

Abstract
A substantial number of new chemical entities and marketed drugs show poor solubility characteristics and amorphisation is one of the favorable approaches to enhance solubility characteristics of such poorly soluble drugs. Formulation efforts in the present study were devoted to investigate amorphisation of a model poorly soluble drug, atorvastatin calcium by molecular complexation with anion exchange resin, Duolite®AP 143/1093 and hence enhancement in its solubility characteristics. Drug resinates in 1:1, 1:2, and 1:4 weight ratios were prepared by simple batch operation and subsequently studied for drug content, residual solvent content, molecular interactions, solid state characterisation and solubility characteristics. During initial characterisation, all the proportions of drug resinates, except 1:1 proportion showed partial amorphisation of the drug, whereas 1:1 proportion showed complete amorphisation of the drug. This proportion reported distinctly enhanced solubility characteristics over pure drug and other proportions. Such amorphisation and solubility enhancement could be attributed to the binding of individual drug molecules to the functional sites of the resin molecules, either partially or completely, resulting in reduction of crystal lattice energy, a main barrier to dissolution. Hydrophilic nature of ion exchange resin matrices also assisted in enhancing dissolution of the drug from the resinates. During accelerated stability study, there was an insignificant decrease in solubility characteristics of the drug and its amorphous form was also found to be stable in 1:1 proportion. Atorvastatin resinates formed in 1:1 weight ratio were in stoichiometric proportion and such drug resinates in stoichiometric proportion showed to have tremendous potential in conversion of crystalline form of drug substances to its amorphous form and subsequent stabilisation. It hence proved to be a very effective, yet simple approach for improving solubility characteristics of poorly soluble actives.
Keywords: Amorphisation, anion exchange resin, atorvastatin calcium, dissolution rate enhancement, poorly water soluble drug, solubility enhancement, stability study
Atorvastatin (ATV), chemically designated as [R-(R*, R*)]-2-(4-fluorophenyl)-β,δ-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino) carbonyl]-1H-pyrrole-1-heptanoic acid is a selective, competitive inhibitor of HMG-CoA reductase. The free acid is prone to lactonisation and hence ATV is prepared as its calcium salt so as to ensure its chemical stability for extended periods of time and to be conveniently formulated in the dosage forms for oral administration[1]. However, as per biopharmaceutical classification system (BCS), ATV calcium belongs to Class II category and has an aqueous solubility of about 100 μg/ml[2]; exhibiting dissolution rate limited oral bioavailability. In order to improve solubility and/or dissolution rate of the drug, strategies like particle size reduction and amorphisation were explored[3,4,5]. Though the abovementioned approaches were appealing in enhancing solubility of the drug, its amorphous form, in fact, tends to revert to the stable polymorph during storage because of its inherent thermodynamic properties and the studies were not found to be effective on stabilisation of metastable amorphous form of the processed drug, which would ensure unaltered solubility characteristics over the period of its storage. Hence, many such poorly soluble drugs pose difficulties to the researchers while formulating them into thermodynamically stable and therapeutically effective dosage forms.
Variety of excipients such as polymers[6,7], plasticizers[6,8], complexing agents[9,10], and ion exchange resins[11,12,13] have been used satisfactorily to enhance solubility of poorly water soluble drugs, out of which formation of drug resinates is the simplest approach. Shukla et al.[14] and Madgulkar et al.[15] reported conversion of risperidone and tramadol hydrochloride, respectively to an amorphous form by monomolecular entrapment within Amberlite® IRP64 and Tulsion 335 resins, respectively. Pisal et al.[16] and Shah et al.[17] reported enhanced solubility characteristics of ciprofloxacin and ondansetron, respectively, as a result of crystal habit modification by forming resinates with Indion grades. In spite of such proven potential of ion exchange resins to enhance solubility characteristics, a little emphasis has been given by the researchers to implement the strategy of formation of drug resinates. Thus, the present study aims at exploring least adopted, but equally appealing approach of solubility enhancement of ATV as a classic model of poorly water soluble drug by formulating thermodynamically stabilised amorphous drug resinates with anion exchange resin, Duolite®AP 143/1093.
MATERIALS AND METHODS
Atorvastatin calcium (ATV) was obtained as a gift sample from Get-Rid Pharmaceuticals, Pune, India. Cholestyramine resin USP, a strongly basic, anion exchange resin as Duolite®AP 143/1093 was a generous gift from Rohm and Haas France S.A.S., Chauny, France. This grade of resin is nontoxic and safe for oral administration and a DMF has been maintained with US FDA[18]. All other chemicals and solvents used were of analytical reagent grade.
Selection of ion exchange resins:
ATV is a polar drug molecule having weakly acidic nature. Hence, a strong base is required to form a salt (strong electrolyte) of a weak acid, which would dissociate into an acid and a base as a result of hydrolysis in the dissolution fluid. Such dissociation is governed by the H+ ion concentration of the dissolution fluid and is favored optimally at neutral pH. However, the physiological fluid, where the drug is supposed to be released rapidly, is acidic in nature (pH for GI fluid is 2-5). Hence, a strongly basic anion exchange resin, which would dissociate ions virtually at any pH was predicted to be suitable. It was also reported that strongly basic anion exchange resin gives a rapid rate of ion exchange facilitating rapid drug release[19]. Hence, a strongly basic anion exchange resin having exchangeable Cl− ions or OH− ions (after conditioning), which can be replaced with an anionic species. Out of the available varieties of anion exchange resins in the market, Duolite®, a reputed brand of resin from Rohm and Haas Company was selected. Low molecular weight of ATV (about 1157.37) is not a constraint during loading into cholestyramine resin (Duolite® AP143/1093 with ion exchange capacity of 1.8-2.2 g/g). Also, lipid lowering property of cholestyramine resins synergistically improves therapeutic efficacy of lipid lowering active pharmaceutical ingredient (API). Complete transfer of the drug from the drug solution is not possible in a single equilibrium stage and due to very fine particle size of the resins, batch technique of resinates preparation is adopted, which allows prolonged equilibration time between the resin substrate and drug molecules.
Conditioning of the resin:
Conditioned resin (DUO) was obtained at room temperature by uniformly dispersing the resin in sufficient quantity of NaOH solution so as to obtain hydroxide form (OH− counter ion) of the resin. The concentration of NaOH solution was changed in a gradient manner that is, from 0.01-1 N NaOH over the period of 4 h. The dispersion was filtered and the residue was washed thoroughly with sufficient quantity of hot purified water (heated to 70-75°). The residue was then dried thoroughly at about 55-60° to constant weight.
Drug loading in the conditioned resins-Batch technique:
5% w/v slurry of the resin was prepared by uniformly dispersing DUO in methanol. The dispersion was stirred for about 45 min, to allow uniform swelling of the resin polymer structure. Required amount of ATV as per predetermined ratios was added to this slurry under stirring over the period of 1 h. Then the dispersion was kept on stirring for 2 h to ensure complete complexation of the drug with the resin. The drug resinates so formed were filtered and washed with methanol. The resinates were dried at room temperature to remove residual solvent content.
Initial characterisation:
ATV, conditioned DUO (to obtain characterisation of the resinates on comparable grounds) and all the proportions of resinates of ATV with DUO (AD) were subjected to initial characterisation within 15 days of preparation, based on the following parameters.
Drug entrapment efficiency:
It was estimated by dispersing 50 mg of AD in 100 ml of 0.1 N HCl solution under continuous ultrasonication. Each of these solutions was further diluted with methanol and the absorbance was measured at λmax.
Residual solvent content:
Residual methanol content of the resinates after drug loading was determined using moisture analyzer (ML-50 Moisture Analyzer, A and D Instruments Ltd., UK) and evaluated according to the ICH guidelines on impurities for residual solvents. An accurately weighed amount of the sample was heated to 75° for 10 min and % LOD was reported.
Fourier transform infrared spectroscopy:
Fourier transform infrared (FT-IR) spectra of ATV and all the proportions of AD were obtained using a FT-IR spectrometer (8400 S, Shimadzu Corporation, Japan). About 2-3 mg of the samples was finely ground with dry KBr and the spectra were scanned over wave number range of 4000-450 cm−1.
Differential scanning calorimetry:
Differential scanning calorimetry (DSC) thermograms of ATV and all the proportions of AD were obtained using differential scanning calorimeter (Mettler Toledo 821e, Mettler Toledo, Switzerland) operated with Star® software (version 9.01). The instrument was equipped with an intra cooler. Accurately weighed quantities of samples (3-5 mg) were analysed in hermetically sealed, pin holed aluminium crucibles. The samples were heated at a constant rate at 10°/min over a temperature range of 30-300°. An inert atmosphere was maintained by purging nitrogen gas at flow rate of 40 ml/min. The instrument was calibrated for temperature and heat flow using indium and zinc standards, respectively. The percent crystallinity was then determined using the following equation: Percent crystallinity=[(ΔHm-ΔHc)/ΔHm°]×100% (1). In Eq. 1, the heat of melting (ΔHm), heat of cold crystallisation (ΔHc) were determined by integrating the areas under the endotherm and represented in terms of J/g. Depending upon the thermal history of a sample, a cold crystallisation exothermic peak may or may not be observed during thermal characterisation. ΔHm0 is a reference value and represents the heat of melting derived from thermal characterisation of the pure, unprocessed drug.
X-Ray Powder Diffraction (XRPD):
X-Ray powder diffraction (XRPD) profiles of ATV and all the proportions of AD were recorded on X-ray diffractometer (Bruker AXS-D8 Advance, Germany). The samples were irradiated with monochromatic Cu-K radiation (1.542 A°) and analysed between 2 and 45° (2θ). The voltage and current of 40 kV and 40 mA were used, respectively. The range and chart speed was 5×103 CPS and 10 mm/° (2θ), respectively.
Saturation Solubility:
To evaluate an increase in solubility of ATV from all the proportions of AD, saturation solubility measurements were carried out. The known excess of ATV and drug resinates were added to 10 ml of 0.1 N HCl solution. The samples were rotated at 80 rpm for 72 h at temperature 37.0±0.5° using an orbital shaking incubator (CIS-24BL, Remi, India). The samples were then filtered, suitably diluted with methanol and analysed spectrophotometrically at λmax.
Dissolution study:
An analytical method to study dissolution rate of pure drug and ATV from all the proportions of AD was developed and validated. It was performed using US. Pharmacopoeia XXXII, Type II Dissolution Test Apparatus (DA-6D, Electrolab, India). The samples equivalent to 10 mg of ATV were placed in the dissolution vessels containing 500 ml of 0.1 N HCl solution maintained at 37.0±0.5° and stirred at 75 rpm±4%. The aliquots of suitable volume (5 ml) were collected at predetermined intervals of time and replaced immediately with equal volumes of fresh dissolution medium, maintained at the same temperature. After filtration, each of the collected aliquots was suitably diluted with methanol and analysed spectrophotometrically at λmax. The data were studied using PCP-Disso software.
Accelerated stability study:
Accelerated stability study of the optimised proportion of AD was carried out at three time points, including initial and final testing time points. Molecular interactions, solid state characterisation, and solubility characteristics of ATV in optimised proportion of AD was observed over the period of 6 months, during its storage at 40±2° and 75±5% RH.
RESULTS AND DISCUSSION
The method of formation of AD was found to be suitable for its use on commercial scale, because of the simplicity in the process, lesser processing steps, use of simple equipments in its manufacturing and higher process yields. Utilisation of time and excess of reagents are of minor importance as compared to completeness of the ion exchange process.
Theoretical quantity of ATV which can be complexed with the resin was anticipated taking into consideration the exchange capacity of the resin and equivalent weight of ATV. Practical determinations (Table 1) showed maximum entrapment efficiency for 1:1 proportion of AD. The binding affinity between two substances could also be predicted from the extent of solubility alteration[20]. The maximum administered daily mass of all the proportions of the drug resinates meet the option 1 limit specified for class 2 solvent (methanol)[21,22].
TABLE 1.
ENTRAPMENT EFFICIENCY OF ALL THE PROPORTIONS OF ATORVASTATIN RESINATES

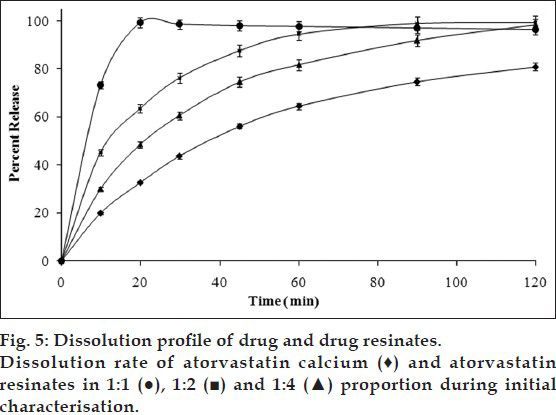
FT-IR spectrum of ATV (fig. 1) showed characteristic peaks at 3364.2 cm−1 (N-H stretching), 3222.8 cm−1 (asymmetric O-H stretching), 3055 cm−1 (symmetric O-H stretching), 1650.8 cm−1 (asymmetric C=O stretching), 1550-1460 cm−1 (four bands related to C-C stretching), 1318.1 cm−1 (CH3/CH2 deformation), 1243.1 cm−1 (C-N stretching) and 1216.9 (C-N/C=O stretching) corresponding to crystalline ATV. However, FT-IR spectra of all the proportions of AD (fig. 1) showed broad band with a shoulder at about 3431, 3330, and 3020 cm−1. Furthermore, stretching vibrations were observed at about 1666, 1515, 1555-1490, 1426, 1415, 1404, 1369, 1313, 1216, 1159, and 1138 cm−1. In addition, crystalline form of ATV showed a characteristic peak at 3670.5 cm−1 corresponding to free O-H stretching vibrations. It was supposed to have appeared because of trihydrate functionality. Such free O-H stretching vibration was found to be absent in FT-IR spectra of all the proportions of AD indicating the presence of an anhydrous form of ATV in the resinates, which also might contribute to enhance the solubility over hydrated form of pure drug. The principal bands in the spectra of resinates were weaker and less sharper compared to those observed in the spectra of ATV. Among the resinates, 1:1 proportion showed weakest principal peaks, whereas 1:4 proportion showed sharpest principal peaks. It provided a slight hint of degree of amorphisation on comparative basis. Similar observations were reported by Kim et al. in the characterisation of crystalline and amorphous form of ATV[3]. Though the intensity of principal bands characteristic of ATV and DUO was found to be lowered in spectra of AD, their positions were only slightly changed. Also, the bands observed in finger print region were similar in position to those observed in their individual spectrum. Thus, FT-IR study indicated that the overall symmetry of ATV molecule in all the proportions of AD was unchanged and it exhibited conversion of crystalline form of ATV molecules to amorphous form, which was further confirmed with the help of DSC and XRPD studies.
Fig. 1.
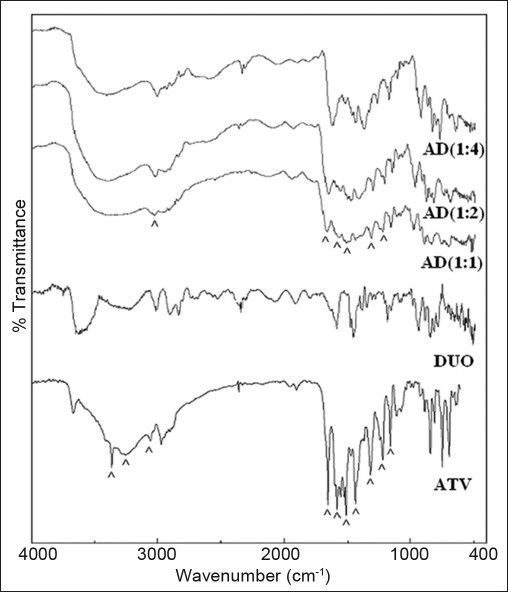
FT-IR spectra of drug, resin and drug resonates.
FT-IR spectrum of atorvastatin calcium (ATV), Duolite®AP 143/1093 (DUO) and all the proportions of atorvastatin resinates during initial characterisation (AD).
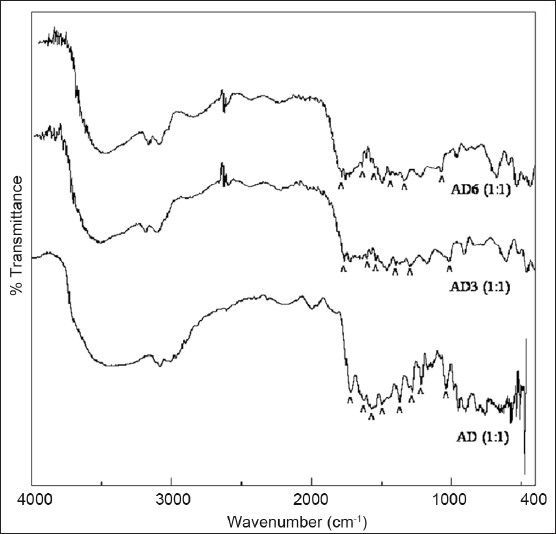
DSC analysis of ATV (fig. 2) showed an endotherm of enthalpy 18.7 J/g in the temperature range of 82-118° corresponding to the loss of loosely bound water. The second sharp endotherm at 149.4° indicated melting of ATV. It had a shoulder peak in the tailing end, which was ascribed to the molecular re-organisation occurring at lower heating rates[5]. This melting endotherm was followed by a significantly broadened endotherm at an onset temperature of 189.2° with enthalpy of 154.2 J/g. Though this endotherm was attributed to complete dehydration of calcium salt of ATV, it also suggested polymorphic impurity and hence served as a useful tool together with melting endotherm in prediction of degree of complexation and subsequent amorphisation, evident by the changes in Tg/peak temperature and changes in heat capacities. DSC thermogram of DUO (fig. 2) also showed a broad endotherm at 90.1° corresponding to the loss of loosely bound water. It was followed by a broad endotherm of enthalpy 138.5 J/g at 254.9° indicating partial amorphous nature of the resin. DSC thermogram of 1:1 proportion of AD (fig. 2) showed the absence of an endotherm at 149.4° corresponding to the melting of ATV. The broad endotherm at 243.3°, which was present in the thermogram of ATV was also found to be absent in 1:1 proportion of AD. It indicated that ATV had totally lost its crystalline nature through formation of drug resinates in 1:1 proportion. On the other hand, DSC thermogram of 1:2 proportion of AD (fig. 2) showed a Tg with an onset at 144° and a change in heat capacity of 33.6 J/g, followed by a broadened endotherm with an onset at 161.1° and an enthalpy of 45.3 J/g. DSC thermogram of 1:4 proportion of AD (fig. 2) also showed similar thermal events such as a Tg with an onset at 166.4° and a change in heat capacity of 33.6 J/g, followed by a broadened endotherm with an onset at 183.3° and an enthalpy of 34.9 J/g. The percent crystallinity calculated from a Tg was 49.58% and 76.76%, respectively. Thus, as evident by thermogram of these two proportions, reduction in Tg followed by an endotherm with decreased intensity and enthalpy suggested that there was only a partial conversion of crystalline ATV to its amorphous state in 1:2 and 1:4 proportions of AD. These predictions by thermal analysis about amorphisation were further confirmed with the help of XRPD study.
Fig. 2.
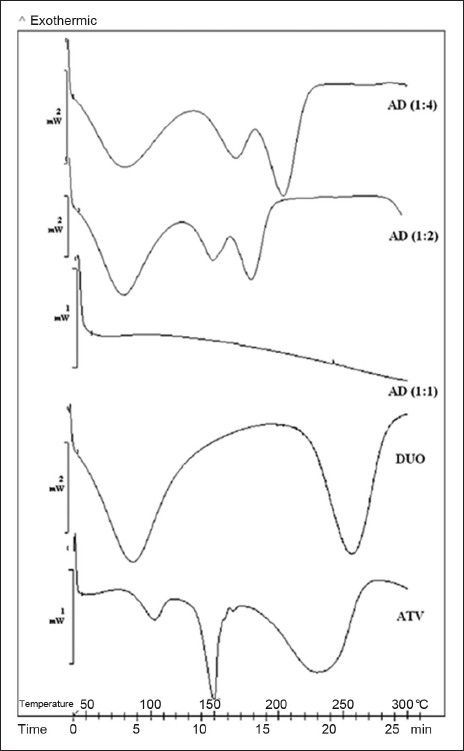
DSC thermograms of drug, resin and drug resonates.
DSC thermogram of atorvastatin calcium (ATV), Duolite®AP 143/1093 (DUO) and all the proportions of atorvastatin resinates during initial characterisation (AD).
XRPD profile of ATV (fig. 3) showed diffraction peaks at 2θ values of 8.9, 10.2, 12.1, 16.9, 18.2, 18.8, 19.4, 21.5, 23.2, 24.3, 26.3, and 29.1° corresponding to crystalline ATV form III[23]. It also showed additional peaks at 2θ values of 9.4, 11.7, 13.3, 22.6, 23.6, 28.2, 30.2, and 37.2°. These peaks were of low intensity and diffused. It hence supported the data obtained from DSC thermograms that ATV had poor crystallinity and was a mixture of polymorphic forms. XRPD profile of 1:1 proportion of AD (fig. 3) distinctly showed a halo pattern corresponding to the diffraction pattern of DUO (fig. 3) and the absence of principal diffraction peaks corresponding to crystalline ATV. These observations confirmed that complete loss of drug crystallinity, as suggested by DSC thermogram was not merely a thermal artifact caused during DSC heating cycle. It could be construed that the formation of drug resinates monomolecularly in 1:1 proportion consequently resulted in complete amorphisation of ATV complexed within macroreticular structure of DUO; causing reduction of crystal lattice energy, a main barrier to dissolution[15,17]. Whereas, it could be observed from the diffraction profile of 1:2 and 1:4 proportions of AD that though almost all the diffraction peaks characteristic of crystalline ATV were absent, a few diffraction peaks were observed at 12.3, 13.3, and 18.4° 2θ with much lower intensity and significant broadening. It supported the data obtained from thermal analysis of corresponding samples that there was only a partial conversion of crystalline ATV to its amorphous form in these proportions of drug resinates. Such a decrease in drug crystallinity was observed by Elgindy et al. also and it was explained by the presence of reciprocal interactions in the solid state between hosts−guest molecules[10] or by physical phenomenon of drug adsorption on the external surfaces of the microfine resin particles.
Fig. 3.
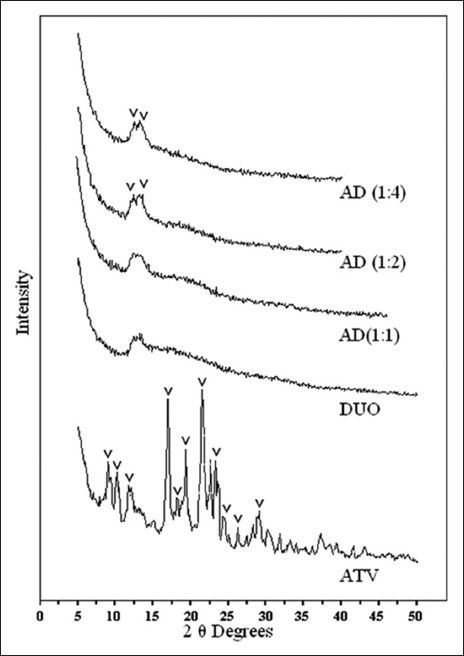
XRPD of drug, resin and drug resonates.
XRPD profile of atorvastatin calcium (ATV), Duolite®AP 143/1093 (DUO) and all the proportions of atorvastatin resinates during initial characterisation (AD).
Molecular resinate formation and subsequent drug release in the physiological system, as depicted in fig. 4 is a reversible equilibrium process, the position of which will depend on many factors including salt concentration in the aqueous phase[19,24]. OH−–N+–(R)3 represents a functional component attached to the styrene-divinyl benzene copolymer structural component of the conditioned strongly basic, anion exchange resin. N+ is an integral ion of the resin and OH− is its counter ion. Competitive affinity of the drug ion (API–COO−) toward resin ion favors a forward reaction and both, the substrate (ATV molecule) and ligand (DUO molecule) are noncovalently bound[20]. When the resinates reach the GI tract, swelling property and hydrophilic nature of ion exchange resin matrices allow aqueous fluids to enter the resinates structure and the exchange process is reverted. Free drug ions are dissociated from the resinates as a result of dilution and competitive displacement (binding of drug and/or ligand molecule to other competing agents, as gastrointestinal H+ and Cl− ions, respectively). This mechanism of liberation of free drug ions occurs at a very fast rate for strong ion exchange resins[19] and irrespective of the pH of surrounding medium[18]. Thus, all the proportions of AD reported quantitative (Table 2), very rapid (fig. 5) and hence predictable dissociation of ATV. The molecules of ATV in AD (1:1) were supposed to have complexed monomolecularly with the functional sites within a three dimensional resin matrix. Thus, it was obvious that such molecular complexation had disturbed strong crystal lattice of ATV molecules leading to the formation of amorphous solid with higher free energy, which was responsible for marked rise in its solubility (~3.5 folds) and dissolution rate (~100% drug release within initial 20 min)[25]. Moreover, such modification in crystal habit can affect the dissolution rate of the drug by a magnitude equivalent to particle size reduction[26] and the particle size of complexed ATV in 1:1 proportion was predicted to have reduced up to the molecular level, hence having distinguished effect on solubility improvement compared to that of pure drug and other two proportions of AD. The dissolution profile of the drug from 1:1 proportion of AD, as seen in fig. 5, showed an initial rapid dissolution and attainment of a peak concentration of the drug in dissolution medium. This rapid drug release was followed by a slight decline of drug concentration in dissolution medium to the solubility limit. Such dissolution pattern could be attributed to the formation of amorphous form of ATV in 1:1 proportion of AD containing only trace amounts of water in equilibrium with surrounding moisture. It has been reported that such forms dissolved more rapidly in water and in all cases yielded drug concentrations in solution substantially higher than the amorphous form containing moisture in equilibrium attained during dissolution[27]. Such observations were also reported by Lin et al. while studying the in situ crystalline transformation during intrinsic dissolution profile of an antihypertensive drug[23]. Amorphisation of ATV in 1:1 proportion AD was predicted by the characteristic DSC thermogram and XRPD profile during initial characterisation. It hence highlighted the influence of extent of complexation between ATV and DUO molecules and hence degree of amorphisation on dissolution. On the other hand, AD (1:2) and AD (1:4) observed lower saturation solubility (325.1 and 301.1 μg/ml, respectively) and dissolution rate (81.82 and 79.69% drug release, respectively within initial 60 min) compared to saturation solubility and dissolution rate of 1:1 proportion; as a result of the lower degree of crystallinity of ATV and its adsorption only on the surface of DUO molecules. Thus, 1:1 proportion of AD was optimised on the basis of maximum degree of amorphisation and improvement in solubility characteristics of ATV from the resinates and was subjected to accelerated stability study.
Fig. 4.
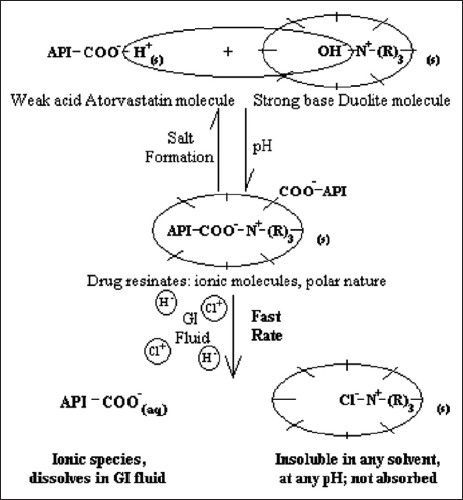
Mechanism of drug dissociation from atorvastatin resinates and in vivo proposition.
TABLE 2.
SATURATION SOLUBILITY OF DRUG AND DRUG RESINATES
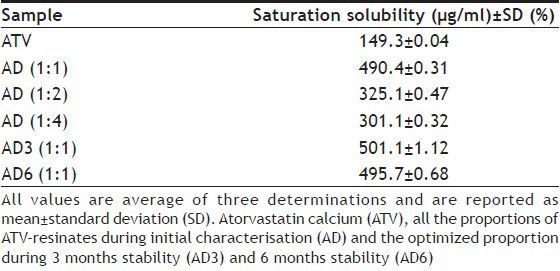
Fig. 5.
Quality attributes of 1:1 proportion of drug resinates that were supposed to be susceptible to change under the influence of environmental conditions of temperature and humidity, such as molecular interactions, amorphous nature, and solubility characteristics were found to be unaltered. Neither compaction nor change in physical appearance was observed. FT-IR spectrum (fig. 6) of the optimised proportion of drug resinates during 3 months stability study (AD3) and 6 months stability study (AD6) revealed that the position and intensity of the principal peaks was only slightly affected. 1:1 proportion of AD3 and AD6 did not involve any further interactions between ATV substrate and DUO ligand molecules during stability testing period. DSC thermogram of 1:1 proportion of AD3 and AD6 (fig. 7) also showed the absence of an endotherm at 149.4° corresponding to the melting of crystalline ATV. However, both of them showed a Tg with negligible change in heat capacity (34.6 J/g) at an onset temperature of about 80° as opposed to corresponding thermogram observed during initial characterisation. It was also followed by a broadened endotherm of enthalpy (51.6 J/g) at about 180°. This could be attributed to the loss of moisture, adsorbed by the resinates during storage at higher relative humidity and its complete dehydration during DSC run. Similar observations were reported by Ambike et al.[28]. Thus, the drug was predicted to have stabilised in its amorphous form in optimised proportion of AD and it was further confirmed with the help of XRPD study. XRPD profile of the optimised proportion of ATV resinates during stability study (fig. 8) showed a halo pattern identical to that of its initial profile. It neither showed appearance of any of the principal peaks characteristic of crystalline nature of pure drug. It was hence concluded that the formation of drug resinates enhanced physical stability of ATV in its metastable amorphous form by the immobilisation or prevention of crystal growth[29]. Such stabilisation of a substrate (drug molecule) may be the result of either a microsolvent effect or a conformational effect[20]. Such stabilisation of amorphous system only in 1:1 stoichiometry have contributed to unaltered saturation solubility (Table 2) of ATV from its resinates. 1:1 proportion of AD3 and AD6 showed different pattern of drug release between 20 and 30 min (fig. 9) compared to that of its initial profile. However, the time required for releasing 90% drug from the resinates was still comparable. Such difference in release pattern could be attributed to the formation of hydrates during accelerated stability study, though, interestingly, recrystallisation of complexed ATV was not observed.
Fig. 6.
FT-IR spectra of stability samples.
FT-IR spectrum of the optimized proportion of atorvastatin resinates during initial characterisation (AD), 3 months stability (AD3) and 6 moths stability (AD6).
Fig. 7.
DSC thermograms of stability samples.
DSC thermogram of the optimised proportion of atorvastatin resinates during initial characterisation (AD), 3 months stability (AD3) and 6 moths stability (AD6).
Fig. 8.
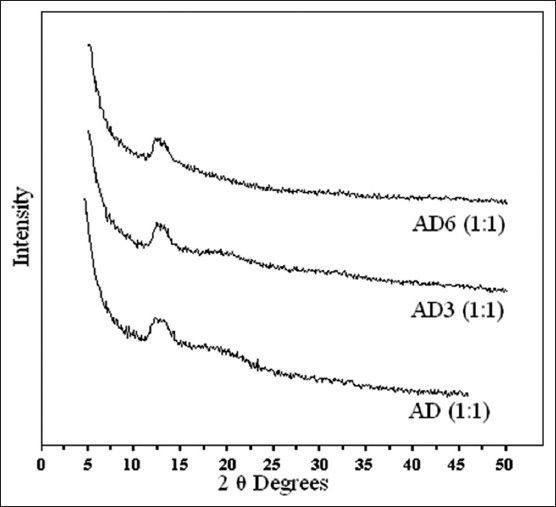
XRPD of Stability samples.
XRPD profile of the optimised proportion of atorvastatin resinates during initial characterisation (AD), 3 months stability (AD3) and 6 moths stability (AD6).
Fig. 9.
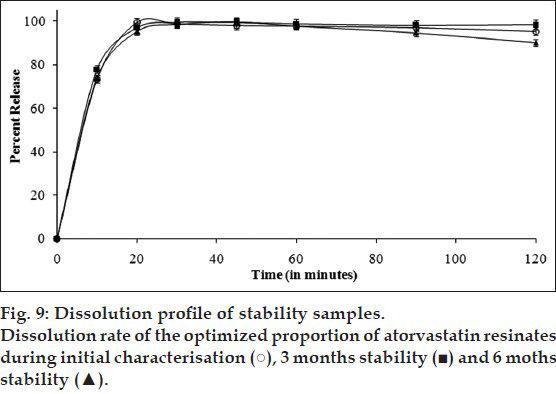
Thus, it can be concluded from the practical observations that all the proportions of the drug resinates showed enhanced solubility and faster dissolution rate of complexed ATV compared to its unprocessed form. It was attributed to reduced particle size and reduced agglomeration because of complexation within and/or adsorption on hydrophilic resin structure. In addition, FT-IR, DSC, and XRPD analysis during initial characterisation indicated that ATV existed as amorphous form typically in 1:1 stoichiometry in AD. Such conversion to amorphous form and molecular complexation of drug molecules had further contributed to better performance in its solubility and dissolution characteristics. Thus, AD (1:1) was optimised and further subjected to an accelerated stability study, which confirmed the stabilisation of complexed amorphous form ATV and an insignificant alteration in solubility characteristics over the period of its storage. Thus, initial characterisation and stability study carried out over the period of 6 months strongly suggested that formation of amorphous ATV resinates is a promising approach to improve its solubility characteristics and can be adopted at an industrial scale. It could further be generalised that the drug resinates may certainly provide an attractive approach for solubility enhancement of even very weakly acidic as well as basic, but ionisable poorly soluble drug molecules with low (preferably <1500) molecular weight; with appropriate consideration to ion exchange capacity of the resin, hydrophobicity/hydrophilicity of the drug molecules, pKa of the drug and resin and concentration of competing ions during equilibration process.
Footnotes
Kulthe and Chaudhari: Drug Resinates for Enhancing Solubility and Physical Stability
REFERENCES
- 1.Mckenzie A. Form III Crystalline [R-(R*, R*)]-2-(4-fluorophenyl)-β,δ-dihydroxy-5-(1-methylethyl)-3-phenyl-4-[(phenylamino) carbonyl]-1H-pyrrole-1-heptanoic acid calcium salt (2:1) US Patent 6,121,461. 2000 [Google Scholar]
- 2.Kasim N, Whitehouse M, Ramachandran C, Bermeja M, Lennerna H, Hussain A, et al. Molecular properties of who essential drugs and provisional biopharmaceutical classification. Mol Pharm. 2004;1:85–96. doi: 10.1021/mp034006h. [DOI] [PubMed] [Google Scholar]
- 3.Kim M, Jin S, Kim J, Park H, Song H, Neubert R, et al. Preparation, characterisation and in vivo evaluation of amorphous atorvastatin calcium nanoparticles using supercritical antisolvent (SAS) process. Eur J Pharm Biopharm. 2008;69:454–65. doi: 10.1016/j.ejpb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Arunkumar N, Deecaraman M, Rani C, Mohanraj K, Kumar K. Preparation and solid state characterisation of atorvastatin nanosuspensions for enhanced solubility and dissolution. Int J Pharm Tech Res. 2009;1:1725–30. [Google Scholar]
- 5.Shete G, Puri V, Kumar L, Bansal A. Solid state characterisation of commercial crystalline and amorphous atorvastatin calcium samples. AAPS PharmSciTech. 2010;11:598–609. doi: 10.1208/s12249-010-9419-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kulthe V, Chaudhari P. Solubility enhancement of etoricoxib by solid dispersions prepared by spray drying technique. Indian J Pharm Educ Res. 2011;45:248–58. [Google Scholar]
- 7.Hulsmann S, Backensfeld T, Keitel S, Bodmeier R. Melt extrusion: An alternative method for enhancing the dissolution rate of 17 β-estradiol hemihydrate. Eur J Pharm Biopharm. 2000;49:237–42. doi: 10.1016/s0939-6411(00)00077-1. [DOI] [PubMed] [Google Scholar]
- 8.Ghebremeskel A, Vemavarapu C, Lodaya M. Use of surfactants as plasticizers in preparing solid dispersions of poorly soluble api: Stability testing of selected solid dispersions. Pharm Res. 2006;23:1928–36. doi: 10.1007/s11095-006-9034-1. [DOI] [PubMed] [Google Scholar]
- 9.Loftsson T, Brewster M. Pharmaceutical applications of cyclodextrins. 1. Drug solubilisation and stabilisation. J Pharm Sci. 1996;85:1017–25. doi: 10.1021/js950534b. [DOI] [PubMed] [Google Scholar]
- 10.Elgindy N, Elkhodairy K, Molokhia A, Elzoghby A. Lyophilisation monophase technique for improvement of the physicochemical properties of an anticancer drug, flutamide. Eur J Pharm Biopharm. 2012;74:397–405. doi: 10.1016/j.ejpb.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 11.Bajpai SK, Bajpai M, Saxena S. Ion exchange resins in drug delivery. In: Sengupta AK, editor. Ion exchange and solvent extraction: A series of advances. New York: CRC Press; 2007. pp. 104–52. [Google Scholar]
- 12.Avari J, Bhalekar M. Cation exchange resins for taste masking and rapid dissolution of sparfloxacin. Indian Drugs. 2003;41:19–23. [Google Scholar]
- 13.Irwin W, Mchale R, Watts P. Drug delivery by ion exchange. Part VII: Releases of acidic drugs from anionic exchange resinate complexes. Drug Dev Ind Pharm. 1990;16:883–98. [Google Scholar]
- 14.Shukla D, Chakraborty S, Singh S, Mishra B. Fabrication and evaluation of taste masked resinate of risperidone and its orally disintegrating tablets. Chem Pharm Bull. 2009;57:337–45. doi: 10.1248/cpb.57.337. [DOI] [PubMed] [Google Scholar]
- 15.Madgulkar A, Bhalekar M, Padalkar R. Formulation design and optimisation of novel tatste masked mouth-dissolving tablets of tramadol having adequate mechanical strength. AAPS PharmSciTech. 2009;10:574–81. doi: 10.1208/s12249-009-9237-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pisal S, Zainnuddin R, Nalawade P, Mahadik K, Kadam S. Molecular properties of ciprofloxacin-Indion 234 complexes. AAPS PharmSciTech. 2004;5:1–8. doi: 10.1208/pt050462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shah S, Pandya S, Bhalekar M. Molecular properties and evaluation of Indion 234-ondansetron resinates. J Young Pharm. 2010;2:247–51. doi: 10.4103/0975-1483.66799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rohm and Haas Research Laboratories-Spring House. Duolite® AP143/1093 [Online] [Last cited on 2010 May 8]. Available from: http://www.dow.com/assets/attachments/business/process_chemicals/amberlite_and_duolite_pharmaceutical_grade_resins/duolite_ap143-1093/tds/doulite_ap143-1093.pdf .
- 19.Anand V, Kandarapu R, Garg S. Ion-exchange resins: Carrying drug delivery forward. Drug Discov Today. 2001;6:905–14. doi: 10.1016/s1359-6446(01)01922-5. [DOI] [PubMed] [Google Scholar]
- 20.Tong WQ, Wen H. Applications of complexation in the formulation of insoluble compounds. In: Liu R, editor. Water-insoluble drug formulation. 2nd ed. New York: CRC Press; 2008. pp. 33–59. [Google Scholar]
- 21.International Conference on Harmonisation; Guidance on (Q3C) R5 Specifications: Impurities: Guideline for Residual Solvents [Online] [Last cited on 2011 Dec 8]. Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q3C/Step4/Q3C_R5_Step4.pdf .
- 22.Asian edition. Rockville MD: United States Pharmacopoeial Convention Inc; 2009. The United States Pharmacopoeia XXXII. [Google Scholar]
- 23.Lin S, Lachman L. In situ crystalline transformation of a new antihypertensive determined by photomicrographic and dissolution methods. J Pharm Sci. 1969;58:377–9. doi: 10.1002/jps.2600580325. [DOI] [PubMed] [Google Scholar]
- 24.Rohm and Haas Research Laboratories-Spring House. Ion Exchange Resins: Unique Solutions to Formulation Problems. [Last cited on 2010 May 8]. Available from: http://www.theartofformulation.com/Formulations_doc/unique_solutions.pdf .
- 25.Chowdary K, Madhavi B. Novel drug delivery technologies for insoluble drugs. Indian Drugs. 2005;42:557–64. [Google Scholar]
- 26.Myrdal PB, Jozwiakowski MJ. Alteration of the solid state of the drug substances: Polymorphs, solvates, and amorphous forms. In: Liu R, editor. Water-insoluble drug formulation. 2nd ed. New York: CRC Press; 2008. pp. 531–66. [Google Scholar]
- 27.Stavchansky SA, McGinity JW. Bioavailability in tablet technology. In: Lieberman HA, Lachman L, Schwartz JB, editors. Pharmaceutical dosage forms: Tablets. 2nd ed. New York: Marcel Dekker Inc; 2005. pp. 349–569. [Google Scholar]
- 28.Ambike A, Mahadik K, Paradkar A. Stability study of amorphous valdecoxib. Int J Pharm. 2004;282:151–62. doi: 10.1016/j.ijpharm.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Widjaja E, Kanaujia P, Lau G, Ng W, Garland M, Saal C, et al. Detection of trace crystallinity in an amorphous system using raman microscopy and chemometric analysis. Eur J Pharm Sci. 2011;42:45–54. doi: 10.1016/j.ejps.2010.10.004. [DOI] [PubMed] [Google Scholar]