

Abstract
In addition to xeroderma pigmentosum (XP), mutations in the human XPG gene cause early onset of Cockayne syndrome (CS) in some patients (XPG/CS). The CS-causing mutations in such patients all produce truncated XPG proteins. To test the hypothesis that the CS phenotype, with characteristics such as growth retardation and a short life span in XPG/CS patients, results from C-terminal truncations, we constructed mutants with C-terminal truncations in mouse XPG (Xpg) (from residue D811 to the stop codon [XpgD811stop] and deletion of exon 15 [XpgΔex15]). In the XpgD811stop and XpgΔex15 mutations, the last 360 and 183 amino acids of the protein were deleted, respectively. To generate Xpg mutant mice, we devised the shortcut knock-in method by replacing genomic DNA with a mutated cDNA fragment (cDNA-mediated knock in). The control mice, in which one-half of Xpg genomic DNA fragment was replaced with a normal Xpg cDNA fragment, had a normal growth rate, a normal life span, normal sensitivity to UV light, and normal DNA repair ability, indicating that the Xpg gene partially replaced with the normal cDNA fragment retained normal functions. The XpgD811stop homozygous mice exhibited growth retardation and a short life span, but the XpgΔex15 homozygous mice did not, indicating that deletion of the last 360 amino acids results in the CS phenotype but deletion of the last 183 amino acids does not. The XpgD811stop homozygous mice, however, exhibited a slightly milder CS phenotype than did the Xpg null mutant mice, indicating that the XpgD811stop protein still retains some Xpg function that affects the severity of the CS phenotype.
Xeroderma pigmentosum (XP) is a rare autosomal recessive disease clinically characterized by hypersensitivity to sunlight, abnormal pigmentation, and a predisposition to skin cancers, particularly at sun-exposed areas. XP patients are classified into eight complementation groups. Cells of patients from seven out of the eight groups (XP-A to XP-G) exhibit a defect in the early steps of the nucleotide excision repair (NER) pathway, and cells from the remaining group (XP variant) are defective in bypass DNA synthesis across DNA lesions induced by UV radiation (1, 23, 34).
Another sun hypersensitivity disorder, Cockayne syndrome (CS), is both clinically and genetically distinct from XP and is characterized by postnatal growth failure, a short life span, and progressive neurological dysfunction, but no predisposition to cancer (29). To date, more than 140 CS cases have been reported (29) that can be classified into five complementation groups. Most patients exhibit only CS symptoms, and they belong to either group CS-A or CS-B. These symptoms arise from mutations in the CSA or CSB gene, both of which are required for the preferential removal of UV light-induced lesions in transcribed strands of genes by the transcription-coupled repair (TCR) process (14, 36).
In very rare cases, complementation analyses have classified some CS patients into XP groups XP-B, XP-D, and XP-G (6, 12, 13, 20, 27, 37, 38). XP-B patients are extremely rare, a fact that presumably reflects the essential role of XPB in transcription initiation (35), and two patients were also found to have CS (34). Two XP-D patients were also reported to have CS, although this was not so for most XP-D patients. The XPB and XPD proteins are helicase components of the basal transcription factor TFIIH required for transcription initiation by RNA polymerase (pol) II, and the role of TFIIH in NER involves the unwinding of DNA around the lesion to allow incision by structure-specific endonucleases (10, 11). A combination of the clinical hallmarks of XP and CS has been observed in several XP-G patients. XPG produces the initial incision 3′ to the lesion, followed by the 5′ incision by the ERCC1/XPF heterodimer (2, 24, 25, 28, 31, 39). The clinical features of CS have not been correlated with the roles of these XP gene products in NER.
As described above, the XPG protein is a DNA endonuclease with structure-specific properties, cleaving near the junction between duplex and single-stranded DNAs with a defined polarity. In its N-terminal (N) and internal (I) regions, XPG shows sequence similarity to a family of other nucleases. These include the 5′ nuclease domains of Escherichia coli DNA pol I and Thermus aquaticus (Taq) DNA pol. This family also includes the human FEN-1 and Saccharomyces cerevisiae Rad27 (Rth1) proteins (3, 17, 22).
To investigate the XPG active site, studies of the XPG protein with site-directed mutations in the conserved region have been carried out (17, 39). Mutation of residue 791 from glutamic acid to alanine (E791A) or mutation of residue 812 from aspartic acid to alanine (D812A) in the human XPG protein completely abolished the junction-cleaving and 3′ incision activities of XPG, but the excision nuclease reconstituted with XPG (E791A or D812A) caused normal 5′ incision at the 15th to the 24th phosphodiester bonds 5′ to a platinum adduct or a 6-4 photoproduct without producing any 3′ incision (7, 39). Although no XP-G patients with the E791A or D812A mutation have been identified, the mutation of residue 792 from alanine to valine (A792V) has been identified in two siblings, XP124LO and XP125LO. The A792V protein, however, retains some residual endonuclease activity, and patients with this mutation exhibit an XP phenotype (7).
Three XP-G patients, XPCS1LV, XPCS2LV, and 94RD27, suffered from severe early-onset CS, and they died at 6.5 years, 20 months, and 7 months of age, respectively. The mutations causing CS in these patients were identified, and they all produced truncated XPG proteins (30). In patient XPCS1LV, the XPG protein showed a truncation after amino acid 659. Patient XPCS2LV carried two XPG mutant alleles, one of which encoded a truncation after amino acid 659, while the other mutant allele encoded a truncation after amino acid 262. In patient 94RD27, the XPG protein had a frame shift after amino acid 925, and another 55 amino acids unrelated to XPG were present before the next in-frame stop codon. Thus, these observations indicate that the mutations that truncate the XPG protein cause severe early-onset CS with growth retardation and early death.
To test the hypothesis that the growth and developmental defects in XP-G/CS patients result from C-terminal truncations, we constructed mouse XPG (Xpg) with C-terminal truncations analogous to the truncation in XP-G/CS patient 94RD27. We also generated a base substitution mutation at an active site for NER to completely abolish the 3′ nuclease activity of Xpg to confirm that the mutation did not result in the CS phenotype, as expected from analyses of mutations in human XP-G patients. To generate the Xpg mutant mouse strains, we adopted the shortcut knock-in method as reported by de Boer et al. (9) with a modification. We extended the replaced region of the genome sequence with a mutated cDNA fragment to about one-half of the gene (cDNA-mediated knock in). The control mice, in which about one-half of the Xpg genomic DNA fragment was replaced with a normal Xpg cDNA fragment, had a normal growth rate, a normal life span, normal sensitivity to UV light, and normal DNA repair ability, indicating that the Xpg gene partially replaced with the normal cDNA fragment retained normal functions. Here we report the cDNA-mediated knock-in procedure and the characteristics of the Xpg mutant mice generated by this method and the conventional method in addition to the previously reported Xpg null mutant mouse strain (16).
MATERIALS AND METHODS
Generation of mutations in Xpg cDNA.
Mutations were generated in the pSV2MER5 plasmid, containing the full-length Xpg cDNA (33). The following oligonucleotides were used to construct the mutants by PCR-based site-directed mutagenesis: D811A, 5′-GCCATCTGGCTGTTTGGGGCCC (22-mer); D811stop, 5′-TAAATCTGGCTGTTTGGGGCCC (22-mer); reverse primer, 5′-ACTATCGTCAGTAATCGTTCCAGA (24-mer). The altered nucleotides are underlined. Nucleotide changes were introduced with an ExSite PCR-based site-directed mutagenesis kit (Stratagene). The mutations were identified by restriction enzyme (EcoRV) analysis and confirmed by DNA sequencing with an automated ABI 373A sequencer (Perkin-Elmer Applied Biosystems Division, Foster City, Calif.). Fragments containing a mutation were excised by cleaving the adjacent restriction sites and were used to replace the corresponding fragment in pSV2MER5.
Construction of targeting vectors.
Mouse genomic clones containing several exons of the Xpg gene were isolated from a lambda DASHII (Stratagene) phage library constructed with genomic DNA from D3 (an embryonic stem [ES] cell line derived from mouse strain 129/Sv). A 6.8-kb EcoRI-ScaI fragment obtained from one of the genomic clones was subcloned into pUC118, which was used to generate the targeting vector. The 6.8-kb EcoRI-ScaI fragment contained exon 15 (the last exon of Xpg), which had the coding and 3′ untranslated region sequences and a poly(A) signal (Fig. 1A). To delete exon 15, a 1.1-kb sequence (from the XhoI site in intron 14 to the SpeI site in exon 15) was removed from the 6.8-kb EcoRI-ScaI fragment and a 1.1-kb neo cassette from pMC1neo (Stratagene) was inserted in its place with the same transcriptional orientation. A 3.5-kb BamHI-EcoRI fragment of the herpes simplex virus thymidine kinase (TK) cassette was positioned at the 3′ end of the construct for negative selection. The targeting vector thus constructed was designated pMER5/Δex15.
FIG. 1.

Generation of Xpg-deficient (Δex15) mice. (A) Schematic of the deletion mutation at the Xpg locus. Exons 14 and 15 of the Xpg gene are represented as boxes. PCR primers are indicated by arrows. The 3′ external probe used for Southern blot analysis is indicated by a solid bar corresponding to the ScaI-PstI fragment on the wild-type map at the top of panel A, and the diagnostic fragments of 4.3 and 8.2 kb are indicated by solid lines at the bottom of panel A. Abbreviations: B, BamHI; E, EcoRI; H, HindIII; X, XhoI. (B) PCR and Southern blot analyses of the targeted ES clone, G138. The size of the predicted PCR product for the mutant allele was 1.8 kb, and no PCR product was amplified from the wild-type allele. Southern blot analysis with the 3′ external probe also detected the predicted restriction fragments shown in panel A. Lanes: M, size markers; ES, ES cells used as a control. (C) Southern blot analyses of offspring from intercrosses between chimeric males and C57BL/6J females. Lanes: +/+, wild type; +/Δex15, heterozygote; Δex15/Δex15, homozygous XpgΔex15 mutant. (D) Northern blot analysis of total RNA from newborn mice derived from a heterozygous intercross, with Xpg cDNA as the probe. As a loading control, the 28S rRNA band is shown.
A 3.8-kb HindIII-PstI fragment obtained from one of the genomic clones was subcloned into pUC119 and used to generate the other targeting vectors (gene-replacing vectors, knock-in vectors). As shown in Fig. 2A, the 3.8-kb fragment contained a part of exon 8 at its 3′ end. Next, 2.45-kb PstI-BamH1 Xpg cDNA fragments (corresponding to exons 8 to 15) obtained from pSV2MER5 with D811A, D811stop, or no mutation were cloned into the PstI and BamHI sites 3′ to the PstI site of the 3.8-kb fragment (Fig. 2A). A PGKneo cassette that contained loxP sequences at both ends (LNLneo) was then ligated 3′ to the BamHI site of the Xpg cDNA fragment in the opposite transcriptional orientation. Finally, a 5.3-kb SpeI-EcoRI fragment from pMER5/Δex15, which contained a 1.8-kb SpeI-ScaI sequence downstream of exon 15 and the herpes simplex virus TK gene for negative selection (Fig. 1A), was ligated to the 3′ end of the neo cassette (Fig. 2A). The constructed targeting vectors were named pMER5/D811A, pMER5/D811stop, and pMER5/cXpg.
FIG. 2.

Generation of a truncation or base substitution mutation in the Xpg gene by the cDNA-mediated knock-in method. (A) Schematic of generation of mutations at the Xpg locus by the cDNA-mediated knock-in method. Exons 8 to 15 are shown. PCR primers are indicated by arrows. The 5′ probe used for Southern blot analysis is indicated by a solid bar on the wild-type map at the top of panel A, and the diagnostic fragments of 5.5 and 8.3 kb are indicated by solid lines at the bottom of panel A. Abbreviations: B, BamHI; E, EcoRI; H, HindIII; P, PstI. (B) PCR and Southern blot analyses of the targeted ES clones, ES-D811A, ES-D811stop, and ES-cXpg. The size of the predicted PCR product for mutant alleles was 1.8 kb, and no PCR product was amplified from the wild-type allele. Southern blot analysis with the 5′ probe also detected the predicted restriction fragments shown in panel A. Lanes: M, size markers; ES, ES cells used as a control. (C) Southern blot analyses of offspring from intercrosses between chimeric males and C57BL/6J females. +/+ represents the wild type (W.T.). cXpg/cXpg, D811A/D811A, and D811stop/D811stop represent homozygous cXpg, XpgD811A, and XpgD811stop mutants, respectively. (D) Northern blot analysis of total RNA from newborn mice, which were derived from heterozygous intercrosses and were determined to be homozygous mutants, with Xpg cDNA as the probe. As a loading control, the 28S rRNA band is shown. (E) Schematic of mutations in the Xpg protein. Arrows indicate the locations of mutations in XPG where the protein terminates in three XPG/CS patients. N and I indicate the highly conserved N-terminal and internal regions. C indicates the moderately conserved C-terminal region. The Xpg and S. cerevisiae (S.c.) Rad2 proteins have 45.5, 52.6, and 34.2% identical residues in the N, I, and C regions, respectively. In the XpgΔex15 and XpgD811stop mutant proteins, the last 183 and 360 amino acids were deleted, respectively. In the XpgD811A mutation, the highly conserved D811 residue of the Xpg protein was replaced with alanine. H.s., Homo sapiens; M.m., Mus musculus.
Gene targeting in ES cells and generation of Xpg mutant mice.
The 129/Sv-derived ES cell line D3 and the (129/Sv/129/SvJ)F1-derived ES cell line R1 were maintained in Dulbecco's modified Eagle's medium (Nissui Seiyaku Co., Ltd., Tokyo, Japan) containing 20% heat-inactivated fetal bovine serum (GIBCO), 20 mM glucose, 0.1 mM 2-mercaptoethanol, 2 mM glutamine, nonessential amino acids (GIBCO), nucleosides (adenosine, guanosine, cytidine, and uridine at 30 μM each and thymidine at 10 μM), and 1,000 U of leukemia-inhibiting factor (GIBCO) per ml. Gene-targeting experiments were carried out with 50 μg of SalI-linearized pMER5/Δex15, pMER5/D811A, pMER5/D811stop, or pMER5/cXpg vector DNA as described previously (16). Homologous recombination between the targeting vector pMER5/Δex15 and mouse chromosome 1, on which Xpg was localized (15), was screened by PCR analysis with the neoS2 and TV3R3 primers. Homologous recombination between the targeting vector pMER5/D811A, pMER5/D811stop, or pMER5/cXpg and mouse chromosome 1 was screened by PCR analysis with the LNLF2 and TV3R3 primers. The sequences of the primers were as follows: 5′-GCGAAGGGGCCACCAAAGAACGG for LNLF2, 5′-GCTGACCGCTTCCTCGTGCTTTAC for neoS2, and 5′-CAGTCCTGGTTGTAGACACAGTACC for TV3R3. The correct targeting event was verified by Southern blot hybridization with a probe containing a 0.7-kb ScaI-PstI fragment (Fig. 1A) for pMER5/Δex15 or with a probe containing a 0.7-kb PstI fragment for pMER5/D811A, pMER5/D811stop, or pMER5/cXpg. Genomic DNA isolated from ES cells or mouse tails was digested with EcoRI or HindIII, separated through 0.8% agarose gel electrophoresis with Tris-acetate-EDTA buffer at pH 7.5, and transferred onto Hybond N+ membranes (Amersham). The absence of additional random integration of the targeting construct was examined with a neo probe.
The targeting event occurred at a frequencies of 26.5% (48/181) for pMER5/Δex15 in D3 cells and 2.6% (8/312) for pMER5/D811A, pMER5/D811stop, and pMER5/cXpg in R1 cells. Chimeras were constructed by targeted ES cell injection into C57BL/6 blastocysts as reported previously (16, 18).
Other methods.
Primary cell culture, cell survival assays, and in vivo repair assays were carried out as described previously (16, 26).
RESULTS
Generation of Xpg mutant mice.
To test the hypothesis that the growth and developmental defects in XP-G/CS patients result from C-terminal truncations, we generated C-terminal truncation mutations in mouse XPG (Xpg) (residue D811 to the stop codon [XpgD811stop] and deletion of exon 15 [XpgΔex15]) with C-terminal truncations, as found also in XP-G/CS patient 94RD27. In the XpgD811stop or XpgΔex15 mutation, the last 360 or 183 amino acids of the XPG protein were deleted, respectively, whereas in 94RD27, the last 261 residues of XPG (corresponding to the last 246 residues of Xpg) were absent (Fig. 2E). We also replaced highly conserved residue D811 in Xpg with alanine (XpgD811A), which corresponds to the D812A mutation in human XPG. As mentioned above, the D812A mutation was reported to completely abolish the 3′ incision activity of the XPG protein (39). The Xpg null mutant (XpgΔ) mouse strain has already been generated, and it failed to grow postnatally and died (16). Mutant mice with the truncation or the base substitution mutation were generated as follows.
The pMER5/Δex15 targeting vector was designed to delete exon 15 from the Xpg gene (Fig. 1A). We also constructed three targeting vectors designed to replace a region of the Xpg genomic DNA (exons 8 to 15) fragment with corresponding Xpg cDNA fragments that contained D811A, D811stop, or no mutation (Fig. 2A). Targeted ES cell clones with the predicted mutations were identified by PCR and Southern blot analyses (Fig. 1B and 2B). The targeted ES cell clones were injected into C57BL/6 blastocysts to generate chimeric mice capable of transmitting the mutant allele to F1 offspring, which were later identified by both PCR and Southern blot analyses (Fig. 1C and 2C). To examine the effects of the deletion of exon 15 or replacement of a genomic DNA fragment with a cDNA fragment on Xpg gene expression, total RNA from the embryos was analyzed by Northern blotting. In XpgΔex15 homozygous mice, two large mRNAs were detected (Fig. 1D). Analysis of these two mRNAs by the 3′ rapid amplification of cDNA ends technique revealed that 6 or 36 amino acids were incorporated after the amino acid residues that corresponded to exon 14. These transcripts could be produced by alternative splicing owing to the lack of exon 15. In contrast, Xpg mRNA of normal size and amount was detected in mice homozygous for Xpg replaced with cDNA fragments with or without mutations (Fig. 2D), indicating that the replacement with cDNA fragments did not affect Xpg gene expression.
UV light sensitivities of cells obtained from Xpg mutant mouse embryos.
As shown in Fig. 3, cells from mice homozygous for Xpg replaced partially with the normal cDNA fragment (cXpg) were as resistant to UV light as were cells from wild-type mice, indicating that the Xpg gene replaced partially with the normal Xpg cDNA fragment functioned normally. Cells from the XpgD811stop homozygotes were highly sensitive to UV light and were as sensitive as Xpg null mutant XpgΔ cells, indicating that the XpgD811stop protein completely lost its function in NER. Cells from the XpgD811A homozygotes were also sensitive to UV light. Although the XpgD811A mutant cells were not expected to have 3′ incision activity, their higher UV light resistance than that of XpgΔ cells suggests that the mutant protein can still promote the removal of some UV light lesions. Biochemical studies of the XPG D812A mutant protein have revealed that although the protein is devoid of any 3′ incision activity during NER, 5′ incisions still occur normally in its presence (39). The residual repair activity in XPGD812A, and also in XpgD811A, may then result from the removal of the 5′-incised DNA strand by the action of nucleases such as FEN1 and other members of this nuclease family. The XpgΔex15 homozygous cells were as sensitive to UV light as the XpgD811A homozygous cells, suggesting that this mutant protein also retains residual ability to remove UV light-induced lesions.
FIG. 3.
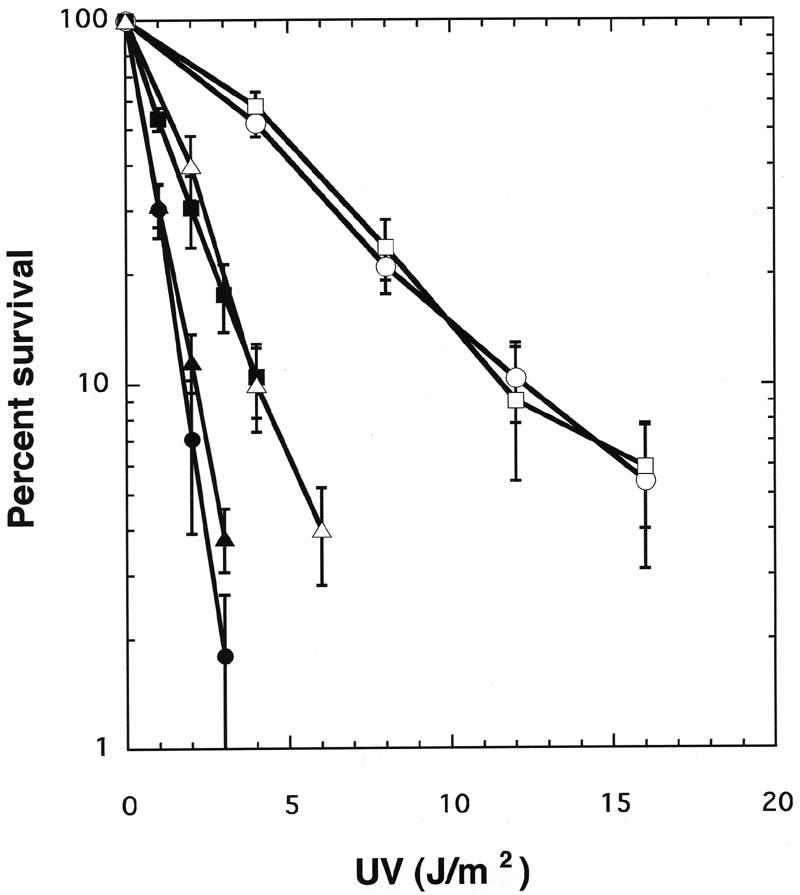
Survival curves of embryonic fibroblasts derived from Xpg mutant mice. Each point represents an average of three independent experiments with wild-type (○), cXpg (□), XpgD811A (▪), XpgD811stop (▴), XpgΔex15 (▵), or XpgΔ (•) homozygous cells. Error bars show the standard deviation derived from at least three independent experiments.
Ability of Xpg mutant mouse cells to repair UV light-induced DNA lesions.
We compared the kinetics of removal of major UV light-induced DNA lesions in the cells obtained from these mutant and control mice as determined by enzyme-linked immunosorbent assay with monoclonal antibodies against cyclobutane-type thymine dimers and 6-4 photoproducts (Fig. 4A and B). Neither thymine dimers nor 6-4 photoproducts were removed in XpgD811stop homozygous cells, but they were removed in wild-type cells and in cells from the cXpg homozygous mice. Lesions were partially removed in moderately UV light-sensitive XpgD811A and XpgΔex15 homozygous cells. These results, together with data from UV light sensitivity tests, indicate that the NER activity of these mutant mice is defective and that their repair abilities reflect their UV light hypersensitivities.
FIG. 4.

Kinetics of UV light-induced DNA damage removal in embryonic fibroblasts derived from wild-type (○), cXpg (□), XpgD811A (▪), XpgD811stop (▴), XpgΔex15 (▵), or XpgΔ (•) homozygous mice. The kinetics of 6-4 photoproduct (6-4PP) (A) and cyclobutane pyrimidine dimer (CPD) (B) removal are presented. Each point is the average of triplicate wells. Error bars show the standard deviation derived from at least three independent experiments.
Growth and life span of mutant homozygous mice.
Since, as already reported, Xpg null mutant (XpgΔ) mice exhibited a failure to grow and died within 23 days of birth (16), we examined the growth rate and life span of the homozygous Xpg mutant mice generated in this study (Fig. 5A and B). Mice heterozygous for each allele were interbred to produce homozygous mutants. Out of the 106 pups born to mice heterozygous for the XpgD811stop allele, 24 (22.6%) exhibited growth failure and most of them (22 out of 24) died within 30 days of birth (Fig. 5A). Two out of the 24 mice died on the 78th and 139th days after birth. PCR analysis revealed that all of the 24 mice were homozygous for the XpgD811stop allele. The 82 survivors exhibiting normal growth were either wild-type mice (27 mice, 25.5%) or heterozygotes (55 mice, 51.9%). These results indicate that the mutation is transmitted in a Mendelian manner and does not interfere with normal embryonic development. Although the sizes of the XpgD811stop homozygotes at birth were not markedly different from those of the wild-type mice or heterozygotes, the growth of the XpgD811stop homozygous mice was severely retarded thereafter, similar to that of the XpgΔ mice (Fig. 5B). Undeveloped small intestines were also observed in the XpgD811stop homozygous mice (data not shown), as in the XpgΔ mice (16). When the life spans and body weights of the D811stop homozygous mice were compared with those of the XpgΔ mice, the XpgD811stop homozygotes showed a 9- to 10-day longer life span and slightly greater body weight than the null mutant mice (Fig. 5A and B), suggesting that the XpgD811stop mutant protein still retains residual Xpg functions required for their growth and survival. In contrast, the cXpg, XpgD811A, or XpgΔex15 homozygous mice exhibited no particular growth abnormalities compared with the wild-type mice. These results indicate that the base substitution mutation at the active site for NER or the C-terminal truncation of 183 amino acid residues in Xpg does not affect the growth or life span of XpgD811A or XpgΔex15 mice.
FIG. 5.

Growth rate and life span of Xpg mutant mice. Survival curves (A) and average body weights (B) of wild-type (○), XpgD811stop (▴), and XpgΔ (•) homozygous mice are shown. Since the survival curves and body weights of cXpg, XpgD811A, and XpgΔex15 homozygous mice are similar to those of wild-type mice, the plots for these mice were omitted.
DISCUSSION
Base substitution mutation that inactivates the 3′ incision activity of Xpg and CS phenotype.
Xpg, as well as XPG, shows sequence similarity in the N and I regions with a family of other nucleases, such as FEN1. Mutations in these regions of human XPG, namely, D77A, E791A, and D812A, have been reported to completely abolish its 3′ incision activity in in vitro assays. Since amino acid residues in these regions of the proteins are highly conserved between humans and mice, mutations of the identical residues in Xpg (D77A, E790A, and D811A), as well as in XPG, may completely eliminate its nuclease activity. Although no XP-G patients have been identified with these mutations, a mutation, A792V, has been identified in two siblings, XP124LO and XP125LO, who exhibited an XP phenotype only. Since the A792V protein retains some residual endonuclease activity, we generated mutant mice that have the D811A mutation in Xpg (XpgD811A) to examine the effect of complete loss of the 3′ incision activity of Xpg on the phenotype of mice. As shown in Fig. 3 to 5, XpgD811A exhibited hypersensitivity to UV light and a defect in the ability to repair UV light-induced DNA damage, as in XP patients, but exhibited no CS features, such as growth retardation and a short life span, suggesting that the mutation that completely abolishes the 3′ incision activity of Xpg results in an XP phenotype only, as expected.
C-terminal truncation of Xpg and CS features.
In addition to the N and I regions, Xpg is significantly homologous with S. cerevisiae Rad2 in the C region, in which they have 34.2% identical residues (Fig. 2E), suggesting that the region plays some role in the function of the protein. The CS-causing mutations in XPG/CS patients have been identified, and they all produce truncated XPG proteins (truncation after amino acid 262, 659, or 925) (30). These findings indicate that mutations that truncate the protein cause a severe form of CS. To examine the notion that the CS phenotype (growth retardation and a short life span) results from C-terminal truncations, we generated mouse strains, XpgD811stop and XpgΔex15, in which the last 360 and 183 amino acids of the protein were deleted, respectively. The XpgD811stop homozygous mice exhibited growth retardation and a short life span, but the XpgΔex15 homozygous mice exhibited normal growth and a normal life span, clearly indicating that deletion of the last 360 amino acids results in the CS phenotype but that deletion of the last 183 amino acids of Xpg does not. Moreover, the C-terminal truncation of the Xpg protein does not always result in the CS phenotype. In one XPG/CS patient, 94RD27, the last 261 amino acid residues of XPG (corresponding to the last 246 amino acid residues of Xpg) are absent (Fig. 2E). Thus, the deletion of the last 246 amino acids of Xpg may cause the CS phenotype in mice. As shown in Fig. 2E, XpgΔex15 retains most of the C region of Xpg, while all or most of the C region is deleted from XpgD811stop or 94RD27, indicating that loss of the integrity of the C region is closely associated with the onset of CS.
Although the XpgD811stop homozygous mice exhibited CS features as described above, when the phenotype of the XpgD811stop mice was compared with that of the Xpg null (XpgΔ) mice, the XpgD811stop homozygotes exhibited a slightly milder CS phenotype, such as a 9- to 10-day longer life span and a slightly rapid growth rate (Fig. 5). The results indicate that XpgD811stop (the Xpg protein that lacks the entire C region) still retains some Xpg function that affects the severity of the CS phenotype.
Molecular bases of CS phenotype expression.
Although the mechanism underlying the growth retardation and neurological defects in CS is unclear, the following molecular bases of CS phenotype expression are proposed. On the basis of the observation that the TCR of oxidative lesions such as those caused by thymine glycol and 8-oxoguanine occurs normally in cells from XP patients that exhibit defects in NER but is defective in cells from CSB, XPB/CS, XPD/CS, and XPG/CS patients. These findings indicate that the TCR of two oxidative lesions (those caused by thymine glycol and 8-oxoguanine) requires CSB, XPB, XPD, and XPG and that the CS-causing mutations in the CSB, XPB, XPD, and XPG genes block the TCR of these lesions. On the basis of these observations, the defective TCR of these oxidative lesions has been suggested to be the underlying cause of CS (8, 21).
The CS phenotype in individuals with mutations in the XPB and XPD helicase subunits of TFIIH may arise from a defect in transcription. However, because of the lack of any evidence linking XPG to a role in transcription, the transcriptional defect model (4, 32) has been noted to be inadequate to account for the association of CS with mutations in the XPG gene. Regarding RAD2, the S. cerevisiae counterpart of XPG, Lee et al. (19) provided evidence for the involvement of RAD2 in promoting efficient RNA pol II transcription. Moreover, the inactivation of RAD26, the S. cerevisiae counterpart of the human CSB gene, also causes a deficiency in transcription, and a synergistic decline in transcription occurs in the absence of both the RAD2 and RAD26 genes. On the basis of these observations, they proposed that transcriptional defects are the underlying causes of CS.
Bradsher et al. (5) have reported that CSB is detected not only in the nucleoplasm but also in the nucleolus inside a complex that contains RNA pol I, TFIIH, and XPG and promotes efficient rRNA synthesis. CSB is active in in vitro RNA pol I transcription and restores rRNA synthesis when transfected into CSB-deficient cells. Bradsher et al. also demonstrated that mutations in CSB, as well as in the XPB and XPD genes, all of which confer the CS phenotype, disturb the RNA pol I-TFIIH interaction within the complex. On the basis of these observations, they proposed that a defect in RNA pol I transcription, in addition to a defect in RNA pol II transcription, may be one of the factors contributing to CS phenotype expression.
These lines of evidence indicate that XPG, as well as CSB, XPB, and XPD, plays important roles, besides NER, in multiple biological processes, including TCR of oxidative base damage and RNA pol I and II transcriptions. Loss of all or a part of these functions may confer the CS phenotype, and the severity of the symptoms may reflect the intensity of the residual function(s) in these processes. Our Xpg mutant mice, XpgD811stop, demonstrated the CS phenotype, but it was slightly milder than that in Xpg null mice. Thus, the XpgD811stop protein may still retain some of these functions. To fully explain the expression of the CS phenotype, further detailed studies are required. For this purpose, our mutant Xpg mouse strains and also the technique of generating such mutant mice described in this study may be powerful tools. In general, a complex phenotype such as the CS phenotype affects the entire body. Our cDNA-mediated knock-in method may be applicable to examining the relationship between mutations and their effects on the entire body.
Acknowledgments
We thank K. Noshiro, K. Sakurai, and T. Tanaka for technical assistance.
This work was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
REFERENCES
- 1.Aboussekhra, A., and R. D. Wood. 1994. Repair of UV-damaged DNA by mammalian cells and Saccharomyces cerevisiae. Curr. Opin. Genet. Dev. 4:212-220. [DOI] [PubMed] [Google Scholar]
- 2.Aboussekhra, A., M. Biggerstaff, M. K. Shivji, J. A. Vilpo, V. Monocollin, V. N. Podust, M. Protic, U. Hubscher, J. M. Egly, and R. D. Wood. 1995. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 80:859-868. [DOI] [PubMed] [Google Scholar]
- 3.Bambara, R. A., R. S. Murante, and L. A. Henricksen. 1997. Enzymes and reactions at the eukaryotic DNA replication fork. J. Biol. Chem. 272:4647-4650. [DOI] [PubMed] [Google Scholar]
- 4.Bootsma, D., and J. H. J. Hoeijmakers. 1993. Engagement with transcription. Nature 363:114-115. [DOI] [PubMed] [Google Scholar]
- 5.Bradsher, J., J. Auriol, L. P. de Santis, S. Iban, J. L. Vonesh, I. Grummt, and J. M. Egly. 2002. CSB is a component of RNA pol I transcription. Mol. Cell 10:819-829. [DOI] [PubMed] [Google Scholar]
- 6.Broughton, B. C., A. F. Thompson, S. A. Harcourt, W. Vermeulen, J. H. J. Hoeijmakers, E. Botta, M. Stefanini, M. D. King, C. A. Weber, and J. Cole. 1995. Molecular and cellular analysis of the DNA repair defect in a patient with xeroderma pigmentosum complementation group D who had the clinical features of xeroderma pigmentosum and Cockayne syndrome. Am. J. Hum. Genet. 56:167-174. [PMC free article] [PubMed] [Google Scholar]
- 7.Constantinou, A., D. Gunz, E. Evans, P. Lalle, P. A. Bates, R. D. Wood, and S. G. Clarkson. 1999. Conserved residues of human XPG protein important for nuclease activity and function in nucleotide excision repair. J. Biol. Chem. 274:5637-5648. [DOI] [PubMed] [Google Scholar]
- 8.Cooper, P. K., T. Nouspikel, S. G. Clarkson, and S. A. Leadon. 1997. Defective transcription-coupled repair of oxidative base damage in Cockayne syndrome patients from XP group G. Science 275:990-993. [DOI] [PubMed] [Google Scholar]
- 9.de Boer, J., J. de Wit, H. van Steeg, R. J. W. Berg, H. Morrreau, P. Visser, A. R. Lehmann, M. Duran, J. H. J. Hoeijmakers, and G. Weeda. 1998. A mouse model for the basal transcription/DNA repair syndrome trichothiodystrophy. Mol. Cell 1:981-990. [DOI] [PubMed] [Google Scholar]
- 10.Evans, E., J. Fellows, A. Coffer, and R. D. Wood. 1997. Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. EMBO J. 16:625-638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Evans, E., J. G. Moogs, J. R. Hwang, J. M. Egly, and R. D. Wood. 1997. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. EMBO J. 16:6559-6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedberg, E. C. 1996. Cockayne syndrome—a primary defect in DNA repair, transcription, both or neither? Bioessays 18:731-738. [DOI] [PubMed] [Google Scholar]
- 13.Hamel, B. C. J., A. Raams, A. R. Shuitema-Dijkstra, P. Simons, I. van der Burgt, N. G. J. Jaspers, and W. J. Kleijer. 1996. Xeroderma pigmentosum-Cockayne syndrome complex: a further case. J. Med. Genet. 33:607-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hanawalt, P. C. 1994. Transcription-coupled repair and human disease. Science 266:1957-1958. [DOI] [PubMed] [Google Scholar]
- 15.Harada, Y.-N., Y. Matsuda, N. Shiomi, and T. Shiomi. 1995. Complementary DNA sequence and chromosomal localization of xpg, the mouse counterpart of human repair gene XPG/ERCC5. Genomics 28:59-65. [DOI] [PubMed] [Google Scholar]
- 16.Harada, Y.-N., N. Shiomi, M. Koike, M. Ikawa, M. Okabe, S. Hirota, Y. Kitamura, M. Kitagawa, T. Matsunaga, O. Nikaido, and T. Shiomi. 1999. Postnatal growth failure, short life span, and early onset of cellular senescence and subsequent immortalization in mice lacking the xeroderma pigmentosum group G gene. Mol. Cell. Biol. 19:2366-2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harrington, J. J., and M. R. Lieber. 1994. Functional domains within FEN-1 and RAD2 define a family of structure-specific endonucleases: implications for nucleotide excision repair. Genes Dev. 8:1344-1355. [DOI] [PubMed] [Google Scholar]
- 18.Kito, S., Y. Noguchi, Y. Ohta, T. Ohhata, M. Abe, N. Shiomi, and T. Shiomi. 2003. Evaluation of developmental competence of vitrified-warmed early cleavage stage embryos and their application for chimeric mouse production. Exp. Anim. 52:179-183. [DOI] [PubMed] [Google Scholar]
- 19.Lee, S. K., S. L. Yu, L. Prakash, and S. Prakash. 2002. Requirement of yeast RAD2, a homolog of human XPG gene, for efficient RNA polymerase II transcription: implications for Cockayne syndrome. Cell 109:823-834. [DOI] [PubMed] [Google Scholar]
- 20.Lehmann, A. R. 1995. Nucleotide excision repair and the link with transcription. Trends Biochem. Sci. 20:402-405. [DOI] [PubMed] [Google Scholar]
- 21.Le Page, F., E. E. Kwoh, A. Avrutskaya, A. Gentil, S. A. Leadon, A. Sarasin, and P. K. Cooper. 2000. Transcription-coupled repair of 8-oxoguanine: requirement for XPG, TFIIH, and CSB and implications for Cockayne syndrome. Cell 101:159-171. [DOI] [PubMed] [Google Scholar]
- 22.Lyamichev, V., M. A. D. Brow, and J. E. Dahlberg. 1993. Structure-specific endonucleolytic cleavage of nucleic acids by eubacterial polymerases. Science 260:778-783. [DOI] [PubMed] [Google Scholar]
- 23.Masutani, C., R. Kusumoto, A. Yamada, N. Dohmae, M. Yokoi, M. Yuasa, M. Araki, S. Iwai, K. Takio, and F. Hanaoka. 1999. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase η. Nature 399:700-704. [DOI] [PubMed] [Google Scholar]
- 24.Matsunaga, T., D. Mu, C. H. Park, J. T. Reardon, and A. Sancar. 1995. Human DNA repair excision nuclease: analysis of the roles of the subunits involved in dual incision by using anti-XPG and anti-ERCC1 antibodies. J. Biol. Chem. 270:20862-20869. [DOI] [PubMed] [Google Scholar]
- 25.Matsunaga, T., C. H. Park, T. Bessho, D. Mu, and A. Sancar. 1996. Replication protein A confers structure-specific endonuclease activities to the XPF-ERCC1 and XPG subunits of human DNA repair excision nuclease. J. Biol. Chem. 271:11047-11050. [DOI] [PubMed] [Google Scholar]
- 26.Mori, T., M. Nakane, T. Hattori, T. Matsunaga, M. Ihara, and O. Nikaido. 1991. Simultaneous establishment of monoclonal antibodies specific for either cyclobutane pyrimidine dimmer or (6-4)photoproduct from the same mouse immunized with ultraviolet-irradiated DNA. Photochem. Photobiol. 54:225-232. [DOI] [PubMed] [Google Scholar]
- 27.Moriwaki, S.-I., M. Stefanini, A. R. Lehmann, J. H. J. Hoeijmakers, J. H. Robins, I. Rapin, E. Botta, B. Tanganelli, W. Vermeulen, B. C. Broughton, and K. H. Kraemer. 1996. DNA repair and ultraviolet mutagenesis in cells from a new patient with xeroderma pigmentosum group G and Cockayne syndrome resemble xeroderma pigmentosum cells. J. Investig. Dermatol. 107:647-653. [DOI] [PubMed] [Google Scholar]
- 28.Mu, D., D. S. Hsu, and A. Sancar. 1996. Replication mechanism of human DNA repair excision nuclease. J. Biol. Chem. 271:8285-8294. [DOI] [PubMed] [Google Scholar]
- 29.Nance, M. A., and S. A. Berry. 1992. Cockayne syndrome: review of 140 cases. Am. J. Med. Genet. 42:68-84. [DOI] [PubMed] [Google Scholar]
- 30.Nouspikel, T., P. Lalle, S. A. Leadon, P. K. Cooper, and S. G. Clarkson. 1997. A common mutational pattern in Cockayne syndrome patients from xeroderma pigmentosum group G: implications for a second XPG function. Proc. Natl. Acad. Sci. USA 94:3116-3121. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31.O'Donnovan, A., A. A. Davis, J. G. Moggs, S. C. West, and R. D. Wood. 1994. XPG endonuclease makes the 3′ incision in human DNA nucleotide excision repair. Nature 371:432-435. [DOI] [PubMed] [Google Scholar]
- 32.Oui, H., E. Park, L. Prakash, and S. Prakash. 1993. The Saccharomyces cerevisiae DNA repair gene RAD25 is required for transcription by RNA polymerase II. Genes Dev. 7:2161-2171. [DOI] [PubMed] [Google Scholar]
- 33.Shiomi, T., Y.-N. Harada, T. Saito, N. Shiomi, Y. Okuno, and M. Yamaizumi. 1994. An ERCC5 gene with homology to yeast RAD2 is involved in group G xeroderma pigmentosum. Mutat. Res. 314:167-175. [DOI] [PubMed] [Google Scholar]
- 34.Thompson, L. H. 1998. Nucleotide excision repair: its relation to human disease, p. 335-393. In J. A. Nickoloff and M. F. Hoekstra (ed.), DNA damage and repair: DNA repair in higher eukaryotes. Humana Press, Totowa, N.J.
- 35.Tirode, F., D. Busso, F. Coin, and J. M. Egly. 1999. Reconstitution of the transcription factor TFIIH: assignment of functions for the three enzymatic subunit, XPB, XPD, and cdk7. Mol. Cell 3:87-95. [DOI] [PubMed] [Google Scholar]
- 36.Van Gool, A. J., G. T. J. van der Horst, E. Citterio, and J. H. J. Hoeijimakers. 1997. Cockayne syndrome: defective repair of transcription? EMBO J. 16:4155-4162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vermeulen, W., J. Jaeken, N. G. Jaspers, D. Bootsma, and J. H. J. Hoeijmakers. 1993. Xeroderma pigmentosum complementation group G associated with Cockayne syndrome. Am. J. Hum. Genet. 53:185-192. [PMC free article] [PubMed] [Google Scholar]
- 38.Vermeulen, W., R. J. Scott, S. Rodgers, H. J. Muller, J. Cole, C. F. Arlett, W. J. Kleijer, D. Bootsma, J. H. J. Hoeijmakers, and G. Weeda. 1994. Clinical heterogeneity within xeroderma pigmentosum associated with mutations in the DNA repair and transcription gene ERCC3. Am. J. Hum. Genet. 54:191-200. [PMC free article] [PubMed] [Google Scholar]
- 39.Wakasugi, M., J. T. Reardon, and A. Sancar. 1997. The noncatalytic function of XPG protein during dual incision in human nucleotide excision repair. J. Biol. Chem. 272:16030-16034. [DOI] [PubMed] [Google Scholar]