):305–8. doi: 10.1161/hh0302.104562. [DOI] [PubMed] [Google Scholar]
- 70.Clement JPt, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of K(ATP) channel subunits. Neuron. 1997;18(5):827–38. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- 71.Ashcroft FM, Harrison DE, Ashcroft SJ. Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature. 1984;312(5993):446–8. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- 72.McTaggart JS, Clark RH, Ashcroft FM. The role of the KATP channel in glucose homeostasis in health and disease: more than meets the islet. J Physiol. 2010;588(Pt 17):3201–9. doi: 10.1113/jphysiol.2010.191767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaxillaire M, Populaire C, Busiah K, Cave H, Gloyn AL, Hattersley AT, Czernichow P, Froguel P, Polak M. Kir6.2 mutations are a common cause of permanent neonatal diabetes in a large cohort of French patients. Diabetes. 2004;53(10):2719–22. doi: 10.2337/diabetes.53.10.2719. [DOI] [PubMed] [Google Scholar]
- 74.Kline CF, Kurata HT, Hund TJ, Cunha SR, Koval OM, Wright PJ, Christensen M, Anderson ME, Nichols CG, Mohler PJ. Dual role of K ATP channel C-terminal motif in membrane targeting and metabolic regulation. Proc Natl Acad Sci U S A. 2009;106(39):16669–74. doi: 10.1073/pnas.0907138106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burgess DL, Kohrman DC, Galt J, Plummer NW, Jones JM, Spear B, Meisler MH. Mutation of a new sodium channel gene, Scn8a, in the mouse mutant ‘motor endplate disease’. Nat Genet. 1995;10(4):461–5. doi: 10.1038/ng0895-461. [DOI] [PubMed] [Google Scholar]
- 76.Cannon SC. Sodium channel defects in myotonia and periodic paralysis. Annu Rev Neurosci. 1996;19:141–64. doi: 10.1146/annurev.ne.19.030196.001041. [DOI] [PubMed] [Google Scholar]
- 77.Escayg A, MacDonald BT, Meisler MH, Baulac S, Huberfeld G, An-Gourfinkel I, Brice A, LeGuern E, Moulard B, Chaigne D, Buresi C, Malafosse A. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24(4):343–5. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 78.Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell. 2001;104(4):569–80. doi: 10.1016/s0092-8674(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 79.Lowe JS, Palygin O, Bhasin N, Hund TJ, Boyden PA, Shibata E, Anderson ME, Mohler PJ. Voltage-gated Nav channel targeting in the heart requires an ankyrin-G dependent cellular pathway. J Cell Biol. 2008;180(1):173–86. doi: 10.1083/jcb.200710107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Papadatos GA, Wallerstein PM, Head CE, Ratcliff R, Brady PA, Benndorf K, Saumarez RC, Trezise AE, Huang CL, Vandenberg JI, Colledge WH, Grace AA. Slowed conduction and ventricular tachycardia after targeted disruption of the cardiac sodium channel gene Scn5a. Proc Natl Acad Sci U S A. 2002;99(9):6210–5. doi: 10.1073/pnas.082121299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA. Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol Rev. 2005;57(4):387–95. doi: 10.1124/pr.57.4.13. [DOI] [PubMed] [Google Scholar]
- 82.Bennett V, Baines AJ. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001;81(3):1353–92. doi: 10.1152/physrev.2001.81.3.1353. [DOI] [PubMed] [Google Scholar]
- 83.Flucher BE, Daniels MP. Distribution of Na+ channels and ankyrin in neuromuscular junctions is complementary to that of acetylcholine receptors and the 43 kd protein. Neuron. 1989;3(2):163–75. doi: 10.1016/0896-6273(89)90029-9. [DOI] [PubMed] [Google Scholar]
- 84.Kordeli E, Lambert S, Bennett V, Ankyrin G. A new ankyrin gene with neural-specific isoforms localized at the axonal initial segment and node of Ranvier. J Biol Chem. 1995;270(5):2352–9. doi: 10.1074/jbc.270.5.2352. [DOI] [PubMed] [Google Scholar]
- 85.Kordeli E, Ludosky MA, Deprette C, Frappier T, Cartaud J. Ankyrin G is associated with the postsynaptic membrane and the sarcoplasmic reticulum in the skeletal muscle fiber. J Cell Sci. 1998;111(Pt 15):2197–207. doi: 10.1242/jcs.111.15.2197. [DOI] [PubMed] [Google Scholar]
- 86.Wood SJ, Slater CR. beta-Spectrin is colocalized with both voltage-gated sodium channels and ankyrinG at the adult rat neuromuscular junction. J Cell Biol. 1998;140(3):675–84. doi: 10.1083/jcb.140.3.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Davis JQ, Lambert S, Bennett V. Molecular composition of the node of Ranvier: identification of ankyrin-binding cell adhesion molecules neurofascin (mucin+/third FNIII domain-) and NrCAM at nodal axon segments. J Cell Biol. 1996;135(5):1355–67. doi: 10.1083/jcb.135.5.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003;300(5628):2091–4. doi: 10.1126/science.1085167. [DOI] [PubMed] [Google Scholar]
- 89.Lemaillet G, Walker B, Lambert S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem. 2003;278(30):27333–9. doi: 10.1074/jbc.M303327200. [DOI] [PubMed] [Google Scholar]
- 90.Brugada J, Brugada R, Brugada P. Right bundle-branch block and ST-segment elevation in leads V1 through V3: a marker for sudden death in patients without demonstrable structural heart disease. Circulation. 1998;97(5):457–60. doi: 10.1161/01.cir.97.5.457. [DOI] [PubMed] [Google Scholar]
- 91.Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392(6673):293–6. doi: 10.1038/32675. [DOI] [PubMed] [Google Scholar]
- 92.Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, Brugada P, Fressart V, Guerchicoff A, Harris-Kerr C, Kamakura S, Kyndt F, Koopmann TT, Miyamoto Y, Pfeiffer R, Pollevick GD, Probst V, Zumhagen S, Vatta M, Towbin JA, Shimizu W, Schulze-Bahr E, Antzelevitch C, Salisbury BA, Guicheney P, Wilde AA, Brugada R, Schott JJ, Ackerman MJ. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. 2010;7(1):33–46. doi: 10.1016/j.hrthm.2009.09.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Razani B, Woodman SE, Lisanti MP. Caveolae: from cell biology to animal physiology. Pharmacol Rev. 2002;54(3):431–67. doi: 10.1124/pr.54.3.431. [DOI] [PubMed] [Google Scholar]
- 94.Stan RV. Structure of caveolae. Biochim Biophys Acta. 2005;1746(3):334–48. doi: 10.1016/j.bbamcr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 95.Glenney JR, Jr, Soppet D. Sequence and expression of caveolin, a protein component of caveolae plasma membrane domains phosphorylated on tyrosine in Rous sarcoma virus-transformed fibroblasts. Proc Natl Acad Sci U S A. 1992;89(21):10517–21. doi: 10.1073/pnas.89.21.10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Scherer PE, Tang Z, Chun M, Sargiacomo M, Lodish HF, Lisanti MP. Caveolin isoforms differ in their N-terminal protein sequence and subcellular distribution. Identification and epitope mapping of an isoform-specific monoclonal antibody probe. J Biol Chem. 1995;270(27):16395–401. doi: 10.1074/jbc.270.27.16395. [DOI] [PubMed] [Google Scholar]
- 97.Scherer PE, Okamoto T, Chun M, Nishimoto I, Lodish HF, Lisanti MP. Identification, sequence, and expression of caveolin-2 defines a caveolin gene family. Proc Natl Acad Sci U S A. 1996;93(1):131–5. doi: 10.1073/pnas.93.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Song KS, Scherer PE, Tang Z, Okamoto T, Li S, Chafel M, Chu C, Kohtz DS, Lisanti MP. Expression of caveolin-3 in skeletal, cardiac, and smooth muscle cells. Caveolin-3 is a component of the sarcolemma and co-fractionates with dystrophin and dystrophin-associated glycoproteins. J Biol Chem. 1996;271(25):15160–5. doi: 10.1074/jbc.271.25.15160. [DOI] [PubMed] [Google Scholar]
- 99.Okamoto T, Schlegel A, Scherer PE, Lisanti MP. Caveolins, a family of scaffolding proteins for organizing “preassembled signaling complexes” at the plasma membrane. J Biol Chem. 1998;273(10):5419–22. doi: 10.1074/jbc.273.10.5419. [DOI] [PubMed] [Google Scholar]
- 100.Galbiati F, Volonte D, Minetti C, Bregman DB, Lisanti MP. Limb-girdle muscular dystrophy (LGMD-1C) mutants of caveolin-3 undergo ubiquitination and proteasomal degradation. Treatment with proteasomal inhibitors blocks the dominant negative effect of LGMD-1C mutanta and rescues wild-type caveolin-3. J Biol Chem. 2000;275(48):37702–11. doi: 10.1074/jbc.M006657200. [DOI] [PubMed] [Google Scholar]
- 101.Minetti C, Sotgia F, Bruno C, Scartezzini P, Broda P, Bado M, Masetti E, Mazzocco M, Egeo A, Donati MA, Volonte D, Galbiati F, Cordone G, Bricarelli FD, Lisanti MP, Zara F. Mutations in the caveolin-3 gene cause autosomal dominant limb-girdle muscular dystrophy. Nat Genet. 1998;18(4):365–8. doi: 10.1038/ng0498-365. [DOI] [PubMed] [Google Scholar]
- 102.Repetto S, Bado M, Broda P, Lucania G, Masetti E, Sotgia F, Carbone I, Pavan A, Bonilla E, Cordone G, Lisanti MP, Minetti C. Increased number of caveolae and caveolin-3 overexpression in Duchenne muscular dystrophy. Biochem Biophys Res Commun. 1999;261(3):547–50. doi: 10.1006/bbrc.1999.1055. [DOI] [PubMed] [Google Scholar]
- 103.Woodman SE, Sotgia F, Galbiati F, Minetti C, Lisanti MP. Caveolinopathies: mutations in caveolin-3 cause four distinct autosomal dominant muscle diseases. Neurology. 2004;62(4):538–43. doi: 10.1212/wnl.62.4.538. [DOI] [PubMed] [Google Scholar]
- 104.Fujimoto T, Nakade S, Miyawaki A, Mikoshiba K, Ogawa K. Localization of inositol 1,4,5-trisphosphate receptor-like protein in plasmalemmal caveolae. J Cell Biol. 1992;119(6):1507–13. doi: 10.1083/jcb.119.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barbuti A, Gravante B, Riolfo M, Milanesi R, Terragni B, DiFrancesco D. Localization of pacemaker channels in lipid rafts regulates channel kinetics. Circ Res. 2004;94(10):1325–31. doi: 10.1161/01.RES.0000127621.54132.AE. [DOI] [PubMed] [Google Scholar]
- 106.Bossuyt J, Taylor BE, James-Kracke M, Hale CC. The cardiac sodium-calcium exchanger associates with caveolin-3. Ann N Y Acad Sci. 2002;976:197–204. doi: 10.1111/j.1749-6632.2002.tb04741.x. [DOI] [PubMed] [Google Scholar]
- 107.Fujimoto T. Calcium pump of the plasma membrane is localized in caveolae. J Cell Biol. 1993;120(5):1147–57. doi: 10.1083/jcb.120.5.1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lockwich TP, Liu X, Singh BB, Jadlowiec J, Weiland S, Ambudkar IS. Assembly of Trp1 in a signaling complex associated with caveolin-scaffolding lipid raft domains. J Biol Chem. 2000;275(16):11934–42. doi: 10.1074/jbc.275.16.11934. [DOI] [PubMed] [Google Scholar]
- 109.Martens JR, Sakamoto N, Sullivan SA, Grobaski TD, Tamkun MM. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations. Targeting of Kv1.5 to caveolae. J Biol Chem. 2001;276(11):8409–14. doi: 10.1074/jbc.M009948200. [DOI] [PubMed] [Google Scholar]
- 110.Teubl M, Groschner K, Kohlwein SD, Mayer B, Schmidt K. Na(+)/Ca(2+) exchange facilitates Ca(2+)-dependent activation of endothelial nitric-oxide synthase. J Biol Chem. 1999;274(41):29529–35. doi: 10.1074/jbc.274.41.29529. [DOI] [PubMed] [Google Scholar]
- 111.Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: regulation of sodium current amplitude. Circ Res. 2002;90(4):443–9. doi: 10.1161/hh0402.105177. [DOI] [PubMed] [Google Scholar]
- 112.Galbiati F, Engelman JA, Volonte D, Zhang XL, Minetti C, Li M, Hou H, Jr, Kneitz B, Edelmann W, Lisanti MP. Caveolin-3 null mice show a loss of caveolae, changes in the microdomain distribution of the dystrophin-glycoprotein complex, and t-tubule abnormalities. J Biol Chem. 2001;276(24):21425–33. doi: 10.1074/jbc.M100828200. [DOI] [PubMed] [Google Scholar]
- 113.Volonte D, Peoples AJ, Galbiati F. Modulation of myoblast fusion by caveolin-3 in dystrophic skeletal muscle cells: implications for Duchenne muscular dystrophy and limb-girdle muscular dystrophy-1C. Mol Biol Cell. 2003;14(10):4075–88. doi: 10.1091/mbc.E03-03-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Hagiwara Y, Sasaoka T, Araishi K, Imamura M, Yorifuji H, Nonaka I, Ozawa E, Kikuchi T. Caveolin-3 deficiency causes muscle degeneration in mice. Hum Mol Genet. 2000;9(20):3047–54. doi: 10.1093/hmg/9.20.3047. [DOI] [PubMed] [Google Scholar]
- 115.Vatta M, Ackerman MJ, Ye B, Makielski JC, Ughanze EE, Taylor EW, Tester DJ, Balijepalli RC, Foell JD, Li Z, Kamp TJ, Towbin JA. Mutant caveolin-3 induces persistent late sodium current and is associated with long-QT syndrome. Circulation. 2006;114(20):2104–12. doi: 10.1161/CIRCULATIONAHA.106.635268. [DOI] [PubMed] [Google Scholar]
- 116.Lossin C, Wang DW, Rhodes TH, Vanoye CG, George AL., Jr Molecular basis of an inherited epilepsy. Neuron. 2002;34(6):877–84. doi: 10.1016/s0896-6273(02)00714-6. [DOI] [PubMed] [Google Scholar]
- 117.Cronk LB, Ye B, Kaku T, Tester DJ, Vatta M, Makielski JC, Ackerman MJ. Novel mechanism for sudden infant death syndrome: persistent late sodium current secondary to mutations in caveolin-3. Heart Rhythm. 2007;4(2):161–6. doi: 10.1016/j.hrthm.2006.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci. 1998;18(1):128–37. doi: 10.1523/JNEUROSCI.18-01-00128.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Gavillet B, Rougier JS, Domenighetti AA, Behar R, Boixel C, Ruchat P, Lehr HA, Pedrazzini T, Abriel H. Cardiac sodium channel Nav1.5 is regulated by a multiprotein complex composed of syntrophins and dystrophin. Circ Res. 2006;99(4):407–14. doi: 10.1161/01.RES.0000237466.13252.5e. [DOI] [PubMed] [Google Scholar]
- 120.Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, Wu Z, Huang F, Xia H, Peters MF, Froehner SC, Bredt DS. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell. 1996;84(5):757–67. doi: 10.1016/s0092-8674(00)81053-3. [DOI] [PubMed] [Google Scholar]
- 121.Williams JC, Armesilla AL, Mohamed TM, Hagarty CL, McIntyre FH, Schomburg S, Zaki AO, Oceandy D, Cartwright EJ, Buch MH, Emerson M, Neyses L. The sarcolemmal calcium pump, alpha-1 syntrophin, and neuronal nitric-oxide synthase are parts of a macromolecular protein complex. J Biol Chem. 2006;281(33):23341–8. doi: 10.1074/jbc.M513341200. [DOI] [PubMed] [Google Scholar]
- 122.Miyagoe-Suzuki Y, Takeda SI. Association of neuronal nitric oxide synthase (nNOS) with alpha1-syntrophin at the sarcolemma. Microsc Res Tech. 2001;55(3):164–70. doi: 10.1002/jemt.1167. [DOI] [PubMed] [Google Scholar]
- 123.Ueda K, Valdivia C, Medeiros-Domingo A, Tester DJ, Vatta M, Farrugia G, Ackerman MJ, Makielski JC. Syntrophin mutation associated with long QT syndrome through activation of the nNOS-SCN5A macromolecular complex. Proc Natl Acad Sci U S A. 2008;105(27):9355–60. doi: 10.1073/pnas.0801294105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Aarnoudse AJ, Newton-Cheh C, de Bakker PI, Straus SM, Kors JA, Hofman A, Uitterlinden AG, Witteman JC, Stricker BH. Common NOS1AP variants are associated with a prolonged QTc interval in the Rotterdam Study. Circulation. 2007;116(1):10–6. doi: 10.1161/CIRCULATIONAHA.106.676783. [DOI] [PubMed] [Google Scholar]
- 125.Arking DE, Pfeufer A, Post W, Kao WH, Newton-Cheh C, Ikeda M, West K, Kashuk C, Akyol M, Perz S, Jalilzadeh S, Illig T, Gieger C, Guo CY, Larson MG, Wichmann HE, Marban E, O’Donnell CJ, Hirschhorn JN, Kaab S, Spooner PM, Meitinger T, Chakravarti A. A common genetic variant in the NOS1 regulator NOS1AP modulates cardiac repolarization. Nat Genet. 2006;38(6):644–51. doi: 10.1038/ng1790. [DOI] [PubMed] [Google Scholar]
- 126.Echt DS, Liebson PR, Mitchell LB, Peters RW, Obias-Manno D, Barker AH, Arensberg D, Baker A, Friedman L, Greene HL, et al. Mortality and morbidity in patients receiving encainide, flecainide, or placebo. The Cardiac Arrhythmia Suppression Trial. N Engl J Med. 1991;324(12):781–8. doi: 10.1056/NEJM199103213241201. [DOI] [PubMed] [Google Scholar]