

Abstract
Ect2, a Rho guanine nucleotide exchange factor (RhoGEF), is atypical among RhoGEFs in its predominantly nuclear localization in interphase cells. One current model suggests that Ect2 mislocalization drives cellular transformation by promoting aberrant activation of cytoplasmic Rho family GTPase substrates. However, in ovarian cancers, where Ect2 is both amplified and overexpressed at the mRNA level, we observed that the protein is highly expressed and predominantly nuclear and that nuclear but not cytoplasmic Ect2 increases with advanced disease. Knockdown of Ect2 in ovarian cancer cell lines impaired their anchorage-independent growth without affecting their growth on plastic. Restoration of Ect2 expression rescued the anchorage-independent growth defect, but not if either the DH catalytic domain or the nuclear localization sequences of Ect2 were mutated. These results suggested a novel mechanism whereby Ect2 could drive transformation in ovarian cancer cells by acting as a RhoGEF specifically within the nucleus. Interestingly, Ect2 had an intrinsically distinct GTPase specificity profile in the nucleus versus the cytoplasm. Nuclear Ect2 bound preferentially to Rac1, while cytoplasmic Ect2 bound to RhoA but not Rac. Consistent with nuclear activation of endogenous Rac, Ect2 overexpression was sufficient to recruit Rac effectors to the nucleus, a process that required a functional Ect2 catalytic domain. Furthermore, expression of active nuclearly targeted Rac1 rescued the defect in transformed growth caused by Ect2 knockdown. Our work suggests a novel mechanism of Ect2-driven transformation, identifies subcellular localization as a regulator of GEF specificity, and implicates activation of nuclear Rac1 in cellular transformation.
Keywords: Ect2, RhoGEF, Rac, ovarian cancer
Introduction
Rho family GTPases are molecular switches that act as signaling nodes to integrate extracellular signals and propagate intracellular signals. They control many normal cellular processes, including actomyosin remodeling, cell polarity, gene expression, and cell cycle progression.1,2 The best-studied members of this family are RhoA, Rac1, and Cdc42. These Rho GTPases are frequently overexpressed or dysregulated in tumors.3-5 Aberrant Rho GTPase activity regulates transformation, invasion, metastasis, and angiogenesis.3-5
GDP-bound Rho proteins are in the off-state, whereas GTP-bound proteins are in the on-state, in which they bind to their effector targets and transmit downstream signals. Guanine nucleotide exchange factors (GEFs) catalyze release of guanine nucleotides from GTPases, thereby enhancing binding of the more abundant cellular GTP.6 While some GEFs activate only specific GTPases, others are more promiscuous.
Ect2 (epithelial cell transforming sequence 2) is a Rho family GEF capable of activating RhoA, Rac1, and Cdc42 in vitro,7 but in cellulo it is more selective, in a context-dependent manner.8-14 Atypically for RhoGEFs, Ect2 contains 2 nuclear localization signals (NLSs) and has a prominent nuclear localization in interphase cells.7 In contrast, Rho proteins are found outside the nucleus.5 It has been proposed that Ect2 is auto-inhibited and sequestered from Rho GTPases in the nucleus of normal interphase cells, but becomes mislocalized to the cytoplasm in tumor cells, where its auto-inhibition is lost, and where it then activates Rho family GTPases to drive transformation.8,15 However, the subcellular localization of Ect2/Rho GTPase interactions has never been directly investigated in tumor cells.
Here we used ovarian tumor cells to further examine the role of Ect2 in transformation. Aberrant Rho GTPase activity has been implicated in this tumor type.16-19 Ect2 is located on chromosome 3q26.1-26.2,20 a common amplicon in ovarian tumors21,22; indeed, ovarian cancer has the second highest frequency of Ect2 amplification among human cancers to date.23 Ect2 is also overexpressed at the mRNA level.22,24,25 However, the protein expression of Ect2 and its functional consequences have not been studied in ovarian tumors. We examined a patient tumor tissue microarray (TMA) and observed that Ect2 protein was strongly expressed predominantly in the nucleus of ovarian cancer cells. We have identified a requirement for Ect2 in ovarian cancer cell transformation, and a novel mechanism whereby Ect2 can activate Rac1 and can drive ovarian tumor cell transformation from within the nucleus.
Results
Nuclear localization of Ect2 correlates with advanced disease in human serous epithelial ovarian cancers
Protein expression and subcellular distribution of Ect2 in ovarian cancers has not been evaluated previously. We evaluated these properties in a previously validated ovarian TMA26-29 containing approximately 400 full-faced cores from ovarian tumors and non-matched normal ovarian cysts. We optimized the immunohistochemical protocol such that <5% of OVCAR8 cells with Ect2 knockdown stained positive for Ect2, using an Ect2 antibody that we had previously validated for specificity by immunoblot analysis (Suppl. Fig. S1A and S1B). Nuclear and cytoplasmic Ect2 expressions were scored independently for each core. Unexpectedly, we found that higher scores for nuclear expression correlated with more advanced serous epithelial tumors (P = 0.0001516; Fig. 1A), whereas those for cytoplasmic expression correlated with less advanced tumors (P = 0.0007163; Fig. 1B). Indeed, in serous cysts Ect2 was expressed at low levels in the cytoplasm but undetectable in the nucleus, whereas for most cells in advanced tumors, Ect2 was concentrated in nuclei (Fig. 1C). These results suggested that nuclear rather than cytoplasmic localization of Ect2 may be important for its oncogenic functions in this tumor type.
Figure 1.

Nuclear localization of Ect2 correlates with advanced disease in human serous epithelial ovarian cancers. Ect2 subcellular localization was analyzed by immunohistochemistry (IHC) using a well-validated26-29 ovarian tissue microarray (TMA). Tumor cores were stained with Ect2 antibody (1:350; Millipore, Billerica, MA) and scored for percentage cells stained as well as intensity of staining in each location. Separate nuclear scores and cytoplasmic scores were calculated as described in Materials and Methods and binned according to disease severity. Quantification of Ect2 localization showed (A) significantly greater nuclear expression (P = 0.0001516) and (B) significantly lower cytoplasmic expression (P = 0.0007163) in advanced disease. SBT = serous borderline tumor. Box and whisker plots include horizontal lines corresponding to median, first and third quartiles (hinges), and extreme points excluding outliers (whiskers). (C) Representative images of a benign serous cyst, a serous borderline tumor, a low-grade and a high-grade serous epithelial tumor stained for Ect2 illustrate increased nuclear staining upon disease progression. Scale bars represent 10 µm.
Ect2 is expressed predominantly in the nucleus of ovarian cancer cell lines
We turned to cell lines to study the localization and function of Ect2 in ovarian tumor cells directly. Ect2 was easily detected by immunoblotting in all 8 ovarian cancer cell lines tested (Fig. 2A). To assess its subcellular localization, we performed immunofluorescence, staining for endogenous Ect2, co-staining for DAPI as a nuclear marker, and imaging the cells using confocal microscopy. Specificity of the Ect2 antibody immunofluorescence signal was confirmed using Ect2 knockdown cells (Suppl. Fig. S1C). We observed a direct overlay of the vast majority of the Ect2 and DAPI signals in all cell lines examined (Fig. 2B and data not shown), indicating that Ect2 is predominantly nuclear, and supporting our observation from the TMA that nuclear Ect2 may be more relevant than cytoplasmic Ect2 for ovarian cancer cells.
Figure 2.
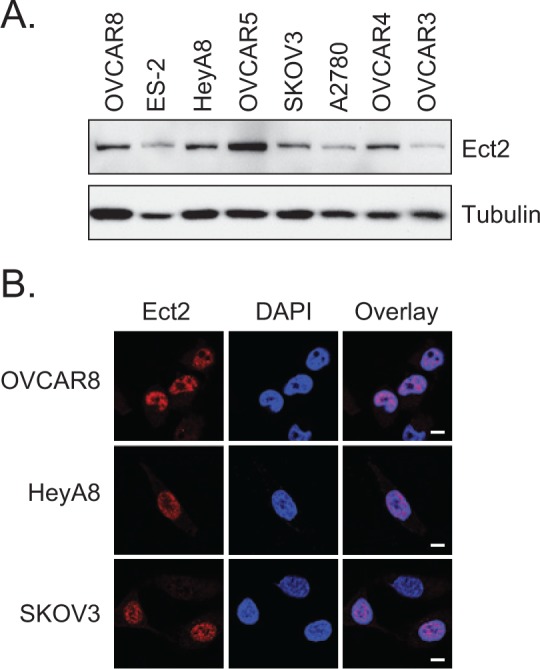
Ect2 is expressed predominantly in the nucleus of ovarian cancer cell lines. (A) Ect2 is highly expressed in ovarian cancer cell lines. Cell lysates from a panel of 8 human ovarian cancer cell lines were probed by immunoblotting with the anti-Ect2 antibody (1:1,000, n = 4). Equal amounts of cell lysate were loaded onto the gel; tubulin (1:1,000, clone DM1A, Sigma-Aldrich, St Louis, MO) served as a loading control. (B) Ect2 is predominantly nuclear in established ovarian cancer cell lines. Cells were stained with anti-Ect2 antibody (1:1,000) and the subcellular distribution of endogenous Ect2 was visualized by confocal immunofluorescence microscopy to detect the Alexafluor 594-coupled secondary antibody. Nuclei were stained with DAPI. Predominantly nuclear staining was seen across cell lines (images representative of n ≥ 4). Scale bars represent 10 µm.
Ect2 is required for transformed growth of ovarian cancer cell lines
Anchorage-independent growth is a hallmark of the transformed phenotype and is classically measured by soft agar colony formation.30 To determine the requirement for Ect2, we stably knocked down Ect2 using 2 independent shRNAs in cell lines that grow vigorously in an anchorage-independent manner. Both shRNAs robustly decreased expression of Ect2 in all cell lines, with Ect2 shRNA#3 showing more complete knockdown than shRNA#2 (98% and 84%, respectively, in OVCAR8 cells) (Fig. 3A). We then compared anchorage-independent growth of cells expressing non-targeted (NT) shRNA or Ect2 shRNA. Knockdown of Ect2 decreased soft agar colony formation of multiple cell lines, with the most striking effects seen in OVCAR8 cells (Fig. 3B, *P < 0.05). Thus, Ect2 is necessary for anchorage-independent growth of multiple ovarian cancer cell lines. We selected OVCAR8 for further study.
Figure 3.

Ect2 is required for transformed growth of ovarian cancer cell lines. Our ovarian cancer cell lines that grow robustly in an anchorage-independent manner were evaluated for the importance of Ect2 in transformed growth. Cells were infected with lentiviruses expressing 1 of 2 independent shRNAs directed against Ect2, or non-targeting (NT) shRNA. Infected cells were selected in puromycin and colonies were pooled for further use. (A) Ect2 knockdown using 2 independent shRNAs was confirmed by immunoblotting. Equal amounts of cell lysate were loaded onto the gel; actin (1:30,000, clone AC-74, Sigma-Aldrich) served as a loading control (n ≥ 3). Percent knockdown shown is based on densitometry performed on each Ect2 blot and normalized to actin. (B) Ect2 knockdown impairs anchorage-independent growth. Cells with or without Ect2 knockdown were grown in soft agar, and colonies were counted after 2 to 5 weeks (n ≥ 3). Statistical significance was evaluated using Student’s t-tests, with the Bonferonni correction (*P < 0.05); error bars represent standard error (SEM). (C) Reduced anchorage-independent growth is not primarily due to a general proliferation defect. Cell growth in monolayer culture was not statistically different whether Ect2 was (Ect2 shRNA#2, #3) or was not (NT shRNA) knocked down, as determined by MTT assay over 4 days (shown: OVCAR8 cells). Error bars represent SEM (n = 3).
MTT proliferation assays revealed no significant differences between NT and Ect2 knockdown ovarian cancer cells (Fig. 3C). This indicates that the observed Ect2 dependence of anchorage-independent growth was not due simply to a general proliferation defect. And although Ect2 is known to play a critical role in cytokinesis of normal cells through activation of RhoA,10,31 its role in some tumor cells is less clear.11,15 Here, the level of multinucleated cells remained low upon knockdown of Ect2 (Suppl. Figs. S2 and S4), indicating that these ovarian tumor cells can still undergo cytokinesis efficiently when Ect2 is depleted.
RhoGEF activity is required for Ect2-mediated transformed growth
Ect2 is a RhoGEF with other functional domains6 that may also play a role in its ability to drive transformed growth. To determine if the GEF activity itself is required, we first generated an shRNA-resistant, putatively GEF-deficient Ect2 mutant. Residues E428 and N608 in the catalytic Dbl homology (DH) domain of Ect2 are conserved among RhoGEFs, interact with the switch I and switch II regions of Rho GTPases, respectively, and are important for nucleotide exchange.6 We mutated these residues to alanines (E428A, N608A). Both wild type (WT) and the DH-mutant Ect2 were tagged with the HA epitope so they could be monitored separately from endogenous Ect2. The localization of the DH-mutant was indistinguishable from WT (Suppl. Fig. S3). Using standard pulldown assays for active Rho GTPases, we then observed in 293T cells that these mutations decreased by 90% the ability of Ect2 to activate RhoA, and completely ablated Rac1 activation (Fig. 4A), validating that this Ect2 mutant is GEF-deficient on these substrates. Interestingly, these mutations did not prevent Ect2-induced activation of Cdc42, indicating that this activation is not dependent on these catalytic residues and is likely indirect.
Figure 4.

RhoGEF activity is required for Ect2-mediated transformed growth. (A) A catalytic DH-domain mutant (E428A/N608A) of Ect2 is impaired in RhoGEF activity on RhoA and Rac1 but not Cdc42. Ectopic expression of HA-tagged wild type (WT) but not mutant Ect2 increased active RhoA-GTP or Rac1-GTP in whole cell lysates, as shown by pulldown assays using the Rho binding domain (RBD) of effectors that selectively bind the active, GTP-bound form of each GTPase (293T cells; anti-HA, 1:1,000, clone 3F10, Roche, Indianapolis, IN; anti-RhoA, 1:1,000, clone 67B9, Cell Signaling, Danvers, MA; anti-Rac1, 1:1,000, clone 23A8, Millipore; anti-Cdc42, 1:500, clone B-8, Santa Cruz, Dallas, TX). In the same assay, overexpression of Ect2 with mutated nuclear localization signals (NLS) caused a dramatic increase in RhoA activity but not Rac activity. Ect2 activation of Cdc42 was unaffected by either DH or NLS mutations. Percent activation is shown based on densitometry performed on each active GTPase blot and normalized to total GTPase (representative of n ≥ 3). (B) Re-expression of WT or the GEF-deficient DH-mutant Ect2 in knockdown cells restores Ect2 levels to similar degrees. The shRNA-resistant mutants indicated above were stably expressed in the Ect2 knockdown OVCAR8 cells and immunoblotted with anti-HA antibody for ectopic Ect2 and with anti-Ect2 antibody for endogenous + ectopic Ect2. Tubulin served as a loading control. (C) Re-expression of WT but not GEF-deficient Ect2 rescues anchorage-independent growth. Error bars represent SEM. The baseline of colony counts was considered to be those seen in cells where Ect2 was not knocked down (NT) and expression was “rescued” with vector-only (VO). Colony counts statistically significant from this baseline are marked with an asterisk * (P < 0.05, with Bonferroni correction). The ability of ectopic Ect2 to rescue Ect2 knockdown was evaluated by comparison to cells expressing Ect2 shRNA#3 and “rescued” with vector-only (^, P < 0.05, with Bonferroni correction; n = 5). (D) Ect2 knockdown results in decreased levels of active RhoA and Rac in whole cell lysate. Standard pulldown assays were performed using the Rho binding domain (RBD) of effectors that selectively bind the active, GTP-bound form of RhoA (Rhotekin-RBD) and Rac (PAK-RBD). Upper panel = representative pulldown; lower panel = average of all assays (n ≥ 4). Error bars represent SEM and * represents P < 0.05, based on a paired t-test in which values were log-transformed for normality.
We next assessed whether GEF-deficient Ect2 could rescue anchorage-independent growth. Ect2 expression was rescued in OVCAR8 Ect2 knockdown cells using either WT or GEF-deficient Ect2 (Fig. 4B). As expected, WT Ect2 rescued soft agar colony numbers to levels similar to those seen in cells without Ect2 knockdown, whereas the GEF-deficient mutant only partially rescued the phenotype (Fig. 4C). Thus, the RhoGEF activity of Ect2 is required to support full anchorage-independent growth in OVCAR8 cells.
We then sought to determine which Rho GTPases are activated by Ect2 in these cells. We compared the levels of active RhoA, Rac, and Cdc42 in whole cell lysates with or without Ect2 knockdown, using standard Rhotekin-RBD (for RhoA) and PAK-RBD (for Rac and Cdc42) pulldown assays. Upon Ect2 knockdown, RhoA activity decreased by about a third (*P < 0.05, Fig. 4D). We also saw a clear but more variable decrease in active Rac, despite the continued presence of multiple other RhoGEFs capable of activating these GTPases. Cdc42 activity remained constant (data not shown), consistent with the lack of effect of Ect2 GEF deficiency on Cdc42-GTP levels. These results indicate that Ect2 GEF activity is required for full RhoA and Rac activation in OVCAR8 cells at steady state and that other GEFs cannot fully compensate for loss of Ect2.
Predominantly nuclear localization is required for Ect2-mediated transformed growth
In ovarian cancer cells, Ect2 is a predominantly nuclear RhoGEF. Although RhoA and Rac1 are predominantly cytosolic proteins,5 both have been observed to lesser extents in the nucleus.32-36 Loss of Ect2 GEF activity impaired ovarian cancer cell transformed growth and also impaired full RhoA and Rac activation. Collectively, these findings suggest that Ect2 could activate nuclear rather than cytoplasmic pools of RhoA and Rac to drive transformation. If so, depleting Ect2 selectively from the nucleus should impair anchorage-independent growth.
To address this possibility, we mutated the arginines in both NLSs of the shRNA-resistant Ect2 to alanines (R348,349,350,370,372A). We first evaluated subcellular localization using immunofluorescence and confocal microscopy. Co-staining for the HA-epitope tag and DAPI confirmed that the NLS-mutant had a greatly decreased nuclear localization and greatly enriched cytoplasmic localization compared to WT Ect2 (Fig. 5A). To quantitate the subcellular distribution of NLS-mutant Ect2, we then fractionated the cells and performed immunoblotting analyses. The results (Fig. 5B) demonstrated that only about 10% of the NLS-mutant was still localized to the nuclear fraction. The NLS-mutant was well-expressed and well-tolerated, such that cell lines stably expressing NLS-mutant Ect2 were easily generated (Suppl. Fig. S4A), without apparent effects on cell cycle distribution (Suppl. Fig. S4D) or multinucleation (Suppl. Fig. S4B and S4C).
Figure 5.

Predominantly nuclear localization is required for Ect2-mediated transformed growth. (A) Disruption of the nuclear localization signals (NLSs) of Ect2 impairs its nuclear localization compared with WT Ect2. Localization of an Ect2 NLS-mutant (R348,349,350,370,372A) in which both NLSs have been disrupted was determined by confocal immunofluorescence microscopy, probing for HA-tagged Ect2 (red, 1:500, clone 16B12, Covance, Princeton, NJ) and the nuclear marker DAPI (blue), n = 3. Scale bars represent 10 µm. (B) NLS-mutant Ect2 is localized predominantly in the cytoplasm. The shift in localization of Ect2 on NLS mutation was confirmed by fractionating cells expressing either NLS-mutant or WT Ect2, and blotting for HA-tagged Ect2 (1:1,000, Covance). Purity of each fraction was confirmed via immunoblot using anti-histone H3 antibody (1:12,000, Ab1791, AbCam, Cambridge, MA) as a nuclear marker and tubulin as a cytoplasmic marker, with equal amounts of the nuclear and cytoplasmic fractions loaded onto the gel. Average percentage of Ect2 in the nucleus was calculated from densitometry values. Whereas 72.3% of WT Ect2 was observed in the nucleus, only 10.4% of NLS-mutant Ect2 was nuclear (n = 2). (C) NLS-mutant Ect2 restored Ect2 expression in knockdown cells to a higher degree than WT. The shRNA-resistant mutants indicated above were stably expressed in Ect2-knockdown OVCAR8 cells. Cell lysates were immunoblotted as in Figure 4B except that actin served as a loading control. (D) Ect2 NLS-mutant failed to rescue colony formation in soft agar. WT but not NLS-mutant Ect2 rescued the impairment of soft agar colony formation caused by Ect2 knockdown (n = 5). Error bars represent SEM. Statistically significant differences are marked with * and ^ as in Figure 4C.
Despite higher levels of expression than WT Ect2 (Fig. 5C), the NLS-mutant was not capable of rescuing the anchorage-independent growth defect of Ect2 knockdown cells (Fig. 5D). Interestingly, its overexpression alone in cells retaining endogenous Ect2 was sufficient to lead to decreased anchorage-independent growth. This suggests that simply localizing Ect2 to the cytoplasm in the presence of nuclear Ect2 is not sufficient to drive transformation in these ovarian cancer cells, but rather may have the opposite effect, consistent with our TMA findings in which higher levels of cytoplasmic Ect2 were found preferentially in the less-advanced lesions.
Some nuclear Ect2 is present in an active conformation that has enhanced specificity for Rac1
It has been proposed that nuclear Ect2 is auto-inhibited,8,37 whereas our results suggest at least a portion is active. To test this directly, we fractionated OVCAR8 cells and performed a pulldown assay38,39 for active Ect2 using a constitutively nucleotide-free form of RhoA(17A), which binds with high affinity to the active form of RhoGEFs.39 Using tubulin as a cytoplasmic marker and PARP as a nuclear marker, we confirmed effective fractionation (Fig. 6A, left). Immunoblotting of endogenous Ect2 demonstrated that, while the vast majority was observably nuclear (NUC), some Ect2 was detectable in the non-nuclear fraction (CYT). This is unlikely to be due to nuclear contamination of the CYT fraction because PARP was not detectable there. In both fractions, Ect2 bound with notably higher affinity to GST-RhoA(17A) compared to the GST-only control (Fig. 6A, right), revealing that some active Ect2 is present in both compartments.
Figure 6.

Some nuclear Ect2 is present in an active conformation that has enhanced specificity for Rac. (A) At least some nuclear Ect2 is present in an active conformation. OVCAR8 cells were fractionated into nuclear (NUC) and non-nuclear (CYT) compartments. Purity of each fraction was confirmed via immunoblot using PARP (1:2,000, clone C2-10, BD Bioscience, San Jose, CA) as a nuclear marker and tubulin as a cytoplasmic marker, with equal amounts of the nuclear and cytoplasmic fractions loaded onto the gel. As expected, Ect2 was predominantly nuclear (left panel). Pulldowns (right panel) were then performed on equal concentrations of each fraction. GST-RhoA(17A) pulled down active Ect2 from each location. GST alone served as a control for nonspecific binding (n = 5). (B) Ect2 preferentially interacts with Rac1 in the nucleus, and with RhoA in the cytoplasm. As above, cells were fractionated, but here the pulldowns were performed on fractions distributed for equal expression of Ect2 as described in Materials and Methods (confirmed by immunoblot, left panel). Pulldown analyses using GST alone, GST-RhoA(17A), GST-Rac1(15A), or GST-Cdc42(15A) were performed on each fraction to determine the GTPase specificity of Ect2 within each fraction (right panel, n = 8). Equal bead loading was confirmed by SDS-PAGE and Coomassie staining. (C) The ratio of Ect2 pulled down by Rac to Ect2 pulled down by Rho in the nucleus is greater than the same ratio in the cytoplasm. The amount of Ect2 pulled down by nucleotide-free Rac1 or RhoA in each subcellular compartment (as shown in B) was quantified by densitometry, and the ratio of the amount of Ect2 pulled down by Rac1 to the amount pulled down by RhoA in the nuclear (NUC) or non-nuclear compartments (CYT) is shown. This ratio switches from >1 in the NUC to <1 in the CYT, indicating that Ect2 associates preferentially with Rac1 in the nucleus and with RhoA in the cytoplasm. The graph shows the average of all experiments (n = 8), and error bars represent SEM. (D) Endogenous Rac and RhoA are present in the nucleus of OVCAR8 cells. Cells were fractionated and immunoblotted for RhoA and Rac1. To allow for detection of the less abundant nuclear GTPases, 6.5× more protein was loaded from the NUC compartment, while still retaining the purity of each compartment. (E) A higher proportion of total Rac is nuclear, compared to RhoA. Densitometry was performed on the immunoblot shown in D and the percentage of nuclear protein was calculated as follows to account for the unequal loading of each compartment: (nuclear intensity)/(nuclear intensity + 6.5× cytoplasmic intensity).
The ability of a GEF to interact with nucleotide-free mutant GTPases is dependent on both the concentration of active GEF38 and the specificity of the GEF for the GTPase.39-41 To determine the ability of Ect2 to interact with other Rho GTPases within each subcellular compartment, we extended these results with GST-RhoA(17A), GST-Rac1(15A), and GST-Cdc42(15A). Equal amounts of GST-Rho proteins were added in excess to each Ect2-containing lysate (Fig. 6B, Coomassie), so the relative amounts of endogenous RhoA and Rac in each cellular compartment were irrelevant and could not contribute to any observed differences in Ect2 interactions. Additionally, to avoid complications from the vastly different amounts of Ect2 present in the nuclear versus non-nuclear compartments, here we pulled down Ect2 from lysates containing equal amounts of total Ect2 rather than equal amounts of total protein.
Unexpectedly, the Rho GTPase interactions of Ect2 in the nucleus were different from those in the cytoplasm (Fig. 6B). In the nucleus, Ect2 interacted with both Rac1(15A) and RhoA(17A), with a distinct preference for Rac1 despite equal exposure to both. In the cytoplasm, Ect2 interacted almost exclusively with RhoA. The minimal levels of Ect2 pulled down with Cdc42(15A) from either compartment are consistent with our proposal that activation of Cdc42 via Ect2 is indirect in these cells. Using densitometry to quantify the amount of Ect2 from each compartment that was pulled down by Rac versus Rho, we calculated that the ratio of Ect2 pulled down by Rac to that of Ect2 pulled down by Rho was 1.7 in the nucleus but only 0.4 in the non-nuclear compartment (Fig. 6C). Because equal amounts of ectopic Rac and Rho were available for Ect2 binding in each case, these different ratios suggest the possibility of an intrinsic conformational difference between nuclear and cytoplasmic Ect2.
We detected both endogenous Rac and endogenous Rho in the nucleus (Fig. 6D), but with unequal distribution: a greater proportion of Rac was distributed to the nucleus (16%) than was Rho (6%) (Fig. 6E). Thus, intrinsic differences detected by the pulldowns using ectopic nucleotide-free GTPases are likely to be amplified by the relative availability of endogenous Rac and Rho in each compartment during cellular signaling.
Ect2 activates endogenous Rac in the nucleus and endogenous RhoA in the cytoplasm
To determine if Ect2 is capable of activating endogenous Rac and/or Rho in the nucleus, we first performed standard Rho binding domain (RBD) recruitment assays.42-46 We used POSH, a downstream effector specific to Rac,47 to detect active Rac-GTP. Under basal conditions, GFP-POSH-RBD was expressed diffusely throughout the cytoplasm and nucleus, but was recruited to the nucleus in the presence of exogenous WT Ect2 (Fig. 7A; quantified in Fig. 7C). Nucleolar exclusion of GFP-POSH-RBD was evident by confocal microscopy (Fig. 7A); thus, GFP-POSH-RBD was recruited into the nucleus and not onto the nuclear envelope. Recruitment was dependent on both GEF activity and nuclear localization of Ect2, because the GEF-deficient and the NLS-mutant each failed to recapitulate the effects of WT Ect2. Consistent with these results, GFP-PAK1-RBD48 was also recruited to the nucleus by Ect2, in a GEF- and NLS-dependent manner (Suppl. Fig. S5A and S5B). These results indicate that WT Ect2 activates endogenous Rac1, and the site of activation is primarily in the nucleus.
Figure 7.

Ect2 recruits downstream effectors of Rac to the nucleus and initiates canonical RhoA signaling in the cytoplasm. To determine if Ect2 was capable of activating endogenous Rho GTPases in the nucleus, we examined GFP-RBD recruitment in OVCAR8 cells expressing empty vector versus exogenous HA-tagged WT, GEF-deficient, or NLS-mutant Ect2. Confocal immunofluorescence microscopy was used to image cells expressing both HA-Ect2 (red) and each GFP-RBD (green); nuclei were stained using DAPI (blue). (A) Ect2 recruits POSH-RBD to the nucleus in a GEF-dependent manner. POSH is a Rac-specific effector, and recruitment of POSH-RBD reveals endogenous Rac activation in the nucleus (n = 7, with an average of 40 cells/condition in each replicate). Scale bars represent 10 µm. (B) Ect2 does not recruit Rhotekin-RBD to the nucleus. Rhotekin is a Rho-specific effector that was not detectably recruited (n = 6, with an average of 30 cells/condition in each replicate); see Results section. Scale bars represent 10 µm. (C) Ect2 recruitment of POSH to the nucleus is robust and statistically significant. Quantification of GFP-RBD localization was performed for each condition, as described in Materials and Methods. The percentage of cells with nuclear-highlighted expression of the GFP-RBD is shown (*P < 0.05 difference from vector with Bonferonni correction; error bars represent SEM). (D) NLS-mutant Ect2 enhances stress fiber formation but not membrane ruffling. Extra-nuclear Rho GTPase signaling was investigated by examining the actin cytoskeleton. Canonical RhoA signal transduction induces stress fiber formation, whereas Rac1 drives membrane ruffling. We compared organization of the actin cytoskeleton in OVCAR8 cells expressing empty vector versus exogenous HA-tagged WT, GEF-deficient, or NLS-mutant Ect2. Confocal immunofluorescence microscopy was used to image cells expressing HA-Ect2 (green, Covance); actin was stained using Alexa Fluor 568-conjugated phalloidin (red, Invitrogen, Grand Island, NY), and nuclei were stained using DAPI (blue). (E) Cells expressing each construct as shown in panel D were quantified for presence or absence of stress fibers or membrane ruffles (n = 3, with an average of 35 cells/condition in each replicate). Statistical significance was evaluated using Student’s t-test, with the Bonferonni correction (*P < 0.05); error bars represent standard error (SEM).
Notably, the cytoplasmically localized NLS-mutant did not activate Rac as measured either by recruitment assay (Fig. 7A, Suppl. Fig. S5A) or by pulldown assay (Fig. 4A). In contrast, we observed that the cytoplasmically localized NLS-mutant Ect2 strongly increased RhoA-GTP levels (50.5-fold, Fig. 4A). These results indicate that Ect2 activity in the cytoplasm preferentially results in activation of Rho. However, GFP-Rhotekin-RBD, a surrogate for RhoA activity,49 was not recruited either to the cytoplasm or to the nucleus (Fig. 7B, quantified in Fig. 7C). One reason for this may be that any shift of the smaller pool of nuclear GFP-Rhotekin-RBD into the cytoplasm may not be detectable in the larger pool of cytoplasmic Rhotekin-RBD. Therefore, to determine if the elevated RhoA-GTP that we detected by pulldown led to a biological consequence in the cytoplasm, we performed phalloidin staining to examine the actin cytoskeleton of OVCAR8 cells exogenously expressing WT or mutant Ect2. The canonical cytoskeletal consequence of RhoA activity is the formation of stress fibers,50 and indeed we found that only the cytoplasmically localized NLS-mutant Ect2 significantly enhanced stress fiber formation, whereas the nuclearly localized WT and GEF-deficient Ect2 did not (Fig. 7D and E). These results indicate that cytoplasmically localized Ect2 can functionally activate RhoA. In contrast, and consistent with its inability to activate Rac (Fig. 4A), NLS-mutant Ect2 did not increase membrane ruffling, a canonical consequence of Rac activity51 (Fig. 7D and E). WT Ect2 also did not enhance membrane ruffling, consistent with a previous report that overexpression of a nuclearly localized, constitutively active Rac1 mutant enhanced cell proliferation without inducing membrane ruffles.34 Collectively, our results indicate that Ect2 preferentially activates Rac1 in the nucleus and RhoA in the cytoplasm. The larger pool of cytoplasmic Rho family GTPases (Fig. 6E) may explain why we observed more striking changes in RhoA activation compared to Rac activation upon Ect2 overexpression (Fig. 4A) and knockdown (Fig. 4D) in whole cell lysate.
Nuclear Rac1 activity is sufficient to rescue defects in Ect2-mediated transformed growth
Since we have demonstrated that both nuclear localization and GEF activity are required for Ect2-mediated transformed growth in ovarian cancer cell lines and that Ect2 preferentially activates Rac1 in the nucleus, our results suggest that Ect2 activation of nuclear Rac may be an important contributor to its transforming ability.
We first attempted to determine directly whether Rac1 is necessary for Ect2-driven anchorage-independent growth by asking whether Rac1 knockdown impaired colony formation in soft agar. However, it was impossible to generate cells lacking Rac1, as transfection with any of 4 independent Rac1-targeted siRNAs (Dharmacon/Thermo Scientific, Pittsburgh, PA) induced widespread cell death (data not shown), leaving us with insufficient Rac1-knockdown cells for analysis and indicating that Rac1 is an essential protein for viability of these cells. Although it is likely that the non-nuclear functions of Rac1 were critical, we were unable to selectively knock down only nuclear Rac1.
Another approach to testing our model is to determine whether nuclear Rac1 can overcome the defect in transformed growth conferred by loss of Ect2 expression. To test this, we generated HA-tagged activated Rac1 mutants that were preferentially localized in the nucleus (Fig. 8A). We used mutants of Rac1 that were activated independently of Ect2 by either the traditional GTP-locked G12V mutation51 or by the fast-cycling F28L mutation.52 Prenylation of Rac1 is required for its plasma membrane association5 and impairs its nuclear import. Non-prenylated Rac1 localizes strongly to the nucleus when GFP-tagged.34,53 However, we determined that an additional N-terminal NLS was required to concentrate unprenylated HA-tagged Rac1 in the nucleus. Therefore, to preferentially localize the activated Rac1 mutants to the nucleus, we both mutated the Rac1 prenylated cysteine to serine (C189S, “SAAX” mutation) and added the classical NLS from SV40T antigen54 to the N-terminus to generate “NLS-Rac1-SAAX” mutants. When expressed stably in cells with Ect2 knocked down (Fig. 8B), each of these nuclearly concentrated, active mutants of Rac1 was able to rescue anchorage-independent growth to the same extent as re-expression of WT Ect2 (Fig. 8C). These results support our model that Ect2 activation of Rac1 in the nucleus is a key component of its transforming function.
Figure 8.

Nuclear Rac1 activity is sufficient to rescue defects in Ect2-mediated transformed growth. (A) Rac1 is targeted to the nucleus upon disruption of its CAAX box and addition of an NLS. The localization of Ect2, NLS-Rac1-G12V-SAAX, and NLS-Rac1-F28L-SAAX was detected by confocal immunofluorescence microscopy, probing for HA-tagged Rac1 or Ect2 (green, Covance) and the nuclear marker DAPI (blue), n = 3. Scale bars represent 10 µm. (B) Stable expression of nuclearly targeted Rac1 mutants in Ect2 knockdown OVCAR8 cells. Lysate was immunoblotted with anti-HA (Covance) for ectopic Rac1 and Ect2 or with anti-Ect2 for endogenous + ectopic Ect2. Actin served as a loading control. (C) Rac1 mutant expression rescued the defect in anchorage-independent growth caused by Ect2 knockdown. Both constitutively active and fast-cycling nuclearly localized Rac1 mutants were able to rescue the defect in soft agar colony formation caused by Ect2 knockdown, to the same extent as re-expression of WT Ect2 (n = 3). Representative images (4× magnification) of colonies are shown. Scale bars represent 100 µm. Error bars represent SEM. Statistically significant differences are marked with * and ^ as in Figure 4C.
Discussion
Despite the largely nuclear localization of the RhoGEF Ect2 in interphase cells,7,13,15,55,56 previous studies have focused on its actions at non-nuclear locations. For example, the most prominent role for Ect2 in normal cells is its requirement during cytokinesis, when the nucleus is not intact and Ect2 activates RhoA at the cleavage furrow and midbody.7,10,11,25,57 But the function of Ect2 in cytokinesis of normal cells can be uncoupled from its role in promoting transformed growth of tumor cells,11,15,58 leaving the mechanisms whereby it promotes transformation less well understood. One model suggests that Ect2 must be cytoplasmically mislocalized to drive transformation.8,15 In contrast, our study indicates a prominent role for a nuclear pool of Ect2 in the transformed phenotype of ovarian cancer cells. Furthermore, we have identified a requirement for Ect2 to act as a RhoGEF within the nucleus to promote growth transformation in ovarian cancer cells.
In both ovarian cancer patient tumor tissues and established cell lines, we found that Ect2 protein was expressed robustly, consistent with its published genomic amplification and mRNA overexpression in ovarian tumors.22,24,25 Ect2 was predominantly localized to the nucleus, particularly in advanced disease. Interestingly, we did not observe significant Ect2 localization to the cytoplasm in advanced tumors; instead, benign ovarian cysts had the most prominent cytoplasmic staining. Although Ect2 was reported to be strongly mislocalized from the nucleus to the cytoplasm in non–small cell lung cancers (NSCLC) compared to normal lung,15 our observations of increased nuclear expression in ovarian cancers are more consistent with other reports where inspection of the data shows that Ect2 increases globally on tumor progression, with extensive increases in nuclear expression and less dramatic increases in cytoplasmic Ect2.13,56,59,60 We show here that both Ect2 nuclear localization and its RhoGEF activity are required for Ect2-driven anchorage-independent growth of ovarian cancer cells, suggesting that Ect2 acts as a GEF from within the nucleus to drive transformation. Indeed, our observations that cytoplasmic Ect2 localization was inversely correlated with advanced disease, and that a cytoplasmically localized Ect2 NLS-mutant reduced anchorage-independent growth, indicate that mislocalization of Ect2 to the cytoplasm does not drive ovarian transformation but rather might oppose the effect of nuclear Ect2. That this NLS-mutant Ect2 was able to act as a GEF to increase RhoA-GTP and promote stress fiber formation suggest the intriguing possibility that Ect2 activation of RhoA in the cytoplasm might even counteract its actions in the nucleus.
In NSCLC cell lines, transformed growth can be driven by Ect2-induced activation of Rac1.15 Rac1 activation was proposed to occur in the cytoplasm, although this was not tested directly.15,58 Since the BRCT domains of Ect2 can auto-inhibit its activity,8,37,61 nuclear Ect2 was proposed to be auto-inhibited and sequestered from Rho GTPases.8 Here, we found that active Ect2 was present in the nucleus. There has been almost no prior investigation of the subcellular distribution of active Ect2, but one study did identify active Ect2 in the nucleus of HEK293 cells.36
While Rho GTPases are described as cytoplasmic proteins,5 this is not the entire story. In particular, Rac132-34 and, to a lesser extent, RhoA35,36 and Cdc4262 have all been detected within the nucleus. We were able to detect both endogenous Rac and some endogenous RhoA in the nucleus of OVCAR8 cells; therefore, both are available in the nucleus as substrates for Ect2 RhoGEF activity. Our work also provides further evidence for nuclear activation of Rho GTPases. Although early reports simply described nucleocytoplasmic shuttling,32,33 there are recent reports that RhoA can be activated from within the nucleus in some contexts.35,36
We observed that Ect2 preferentially activates Rac in the nucleus. We also identified at least 2 likely contributors to this finding: (a) the presence of more Rac than RhoA in the nuclear compartment and (b) an intrinsic substrate specificity, such that nuclear Ect2 preferentially binds to Rac1 whereas cytoplasmic Ect2 preferentially binds to RhoA. Consistent with the latter, the NLS-mutant, which is defective for nuclear localization and is distributed well into the cytoplasm, robustly activated RhoA but not Rac.
Modulation of intrinsic substrate specificity may be complex. Phosphorylation is required to optimally activate Rac both in vitro7 and in cells.63 We have observed that bacterially expressed, that is, unphosphorylated Ect2 can activate RhoA but not Rac1 in vitro.57 It is tempting to speculate that nuclear Ect2 is appropriately phosphorylated; however, despite repeated attempts we have not observed mobility differences between nuclear and cytoplasmic Ect2 (as observed previously for phosphorylated versus unphosphorylated Ect27,10). Alternatively, as-yet-undefined protein-protein interactions that occur in the nucleus may promote interaction with Rac, perhaps involving the Ect2 C-terminus, which is required to activate Rac in cells.64 Similarly, cytoplasmic scaffolding may promote RhoA but not Rac activation. Protein-protein interactions have been speculated to explain reports of varying Ect2 specificity throughout cytokinesis/mitosis.65 Regardless of the mechanism, to our knowledge, this study is the first to demonstrate altered specificity of a RhoGEF based on its subcellular localization.
Our work is also the first to show that active Rac1 is capable of driving transformation when concentrated in the nucleus (and excluded from the plasma membrane). The precise function of nuclear Rac in these cells is unknown. Forced expression of a constitutively active but unprenylated Rac1 mutant in the nucleus of NIH3T3 cells accelerated cell cycle progression.34 However, this mutant did not induce lamellipodia formation, a primary function of cytoplasmic Rac1,34 suggesting that nuclear and cytoplasmic functions of active Rac may be distinct. Similarly, although overexpression of Ect2 was capable of recruiting the RBD of Rac effectors to the nucleus, we did not observe an increase in lamellipodia or membrane ruffling on expression of Ect2. Still, negative regulators66,67 and downstream signaling molecules68-72 of Rac can be detected in the nucleus, suggesting that some cytoplasmic signaling pathways could remain intact there, albeit with distinct stoichiometry. Some Rac effectors directly regulate transcription from the nucleus,68-70 but we did not observe Ect2-mediated alterations in transcriptional activation (data not shown). The speckled pattern of Ect2 localization in the nucleus suggests that Ect2 localizes to distinct subnuclear structures. Nuclear speckles regulate mRNA splicing,73 and Ect2 interacts with the SNRNP200 gene product, a component of the spliceosome,74 so it is possible that Ect2 signaling to Rac plays a role in this process.
In conclusion, here we identified Ect2 as necessary for cellular transformation in ovarian tumor cells. We also identified a novel mechanism through which Ect2 can drive cellular transformation by acting as a GEF from within the nucleus, in a manner distinct from its actions in the cytoplasm. We showed that Ect2 preferentially activates Rac in the nucleus of ovarian cancer cells and RhoA in the cytoplasm. In addition to Ect2, the RhoGEFs Net1, LARG, and Vav3 have also been identified in the nucleus.75-77 It would be interesting to determine if subcellular localization also alters the specificity of these and other RhoGEFs. Our results suggest that nuclear Rac1 activity is biologically relevant for transformed growth and implicate activation of nuclear Rac as a mediator of Ect2-driven transformation. In the future it may become possible to selectively ablate Ect2-activated nuclear Rac. If so, this would allow more rigorous testing of a requirement for nuclear Rac activity in Ect2-driven ovarian cancer cell transformation. As physiologically relevant substrates of nuclear Ect2 and Rac1 are identified, it will also be interesting to uncover their roles in transformation.
Materials and Methods
Molecular constructs and transfections
Short hairpin RNA (shRNA) against human Ect2, non-targeting (NT) shRNA, and shRNA-insensitive full length Ect2 were generously provided by Alan Fields (Mayo Clinic, FL).15 Quikchange II site-directed mutagenesis kit (Stratagene, Santa Clara, CA) was used to create NLS- and DH-mutant Ect2 from shRNA-insensitive full length Ect2 (primers, Suppl. Fig. S6). NLS-Rac1-SAAX mutants were generated using PCR (primers, Suppl. Fig. S6) to amplify Rac1 F28L57 and Rac1 G12V (gift of Peter Hordijk, Sanquin). All Ect2 and Rac1 expression constructs were cloned into pFugW-HA-blasticidin (gift of Jeran Stratford, UNC-CH). Viral psPAX2 (packaging) and pMDG.2 (envelope) vectors were from Addgene. pGEX-2T and RBD constructs45,78,79 were gifts from Keith Burridge (UNC-CH). Transfections were performed using TransIT-LTI (Mirus, Madison, WI) according to the manufacturer’s instructions.
Cell culture, lentiviral infection, and generation of stable transfectants
Human ovarian epithelial cancer cell lines were grown in RPMI-1640 with 10% fetal bovine serum (FBS; Sigma, St. Louis, MO). 293T cells were grown in DMEM-H + 10% FBS. Lentiviruses were generated as described,80 except virus was collected 24 and 48 hours posttransfection. Lentivirus-infected cells80 were selected in puromycin (1 µg/mL, Cellgro, Manassas, VA) or blasticidin (10 µg/mL, Invitrogen, Grand Island, NY).
Flow cytometry
Flow cytometry was performed exactly as we have described previously.57
Immunoblotting, immunofluorescence, phalloidin staining, and microscopy
Immunoblotting was performed from RIPA cell lysates by our standard methods.45 For immunofluorescence and phalloidin staining, cells were plated on fibronectin, fixed in paraformaldehyde, exposed to antibodies or to Alexa Fluor568-conjugated phalloidin (Invitrogen), and confocal microscopy imaging performed as we have done previously.45,81 A Nikon (Melville, NY) Eclipse TS100 microscope was used to image cells and colonies in bright field.
Ovarian tumor tissue microarray (TMA) immunohistochemistry
A previously validated TMA,26-29 containing formalin-fixed, paraffin-embedded primary epithelial ovarian carcinomas or benign cysts, was stained for Ect2 by the UNC-Translational Pathology Laboratory Core. We used a standard method of scoring the TMA,29 except each core was scored separately for nuclear and cytoplasmic staining. Each compartment was scored separately on a scale of 0 to 100 for percent cyst or tumor cells stained, and on a scale of 0 to 3 for average intensity of staining, where 0 was no staining and 3 was maximal staining. The score for each compartment was the product of percent cells staining in the nucleus/cytoplasm multiplied by the average intensity of nuclear/cytoplasmic staining for each core.
Previously collected patient data26-29 allowed us to classify the serous tumors by degree of malignancy (cysts, serous borderline tumors, serous low-grade tumors, and serous high-grade tumors). A Kruskal-Wallis test was performed to compare both nuclear and cytoplasmic scores among these groups; P < 0.05 was considered significant. Institutional review board approval was through UNC-CH IRB 08-0242.
Anchorage-dependent and -independent growth assays
Soft agar assays to assess anchorage-independent growth were performed and counted as we have described.45 A 2-tailed Student’s t-test assuming unequal variance was performed on colony counts. The P-values were adjusted for multiple tests using the Bonferroni correction, and P < 0.05 was considered significant. Standard MTT proliferation assays were performed.82 Cells were normalized to the average value for day 0 and growth curves were generated from the average of all experiments. Slopes of the log-transformed growth curves were compared using likelihood ratio tests to determine statistical significance, and a Bonferroni corrected P-value <0.05 was considered significant.
Cell fractionation
Cell fractionation into nuclear and postnuclear compartments was performed by hypotonic swelling, Dounce homogenization, centrifugation through 28.5% iodixanol (Sigma), and sonication as described in detail previously.38
Pulldown and recruitment assays for active Ect2 and Rho GTPases
Active Ect2 was assessed by incubating fractionated cell lysates with nucleotide-free Rho GTPases isolated from bacteria and bound to agarose beads, resolved on SDS-PAGE, and immunoblotted for Ect2.38,39 Prior to performing pulldowns, fractions were subjected to Bradford analysis for total protein content. For some experiments (Fig. 6A), equal amounts of total protein were added to the beads. Alternatively (Fig. 6B), loading of fractionated cell lysates was normalized for equal expression of Ect2. This was done by comparing the distribution of Ect2 in nuclear versus cytosolic fractions using densitometry of western blots of the fractionated lysates, which showed that loading 2.6× more of the cytosolic fraction resulted in approximately equal loading of Ect2 from each fraction. Loading of Ect2 was examined by western blot following each of these assays. Beads were blocked in 5% BSA for 30 minutes to minimize nonspecific binding of Ect2 to GST. Equal loading of the beads was confirmed by SDS-PAGE and Coomassie blue staining.
To determine levels of active, GTP-bound RhoA, Rac, and Cdc42, pulldowns using GST-Rhotekin-RBD or -PAK-PBD, respectively,78 were performed from whole cell lysates. Densitometry on immunoblots was performed using ImageJ and normalized as described in the Results section.
Recruitment assays42-46 using GFP-tagged RBDs for Rhotekin, POSH, and PAK were used to monitor changes in the subcellular distribution of active endogenous Rho GTPases. Fixed cells expressing HA-Ect2 in each specific location (vector: diffuse; WT and DH-mutant: nucleus; NLS-mutant: cytoplasm) that co-expressed GFP-RBD were scored for GFP-RBD localization based on the following categories: nuclear-excluded, plasma membrane-highlighted, diffuse nuclear and cytoplasmic localization, nuclear-highlighted. The average percentage of cells nuclear-highlighted out of total cells counted is shown. Statistical significance was determined as described for the soft agar assays.
Acknowledgments
We thank Kent Rossman for advice on designing the GEF-deficient Ect2 mutant; Alan Fields for the gift of Ect2 plasmids; Keith Burridge for the gift of RBD plasmids and helpful advice; Marc Olorvida of the UNC-Translational Pathology Core Laboratory for assistance with immunohistochemistry; Robert Bagnell of the UNC-Microscopy Services Laboratory for assistance with microscopy; Rafael Garcia-Mata for advice on fractionation-pulldown assays; and Jim Fiordalisi for assistance with plasmid constructs.
Footnotes
Supplementary material for this article is available on the Genes & Cancer website at http://ganc.sagepub.com/supplemental.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Institutes of Health (Grant Numbers CA109550, CA042978, F31-CA165844, T32-GM00 7040, F31-CA159821, T32-CA71341), the Howard Hughes Medical Institute (Med-Into-Grad Program), and the Office of Undergraduate Research at the University of North Carolina at Chapel Hill (Summer Undergraduate Research Fellowship).
References
- 1. Hall A. Rho family GTPases. Biochem Soc Trans. 2012;40(6):1378-82 [DOI] [PubMed] [Google Scholar]
- 2. Vega FM, Ridley AJ. SnapShot: Rho family GTPases. Cell. 2007;129(7):1430 [DOI] [PubMed] [Google Scholar]
- 3. Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2(2):133-42 [DOI] [PubMed] [Google Scholar]
- 4. Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582(14):2093-101 [DOI] [PubMed] [Google Scholar]
- 5. Ellenbroek SI, Collard JG. Rho GTPases: functions and association with cancer. Clin Exp Metastasis. 2007;24(8):657-72 [DOI] [PubMed] [Google Scholar]
- 6. Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6(2):167-80 [DOI] [PubMed] [Google Scholar]
- 7. Tatsumoto T, Xie X, Blumenthal R, Okamoto I, Miki T. Human ECT2 is an exchange factor for Rho GTPases, phosphorylated in G2/M phases, and involved in cytokinesis. J Cell Biol. 1999;147(5):921-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saito S, Liu XF, Kamijo K, et al. Deregulation and mislocalization of the cytokinesis regulator ECT2 activate the Rho signaling pathways leading to malignant transformation.J Biol Chem. 2004;279(8):7169-79 [DOI] [PubMed] [Google Scholar]
- 9. Liu XF, Ishida H, Raziuddin R, Miki T. Nucleotide exchange factor ECT2 interacts with the polarity protein complex Par6/Par3/protein kinase Czeta (PKCzeta) and regulates PKCzeta activity. Mol Cell Biol. 2004;24(15):6665-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kimura K, Tsuji T, Takada Y, Miki T, Narumiya S. Accumulation of GTP-bound RhoA during cytokinesis and a critical role of ECT2 in this accumulation. J Biol Chem. 2000;275(23):17233-6 [DOI] [PubMed] [Google Scholar]
- 11. Kanada M, Nagasaki A, Uyeda TQ. Novel functions of Ect2 in polar lamellipodia formation and polarity maintenance during “contractile ring-independent” cytokinesis in adherent cells. Mol Biol Cell. 2008;19(1):8-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fields AP, Justilien V. The guanine nucleotide exchange factor (GEF) Ect2 is an oncogene in human cancer. Adv Enzyme Regul. 2009;50(1):190-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Weeks A, Okolowsky N, Golbourn B, Ivanchuk S, Smith C, Rutka JT. ECT2 and RASAL2 mediate mesenchymal-amoeboid transition in human astrocytoma cells. Am J Pathol. 2012;181(2):662-74 [DOI] [PubMed] [Google Scholar]
- 14. Fortin SP, Ennis MJ, Schumacher CA, et al. Cdc42 and the guanine nucleotide exchange factors Ect2 and trio mediate Fn14-induced migration and invasion of glioblastoma cells. Mol Cancer Res. 2012;10(7):958-68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Justilien V, Fields AP. Ect2 links the PKCiota-Par6alpha complex to Rac1 activation and cellular transformation. Oncogene. 2009;28(41):3597-607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang H, Linghu H, Wang J, et al. The role of Crk/Dock180/Rac1 pathway in the malignant behavior of human ovarian cancer cell SKOV3. Tumour Biol. 2010;31(1):59-67 [DOI] [PubMed] [Google Scholar]
- 17. Bourguignon LY, Gilad E, Rothman K, Peyrollier K. Hyaluronan-CD44 interaction with IQGAP1 promotes Cdc42 and ERK signaling, leading to actin binding, Elk-1/estrogen receptor transcriptional activation, and ovarian cancer progression. J Biol Chem. 2005;280(12):11961-72 [DOI] [PubMed] [Google Scholar]
- 18. Zhao Y, Zong ZH, Xu HM. RhoC expression level is correlated with the clinicopathological characteristics of ovarian cancer and the expression levels of ROCK-I, VEGF, and MMP9. Gynecol Oncol. 2009;116(3):563-71 [DOI] [PubMed] [Google Scholar]
- 19. Horiuchi A, Kikuchi N, Osada R, et al. Overexpression of RhoA enhances peritoneal dissemination: RhoA suppression with Lovastatin may be useful for ovarian cancer. Cancer Sci. 2008;99(12):2532-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Takai S, Long JE, Yamada K, Miki T. Chromosomal localization of the human ECT2 proto-oncogene to 3q26.1→q26.2 by somatic cell analysis and fluorescence in situ hybridization. Genomics. 1995;27(1):220-2 [DOI] [PubMed] [Google Scholar]
- 21. Suzuki S, Moore DH, 2nd, Ginzinger DG, et al. An approach to analysis of large-scale correlations between genome changes and clinical endpoints in ovarian cancer. Cancer Res. 2000;60(19):5382-5 [PubMed] [Google Scholar]
- 22. Haverty PM, Hon LS, Kaminker JS, Chant J, Zhang Z. High-resolution analysis of copy number alterations and associated expression changes in ovarian tumors. BMC Med Genomics. 2009;2:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cerami E, Gao J, Dogrusoz U, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Integrated genomic analyses of ovarian carcinoma Nature. 2011;474(7353):609-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Saito S, Tatsumoto T, Lorenzi MV, et al. Rho exchange factor ECT2 is induced by growth factors and regulates cytokinesis through the N-terminal cell cycle regulator-related domains. J Cell Biochem. 2003;90(4):819-36 [DOI] [PubMed] [Google Scholar]
- 26. Rosen DG, Cai KQ, Luthra R, Liu J. Immunohistochemical staining of hMLH1 and hMSH2 reflects microsatellite instability status in ovarian carcinoma. Mod Pathol. 2006;19(11):1414-20 [DOI] [PubMed] [Google Scholar]
- 27. Rosen DG, Mercado-Uribe I, Yang G, et al. The role of constitutively active signal transducer and activator of transcription 3 in ovarian tumorigenesis and prognosis. Cancer. 2006;107(11):2730-40 [DOI] [PubMed] [Google Scholar]
- 28. Rosen DG, Yang G, Deavers MT, et al. Cyclin E expression is correlated with tumor progression and predicts a poor prognosis in patients with ovarian carcinoma. Cancer. 2006;106(9):1925-32 [DOI] [PubMed] [Google Scholar]
- 29. Stevens EV, Banet N, Onesto C, et al. RhoGDI2 antagonizes ovarian carcinoma growth, invasion and metastasis. Small GTPases. 2011;2(4):202-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Freedman VH, Shin SI. Cellular tumorigenicity in nude mice: correlation with cell growth in semi-solid medium. Cell. 1974;3(4):355-9 [DOI] [PubMed] [Google Scholar]
- 31. Li J, Wang J, Jiao H, Liao J, Xu X. Cytokinesis and cancer: polo loves ROCK‘n’ Rho(A). J Genet Genomics. 2010;37(3):159-72 [DOI] [PubMed] [Google Scholar]
- 32. Williams CL. The polybasic region of Ras and Rho family small GTPases: a regulator of protein interactions and membrane association and a site of nuclear localization signal sequences. Cell Signal. 2003;15(12):1071-80 [DOI] [PubMed] [Google Scholar]
- 33. Lanning CC, Ruiz-Velasco R, Williams CL. Novel mechanism of the co-regulation of nuclear transport of SmgGDS and Rac1. J Biol Chem. 2003;278(14):12495-506 [DOI] [PubMed] [Google Scholar]
- 34. Michaelson D, Abidi W, Guardavaccaro D, et al. Rac1 accumulates in the nucleus during the G2 phase of the cell cycle and promotes cell division. J Cell Biol. 2008;181(3):485-96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Borikova AL, Dibble CF, Sciaky N, et al. Rho kinase inhibition rescues the endothelial cell cerebral cavernous malformation phenotype. J Biol Chem. 2010;285(16):11760-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dubash AD, Guilluy C, Srougi MC, Boulter E, Burridge K, Garcia-Mata R. The small GTPase RhoA localizes to the nucleus and is activated by Net1 and DNA damage signals. PLoS One. 2011;6(2):e17380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hara T, Abe M, Inoue H, et al. Cytokinesis regulator ECT2 changes its conformation through phosphorylation at Thr-341 in G2/M phase. Oncogene. 2006;25(4):566-78 [DOI] [PubMed] [Google Scholar]
- 38. Guilluy C, Dubash AD, Garcia-Mata R. Analysis of RhoA and Rho GEF activity in whole cells and the cell nucleus. Nat Protoc. 2011;6(12):2050-60 [DOI] [PubMed] [Google Scholar]
- 39. Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 2006;406:425-37 [DOI] [PubMed] [Google Scholar]
- 40. Arthur WT, Ellerbroek SM, Der CJ, Burridge K, Wennerberg K. XPLN, a guanine nucleotide exchange factor for RhoA and RhoB, but not RhoC. J Biol Chem. 2002;277(45):42964-72 [DOI] [PubMed] [Google Scholar]
- 41. Ellerbroek SM, Wennerberg K, Arthur WT, et al. SGEF, a RhoG guanine nucleotide exchange factor that stimulates macropinocytosis. Mol Biol Cell. 2004;15(7):3309-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chiu VK, Bivona T, Hach A, et al. Ras signalling on the endoplasmic reticulum and the Golgi. Nat Cell Biol. 2002;4(5):343-50 [DOI] [PubMed] [Google Scholar]
- 43. Bivona TG, Quatela S, Philips MR. Analysis of Ras activation in living cells with GFP-RBD. Methods Enzymol. 2006;407:128-43 [DOI] [PubMed] [Google Scholar]
- 44. Bivona TG, Perez De Castro I, Ahearn IM, et al. Phospholipase Cgamma activates Ras on the Golgi apparatus by means of RasGRP1. Nature. 2003;424(6949):694-8 [DOI] [PubMed] [Google Scholar]
- 45. Alan JK, Berzat AC, Dewar BJ, Graves LM, Cox AD. Regulation of the Rho family small GTPase Wrch-1/RhoU by C-terminal tyrosine phosphorylation requires Src. Mol Cell Biol. 2010;30(17):4324-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chenette EJ, Mitin NY, Der CJ. Multiple sequence elements facilitate Chp Rho GTPase subcellular location, membrane association, and transforming activity. Mol Biol Cell. 2006;17(7):3108-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tapon N, Nagata K, Lamarche N, Hall A. A new rac target POSH is an SH3-containing scaffold protein involved in the JNK and NF-kappaB signalling pathways. EMBO J. 1998;17(5):1395-404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Molli PR, Li DQ, Murray BW, Rayala SK, Kumar R. PAK signaling in oncogenesis. Oncogene. 2009;28(28):2545-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blumenstein L, Ahmadian MR. Models of the cooperative mechanism for Rho effector recognition: implications for RhoA-mediated effector activation. J Biol Chem. 2004;279(51):53419-26 [DOI] [PubMed] [Google Scholar]
- 50. Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70(3):389-99 [DOI] [PubMed] [Google Scholar]
- 51. Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70(3):401-10 [DOI] [PubMed] [Google Scholar]
- 52. Lin R, Bagrodia S, Cerione R, Manor D. A novel Cdc42Hs mutant induces cellular transformation. Curr Biol. 1997;7(10):794-7 [DOI] [PubMed] [Google Scholar]
- 53. Joyce PL, Cox AD. Rac1 and Rac3 are targets for geranylgeranyltransferase I inhibitor-mediated inhibition of signaling, transformation, and membrane ruffling. Cancer Res. 2003;63(22):7959-67 [PubMed] [Google Scholar]
- 54. Kalderon D, Roberts BL, Richardson WD, Smith AE. A short amino acid sequence able to specify nuclear location. Cell. 1984;39(3 Pt 2):499-509 [DOI] [PubMed] [Google Scholar]
- 55. Ratheesh A, Gomez GA, Priya R, et al. Centralspindlin and alpha-catenin regulate Rho signalling at the epithelial zonula adherens. Nat Cell Biol. 2012;14(8):818-28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Iyoda M, Kasamatsu A, Ishigami T, et al. Epithelial cell transforming sequence 2 in human oral cancer. PLoS One. 2010;5(11):e14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cook DR, Solski PA, Bultman SJ, et al. The ect2 rho Guanine nucleotide exchange factor is essential for early mouse development and normal cell cytokinesis and migration. Genes Cancer. 2011;2(10):932-42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Justilien V, Jameison L, Der CJ, Rossman KL, Fields AP. The oncogenic activity of ECT2 is regulated through protein kinase C{iota} mediated phosphorylation. J Biol Chem. 2011;286(10):8149-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hirata D, Yamabuki T, Miki D, et al. Involvement of epithelial cell transforming sequence-2 oncoantigen in lung and esophageal cancer progression. Clin Cancer Res. 2009;15(1):256-66 [DOI] [PubMed] [Google Scholar]
- 60. Salhia B, Tran NL, Chan A, et al. The guanine nucleotide exchange factors trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am J Pathol. 2008;173(6):1828-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kim JE, Billadeau DD, Chen J. The tandem BRCT domains of Ect2 are required for both negative and positive regulation of Ect2 in cytokinesis. J Biol Chem. 2005;280(7):5733-9 [DOI] [PubMed] [Google Scholar]
- 62. Lagana A, Dorn JF, De Rop V, Ladouceur AM, Maddox AS, Maddox PS. A small GTPase molecular switch regulates epigenetic centromere maintenance by stabilizing newly incorporated CENP-A. Nat Cell Biol. 2010;12(12):1186-93 [DOI] [PubMed] [Google Scholar]
- 63. Niiya F, Tatsumoto T, Lee KS, Miki T. Phosphorylation of the cytokinesis regulator ECT2 at G2/M phase stimulates association of the mitotic kinase Plk1 and accumulation of GTP-bound RhoA. Oncogene. 2006;25(6):827-37 [DOI] [PubMed] [Google Scholar]
- 64. Solski PA, Wilder RS, Rossman KL, et al. Requirement for C-terminal sequences in regulation of Ect2 guanine nucleotide exchange specificity and transformation. J Biol Chem. 2004;279(24):25226-33 [DOI] [PubMed] [Google Scholar]
- 65. Oceguera-Yanez F, Kimura K, Yasuda S, et al. Ect2 and MgcRacGAP regulate the activation and function of Cdc42 in mitosis. J Cell Biol. 2005;168(2):221-32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kawashima T, Bao YC, Minoshima Y, et al. A Rac GTPase-activating protein, MgcRacGAP, is a nuclear localizing signal-containing nuclear chaperone in the activation of STAT transcription factors. Mol Cell Biol. 2009;29(7):1796-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang SM, Ooi LL, Hui KM. Upregulation of Rac GTPase-activating protein 1 is significantly associated with the early recurrence of human hepatocellular carcinoma. Clin Cancer Res. 2011;17(18):6040-51 [DOI] [PubMed] [Google Scholar]
- 68. Tao J, Oladimeji P, Rider L, Diakonova M. PAK1-Nck regulates cyclin D1 promoter activity in response to prolactin. Mol Endocrinol. 2011;25(9):1565-78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Obrdlik A, Percipalle P. The F-actin severing protein cofilin-1 is required for RNA polymerase II transcription elongation. Nucleus. 2011;2(1):72-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kasai M, Guerrero-Santoro J, Friedman R, Leman ES, Getzenberg RH, DeFranco DB. The Group 3 LIM domain protein paxillin potentiates androgen receptor transactivation in prostate cancer cell lines. Cancer Res. 2003;63(16):4927-35 [PubMed] [Google Scholar]
- 71. Siu MK, Chan HY, Kong DS, et al. p21-activated kinase 4 regulates ovarian cancer cell proliferation, migration, and invasion and contributes to poor prognosis in patients. Proc Natl Acad Sci U S A. 2010;107(43):18622-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Siu MK, Wong ES, Chan HY, et al. Differential expression and phosphorylation of Pak1 and Pak2 in ovarian cancer: effects on prognosis and cell invasion. Int J Cancer. 2010;127(1):21-31 [DOI] [PubMed] [Google Scholar]
- 73. Mao YS, Zhang B, Spector DL. Biogenesis and function of nuclear bodies. Trends Genet. 2011;27(8):295-306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Woods NT, Mesquita RD, Sweet M, et al. Charting the landscape of tandem BRCT domain-mediated protein interactions. Sci Signal. 2012;5(242):rs6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Srougi MC, Burridge K. The nuclear guanine nucleotide exchange factors Ect2 and Net1 regulate RhoB-mediated cell death after DNA damage. PLoS One. 2011;6(2):e17108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Grabocka E, Wedegaertner PB. Disruption of oligomerization induces nucleocytoplasmic shuttling of leukemia-associated rho Guanine-nucleotide exchange factor. Mol Pharmacol. 2007;72(4):993-1002 [DOI] [PubMed] [Google Scholar]
- 77. Rao S, Lyons LS, Fahrenholtz CD, et al. A novel nuclear role for the Vav3 nucleotide exchange factor in androgen receptor coactivation in prostate cancer. Oncogene. 2012;31(6):716-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Fiordalisi JJ, Keller PJ, Cox AD. PRL tyrosine phosphatases regulate rho family GTPases to promote invasion and motility. Cancer Res. 2006;66(6):3153-61 [DOI] [PubMed] [Google Scholar]
- 79. Benink HA, Bement WM. Concentric zones of active RhoA and Cdc42 around single cell wounds. J Cell Biol. 2005;168(3):429-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Abdi KM, Bennett V. Adducin promotes micrometer-scale organization of beta2-spectrin in lateral membranes of bronchial epithelial cells. Mol Biol Cell. 2008;19(2):536-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Brady DC, Alan JK, Madigan JP, Fanning AS, Cox AD. The transforming Rho family GTPase Wrch-1 disrupts epithelial cell tight junctions and epithelial morphogenesis. Mol Cell Biol. 2009;29(4):1035-49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Hanker AB, Morita S, Repasky GA, Ross DT, Seitz RS, Der CJ. Tools to study the function of the Ras-related, estrogen-regulated growth inhibitor in breast cancer. Methods Enzymol. 2008;439:53-72 [DOI] [PubMed] [Google Scholar]