

Abstract
Reactive oxygen species generate ~20,000 oxidative lesions in the DNA of every cell, every day. Most of these lesions are located within nucleosomes, which package DNA in chromatin and impede base excision repair (BER). We demonstrated previously that periodic, spontaneous partial unwrapping of DNA from the underlying histone octamer enables BER enzymes to bind to oxidative lesions that would otherwise be sterically inaccessible. In the present study, we asked if these periodic DNA unwrapping events are frequent enough to account for the estimated rates of BER in vivo. We measured rates of excision of oxidative lesions from sites in nucleosomes that are accessible only during unwrapping episodes. Using reaction conditions appropriate for presteady-state kinetic analyses, we derived lesion exposure rates for both 601 and 5S rDNA-based nucleosomes. Although DNA unwrapping-mediated exposure of a lesion ~16 NT from the nucleosome edge occurred ~7–8 times per minute, exposure rates fell dramatically for lesions located 10 or more NT further in from the nucleosome edge. The rates likely are too low to account for observed rates of BER in cells. Thus, chromatin remodeling, either BER-specific or that associated with transcription, replication, or other DNA repair processes, probably contributes to efficient BER in vivo.
Keywords: Base excision repair, Human NTH1 kinetics, Chromatin, Nucleosome dynamics, DNA unwrapping kinetics
1. Introduction
Endogenous reactive oxygen species (ROS) are produced during ordinary oxidative metabolism and serve in various functions, ranging from metabolic signaling to the destruction of microbes ingested by phagocytes. Despite the presence of ROS-neutralizing enzymes, endogenously generated ROS produce ~20,000 oxidative lesions in the DNA of every nucleated human cell, every day. Some oxidatively damaged bases mispair upon replication and are thus mutagenic. Other base lesions, and abasic sites, block progression of replicative polymerases, and may trigger apoptosis. Most oxidatively damaged bases are shunted into the short patch base excision repair (BER) pathway, which begins with the removal of a damaged base by one of several DNA glycosylases [1–5]. DNA glycosylases bind to the minor groove of DNA, flip the damaged base approximately 180° out of the helix, and hydrolyse the glycosidic bond, leaving an abasic site. Bifunctional DNA glycosylases possess an AP lyase activity as well, which cleaves the DNA backbone 3′ to the abasic site. Various polymerase-blocking moieties are removed from the 3′ end by apurinic endonuclease (APE1) or PNK (polynucleotide kinase), which produce a 3′ hydroxyl group that can be used by DNA pol β to fill in the correct base. The resulting nick is sealed by DNA ligase IIIα, to complete the repair.
Most of the DNA in eukaryotes is packaged into regular nucleosome arrays that, together with numerous DNA and nucleosome-associated enzymes and regulatory factors, make up chromatin. Nucleosomes only marginally protect DNA from oxidative damage but substantially impede BER [6]. The underlying histone octamer and close positioning of adjacent DNA gyres hinder the binding of repair enzymes. Additional impediments to the binding of BER factors may include the distortion of the major and minor grooves of DNA wound about the histone octamer, and the restricted mobility of nucleosomal DNA, due to discrete interactions between the histone core and DNA every helical turn [7]. Chromatin-remodeling agents help circumvent nucleosome-related impediments to DNA replication, transcription, nucleotide excision repair and double strand DNA break repair [8–15]. The remodeling complexes SWI/SNF, RSC and ISW1 and ISW2 have all been reported to enhance BER of oxidative lesions in nucleosomes in vitro [16–18], however, it is not yet clear that any of these agents promote BER in vivo. Individual chromatin remodeling factors commonly act in multiple processes, making it experimentally difficult to probe their effects on BER without generating more global cellular effects. Also, treatment of cells with ROS generates multiple classes of lesions, including single and double strand DNA breaks; double strand DNA break repair pathways are known to involve chromatin-remodeling agents [6,19].
We reported previously that the DNA glycosylase hNTH1 efficiently excised thymine glycol (Tg) residues from nucleosomal DNA, provided the Tg lesion was oriented in a manner that enabled hNTH1 to bind without clashing with the histone octamer. At higher concentrations, hNTH1 was also able to excise Tg residues from sites that we predicted would be sterically occluded; excision of lesions from sterically occluded sites near the nucleosome edge was more efficient than for lesions at occluded sites closer to the dyad axis. None of these excision reactions required or induced the irreversible disruption of the Tg-containing nucleosomes, nor did they alter the helical or translational position of DNA relative to the underlying histone octamer. Collectively, these observations provided strong support for what we termed the unwrapping hypothesis, which states that spontaneous, partial unwrapping of DNA from the histone octamer periodically exposes BER substrates, enabling the binding of repair enzymes [6,20,21]. Extensive earlier studies indicate that these periodic unwrapping events occur both in vitro and in vivo, and may facilitate several chromatin-related activities [22–24]. Critically, even though the hNTH1 concentrations needed for the efficient processing of sterically occluded lesions were much higher than those needed to excise more optimally oriented lesions, the hNTH1 concentrations were never higher than those found in vivo [25]. These observations raised the formal possibility that spontaneous nucleosome unwrapping by itself can account for the observed rates of BER in vivo. Estimates of rates of BER in vivo are based on the rate of return to steady-state levels of oxidative DNA damages after exposing cells to exogenous ROS. In such assays, oxidative DNA damages of the kind subject to short patch BER return to steady-state levels within 1 h [26,27]. In the present study, we asked if the frequency of transient unwrapping episodes that enable hNTH1 to excise lesions from occluded sites in nucleosomes is high enough to enable the repair of most lesions within this ~1 h time-frame. By establishing appropriate reaction conditions and using multiple nucleosome constructs, we have calculated lesion exposure rates due to nucleosome unwrapping, as a function of both DNA sequence and distance from the nucleosome edge. The inferred rates of unwrapping are consistent with those measured by Widom and colleagues (c.f. Ref. [28] and Section 4), and suggest that spontaneous unwrapping alone can not account for the efficient repair of oxidative lesions in vivo.
2. Materials and methods
2.1. DNA and nucleosome substrates
Model nucleosomes were assembled with either of two nucleosome positioning sequences. 5S nucleosomes contained Lytechinus variegatus 5S rDNA, with a single, discretely positioned thymine glycol [Tg] in place of an ordinary thymine residue, and these were assembled into nucleosomes by octamer transfer, as described [20]. The mapping of the helical orientation and translational positions of lesions in these nucleosomes is shown in [20]. The replacement of a single dT residue with a Tg had no detectable impact on nucleosome stability or positioning. 601 nucleosomes contained the synthetic 601 DNA nucleosome positioning sequence, again with a single discretely positioned Tg; these were assembled into nucleosomes, using purified histone octamers, containing recombinant Xenopus laevis histones, as described [21]. In all cases, the efficiency of nucleosome reconstitution was quantified as described in [20].
2.2. Enzymes and enzyme assays
hNTH1 is one of five known bifunctional DNA glycosylases in humans, and can excise oxidized pyrimidines, including the polymerase-blocking lesion, thymine glycol (Tg) [29,30]. Human NTH1, and a variant which lacks the first 55 amino acid residues (hNTH1-d55), were prepared as described [25]. Removal of this N-terminal segment reduces the propensity of hNTH1 to dimerize but does not change its base excision activity [31], which simplified the measurement of hNTH1-lesion binding constants in Fig. 2. In single-turnover reactions, full-length hNTH1 and hNTH1-d55 exhibited virtually identical activities on nucleosomal substrates.
Fig. 2.
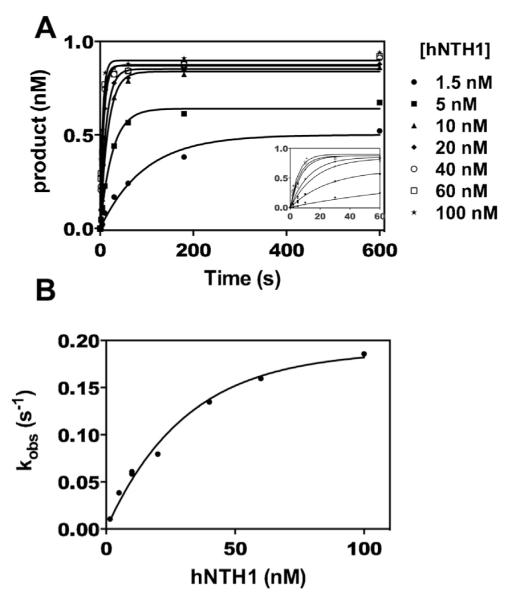
Kinetics of hNTH1-mediated excision of thymine glycol (Tg) from naked DNA. To determine the maximum rate of lesion exposure that could be detected in the assays shown in Figs. 3 and 4, we measured the rate constant for excision of Tg by hNTH1 (k+3 in Fig. 1). Panel A shows product formation as a function of time for excision of Tg from a synthetic, 32P-end-labeled 35 bp DNA fragment; the inset shows early time points on an expanded time scale. Reactions contained 1 nM DNA and 0–100 nM hNTH1(d55). Aliquots from the reactions were removed at varying times, quenched in 0.1 M NaOH, and heated at ~100 °C for 2–3 min; this treatment drives beta and delta elimination at abasic sites, generating single strand DNA cleavage products that can be readily assayed on sequencing gels. Thus the kinetic constants derived from this analysis reflect only the first of the two enzymatic activities in hNTH1, namely hydrolysis of the glycosidic bond. Note that hNTH1 converted all of the available substrate into product at high enzyme concentrations but not at low, subsaturating concentrations. This most likely reflects product inhibition of the enzyme. Panel B: data in Panel A were fit to Eq. (1) (solid lines) and observed rate constants (kobs) were plotted as a function of hNTH1(d55) concentration. In addition to determining the k+3 we used these data to calculate the lesion binding constants (k+2, k−2 and Kd), as described in Section 2. These values indicated the general enzyme concentrations needed to ensure that lesion binding (k−2 and k+2) would not interfere with the lesion exposure measurements in Figs. 3 and 4.
DNA glycosylase reactions were conducted at 37 °C in 100 mM NaCl, 25 mM HEPES, pH 8.0, 1 mM EDTA, and 0.05% NP-40. All enzyme concentrations indicated in the text and figure legends refer to the active enzyme fraction (generally 20–30%, as determined using a Schiff-base trap assay [32]). Reactions with 5S rDNA nucleosomes or the corresponding naked DNA contained 0.33 nM specific substrate and 50–100 nM donor chromatin. In the naked DNA reactions, donor chromatin was added just prior to addition of enzyme, so that its concentration matched that in reactions with nucleosome substrates. Reactions with 601 nucleosomes or the corresponding naked DNA contained 4.5 nM specific substrate and ~45 nM unlabeled, lesion-free nucleosomes. All reactions were quenched in 0.1 M NaOH and incubated at ~100 °C for 2–3 min, to cleave DNA at hNTH1-generated abasic sites. Reaction products were then fractionated, visualized, and quantified as described for measurement of DNA glycosylase activity [20]. Data from enzyme assays with nucleosome substrates were computationally corrected for the contribution of small amounts of contaminating naked DNA, as described [20].
2.3. Data analysis
Previously mechanisms by which proteins access DNA sequences within nucleosomes have been studied using steady-state conditions [22]. More recently partial DNA unwrapping from the histone octamer has been studied using presteady-state conditions [28,33]. We have taken a similar approach here in that all experiments presented here were performed under single-turnover, pseudo-first order conditions. Enzyme reactions summarized in Fig. 2 contained 1 nM of a gel-purified, 32P-end-labeled 35 NT oligonucleotide with a centrally positioned Tg residue, annealed to a complementary oligonucleotide, and treated with hNTH1 concentrations noted in the figure. Each reaction progress curve was fit to Eq. (1) to determine an observed rate constant (kobs). These observed rate constants were graphed as a function of enzyme concentration (Fig. 2B). Under the pseudo-first-order conditions, the slope of the initial linear portion of the resulting curve is equal to the enzyme–substrate association rate constant and the y-intercept is equal to the enzyme–substrate dissociation rate constant. Thus, values for k+2 and k−2 in the reaction scheme shown in Fig. 1 were determined by fitting data from the 5 to 40 nM [hNTH1] reactions in Fig. 2B to Eq. (2). (The 1.5 nM reaction curve data were excluded because they did not meet the pseudo-first-order criterion.) The value for k+3 in the reaction scheme shown in Fig. 1 was determined by extrapolation to a maximum kobs by fitting data in Fig. 2B to a single exponential function [34]. Nucleosome unwrapping rates were determined from kobs measured in Figs. 3 and 4. The value for k+1 in the reaction scheme shown in Fig. 1 was determined by using the expression for a system approaching equilibrium after perturbation (Eq. (4)), in which the observed rate constant for the approach to equilibrium is equal to the sum of the rate constants for the forward and reverse reactions. At saturating amounts of glycosylase, rewrapping of DNA is inhibited (k−1 does not contribute to kobs) and kobs = k+1 [35].
Fig. 1.
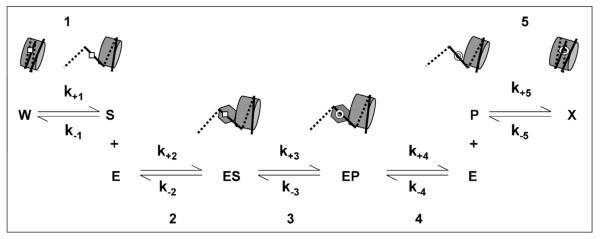
Minimal mechanism for the excision of an oxidatively damaged base from a sterically occluded position within the nucleosome. The five steps and associated kinetic constants depict [1] a spontaneous conformation change (here, partial unwrapping of nucleosomal DNA) that exposes the damaged base (square) to solvent; [2] binding of a DNA glycosylase (hexagon) to the damaged base to form an enzyme–substrate complex; [3] excision of the damaged base by the DNA glycosylase to form an apurinic site (open circle); [4] dissociation of the DNA glycosylase and [5] re-wrapping of the processed DNA. Lesion exposure rates were measured (in Figs. 3 and 4) in reactions in which Step 1 was rate-limiting.
Fig. 3.

Thymine glycol (Tg) exposure rates in 601 nucleosomes. 32P-end-labeled 601 DNA containing a single Tg (DNA), and the same DNA after assembly into nucleosomes (NUC), were treated separately with varying concentrations of hNTH1, as outlined in Section 2. Aliquots from the reactions were collected, treated as described in the legend of Fig. 2, and fractionated on sequencing gels. Naked DNA reactions were used to computationally correct for small amounts of naked DNA contamination present in the nucleosome reactions as described previously [20]. The resulting excision data were normalized to the total naked DNA excised, were graphed as a function of time and fit to Eq. (1) (solid lines). The naked DNA and corrected nucleosome reaction progress curves are shown in the top panel. For added clarity, the bottom panel shows the same nucleosomal curves, graphed with an expanded vertical axis. The single kinetic constant for excision of Tg from 601 nucleosomes is consistent with a single conformational change and thus the single discrete translational position that 601 DNA adopts when assembled into nucleosomes.
Fig. 4.

Multiple thymine glycol (Tg) exposure rates in 5S rDNA nucleosomes. (A) The translational positions of the 5S rDNA nucleosomes were mapped and quantified using restriction enzyme digestions [20]. The positions denoted as −1 and +1 are identical to the two major positions mapped in [37]; position 0 is identical to that seen in [37] after incubating nucleosomes at elevated temperatures. The restriction enzymes used to map the translational variants are indicated [20]. The shaded boxes indicate the position of the histone octamer with respect to the DNA, with the dyad axis indicated by the vertical black bar at the center. The asterisk indicates the position of the Tg within the DNA, which is differentially close to the nucleosome edge in the different translational subpopulations. (B) 32P-end-labeled 5S rDNA containing a single Tg (DNA), and the same DNA after assembly into nucleosomes (NUC), were separately treated with varying concentrations of hNTH1(d55), as outlined in Section 2. Aliquots from the reactions were collected, treated as described in the legend of Fig. 2, and fractionated on sequencing gels. The resulting excision data were normalized to total naked DNA excised and were graphed and fit to Eq. (3) (solid lines). The two kinetic constants observed for excision of Tg from 5S rDNA nucleosomes are consistent with the multiple translational positions that 5S rDNA adopts when assembled into nucleosomes. For clarity, the nucleosomal curves were graphed with an expanded vertical axis (bottom panel). Note that the reaction curve shown for 75 nM hNTH1-d55 is only marginally faster than that obtained with 15 nM d55-hNTH1 as expected for enzyme–saturating conditions.
| (1) |
| (2) |
| (3) |
| (4) |
3. Results
3.1. Experimental design
Early studies of dynamic events that enable proteins to bind DNA within nucleosomes employed steady-state reaction conditions [22]. In later studies, the kinetics of partial unwrapping of DNA from the histone octamer were investigated using pre-steady-state, pseudo-first order conditions [28,33]. We chose this latter methodology as well, as it enabled us to isolate discrete steps leading to formation of enzyme–substrate complexes in nucleosomes, as depicted in Fig. 1. Step 1 in Fig. 1 depicts a conformational change that transiently exposes the lesion, making it available for capture by a DNA glycosylase. In the absence of enzyme, nucleosomes are in a constant dynamic equilibrium between lesion exposure (k+1) and occlusion (k−1). The addition of a DNA glycosylase that can bind a transiently exposed lesion will prevent DNA rewrapping. At glycosylase concentrations high enough to prevent all rewrapping, the observed rate of product formation will be limited by the rate of lesion exposure (k+1). The lesion exposure rates measured will thus reflect nucleosome unwrapping events that are extensive enough to permit the binding of hNTH1. If lesion exposure rates were to exceed the rate of glycosidic bond hydrolysis (i.e. k+1 > k+3 in Fig. 1) then nucleosome substrates could not be differentiated from naked DNA (formation of product in both cases is limited by glycosidic bond hydrolysis (Step 3 in Fig. 1)). In the studies described below, lesion exposure rates proved to be rate-limiting.
Fig. 2 summarizes experiments that enabled us to establish appropriate reaction conditions for the measurement (in Figs. 3 and 4) of exposure rates for lesions in nucleosomes. As described above, it was necessary first to measure k+3 for hNTH1, since this rate constant governs the maximum lesion exposure rate that can be resolved in this study. We therefore conducted a series of reactions with varying concentrations of hNTH1 and a fixed concentration (1 nM) of a 32P-end-labeled 35 bp oligonucleotide containing a centrally positioned Tg (c.f. Section 2). Fig. 2A shows formation of product as a function of time, for increasing concentrations of hNTH1. Data for each reaction were fit to Eq. (1) (c.f. Section 2), to determine an observed rate constant (kobs). The rate constants were then graphed as a function of enzyme concentration (Fig. 2B). The initial linear portion of this graph was fit to a line (Eq. (2); c.f. Section 2), in which the slope is equal to k+2 (2.6 × 106 ± 0.16 × 106 M−1 s−1) and the y-intercept is equal to k−2 (0.03 ± 0.003 s−1). These values allowed us to calculate an equilibrium dissociation constant (Kd) of about 11 nM. Knowing the approximate equilibrium constant enabled us to choose enzyme concentrations for nucleosome reactions that are high enough to be saturating, so that the overall observed rate of lesion processing would not be affected by k+2 and k−2, and would instead directly reflect lesion exposure rates (k+1). Recognizing that k+2 and k−2 may vary with substrate length or local sequence, we also conducted base excision reactions on nucleosomes at various hNTH1 concentrations to demonstrate saturation. These measurements (c.f.Figs. 3 and 4) provided assurance that hNTH1 concentrations used to measure lesion exposure rates were saturating. The data in Fig. 2B also enabled us to determine the rate constant for hydrolysis of the glycosidic bond between Tg and its associated deoxyribose sugar (k+3 = 0.19 ± 0.01 s−1). This hydrolysis rate constant sets the upper limit of detection (in this study) for the conformational changes associated with lesion exposure. As the value of k+3 might also vary as a function of local sequence context, we also measured the base excision rates for naked 5S and 601 DNA substrates (c.f.Figs. 3 and 4), to define more precisely the limits of our assays.
3.2. Lesion exposure rates in 601 nucleosomes
Synthetic 601 DNA assembles into a highly stable nucleosome with a single discrete translational and helical position with respect to the underlying histone octamer [36]. This enabled us to assemble a homogenous population of “Tg-601” nucleosomes, all containing a single Tg residue at a single site (47 NT from the dyad axis). This site is inaccessible to hNTH1, except when exposed by spontaneous partial unwrapping of nucleosomal DNA. Fig. 3 shows reaction progress curves for Tg-601 nucleosomes and naked DNA, incubated separately with increasing concentrations of hNTH1. To determine kobs for the nucleosome reactions, we graphed product formation as a function of time and fit to Eq. (1). In reactions with saturating amounts of hNTH1, the observed rate constant is 0.0014 ± 0.0004 s−1. This is far slower than the rate for hydrolysis at the hNTH1 active site (k+3 = 0.19 s−1), and should therefore reflect the conformational change required for lesion exposure, i.e. DNA unwrapping (k+1). At saturating levels of hNTH1, the observed rate constant for the excision of Tg from naked 601 DNA (0.03 ± 0.005 s−1) is far lower than that measured for the oligonucleotide. We suggest possible reasons for this result in Section 4. Regardless of the reason, however, the lower than expected rate constant means that the upper limit for the detection of 601 nucleosome unwrapping rates is correspondingly lower. Still, this revised upper limit is 20-fold higher than the kobs measured on the nucleosomes (i.e. k+3 ≈ 20 × k+1 in Fig. 1), indicating that kobs reflects unwrapping-mediated lesion exposure rates. The average lifetime of the lesion in its occluded state(s), equal to the inverse of the observed rate of exposure, is approximately 12 min.
3.3. Lesion exposure rates in 5S rDNA nucleosomes
Competitive nucleosome reconstitution studies indicate that 601 nucleosomes are far more stable than most naturally occurring nucleosomes [36], which may explain why it is virtually impossible to propagate 601 DNA in eukaryotic hosts. To the extent that nucleosome unwrapping facilitates access to DNA targets in vivo, it was important to also measure lesion exposure rates in naturally-occurring nucleosomes. 5S rDNA from the sea urchin L. variegatus adopts a discrete helical position relative to the histone octamer, but occupies several translational positions that differ from one another by multiples of 10 bp. The relative abundance of the different translational variants varies somewhat with reconstitution and assay conditions. For example, when 5S rDNA nucleosomes are assembled by dialysis from high to very low salt (final ionic strength ~10 mM), and analyzed in low salt buffers, most exhibit two preferred translational positions, separated by 20 bp [37,38] and corresponding to what we refer to in Fig. 4A as positions +1 and −1. These same nucleosomes, when incubated at 37 °C or higher, for ~40 min or more, migrate to a single dominant translational position [37] which, in Fig. 4A, we refer to as “position 0”. In the present study, 5S rDNA nucleosomes were assembled by octamer transfer in high salt, diluted in a stepwise fashion to near-physiological salt concentrations (final ionic strength ~125 mM), and stored and assayed in buffers of similar ionic strength. DNase I and restriction enzyme analyses [20,25] indicate that approximately half (~48%) of the nucleosomes assembled in this manner formed at position 0, while the other half occupied sites located 10 bp to either side of position 0 (i.e. positions −1 and +1 in Fig. 4A). The Tg lesion in the 5S rDNA nucleosomes at position 0, was located ~46 NT from the dyad axis. In the ~34% of nucleosomes at position −1, the Tg lesion was located 10 NT closer to the dyad axis. In the final ~18% of nucle osomes, which formed at position +1, the Tg lesion was positioned 10 NT further from the dyad axis. (We cannot rule out minor additional translational variants, such as those reported in [38], but such variants would have only a marginal impact on the results reported here.) The helical orientation of the Tg lesion appeared to be the same for each of the translational variants [20]. Thus in all cases lesion excision depended on an unwrapping-mediated exposure event.
Fig. 4 shows reaction curves for the excision of Tg from 5S rDNA nucleosomes, at varying hNTH1 concentrations. Strikingly different from 601, are the biphasic kinetics of lesion excision from 5S rDNA nucleosomes. To determine kobs1 and kobs2, we graphed product formation as a function of time, and fit the data to Eq. (3). In reactions with saturating amounts of hNTH1, kobs1 was 0.13 ± 0.03 s−1 and kobs2 was 0.003 ± 0.0009 s−1. The reaction curves for naked 5S DNA were monophasic and fit to Eq. (1), giving an observed rate constant (0.21 ± 0.03 s−1), very close to that measured for the 35 bp DNA fragment in Fig. 2. As was the case for the Tg-containing 601 nucleosomes, the observed rate constants for excision of Tg from 5S rDNA nucleosomes were less than those observed for naked DNA substrates, indicating that lesion exposure is rate-limiting in the nucleosome reactions. The amplitude of the faster of the two phases closely matches the fraction of the nucleosomes at position +1 (Fig. 4A). Hence, the simplest interpretation is that kobs1 reflects excision of Tg from nucleosomes in the position +1 subpopulation. This interpretation is also consistent with the close proximity of Tg lesions in the +1 subpopulation to the edge of the nuclesome (~17 NT), where we would predict relatively high rates of unwrapping and lesion exposure. The remainder of the substrate processed during the time frame of the reaction occurred at a slower rate (kobs2), and likely reflects the excision of Tg from nucleosomes in translational subpopulation 0 (Fig. 4A). Based on kobs2 we would predict that processing of lesions in this nucleosome fraction would largely be complete in ~20 min. In principle, we should then have seen a third phase, reflecting excision of lesions from nucleosome subgroup −1. However, the kinetic differences between subgroups +1 and 0 led us to predict that the theoretical kobs3 would be so low as to make it impractical to measure using these methods: specifically, we were concerned that extending reaction times to several hours at 37 °C might compromise enzyme activity or substrate stability.
4. Discussion
In this study we have measured the kinetics with which oxidatively damaged DNA at sterically occluded sites in nucleosomes is made available for repair by the transient partial unwrapping of DNA from the histone octamer. Extensive studies by Widom and colleagues indicate that unwrapping of nucleosomal DNA begins at the nucleosome edge and proceeds toward the dyad axis; the extent and duration of unwrapping appear stochastic. Using a LexA binding assay and ensemble FRET measurements, Widom and colleagues estimated that nucleosome unwrapping events expose sites within but near the nucleosome edge several times per second, and that such sites remain in an exposed configuration for 10–50 ms[28,33,39]. Subsequent, single molecule FRET studies suggested that transitions between wrapped and unwrapped states occur on a much faster time scale [40]. These apparently very rapid transitions were later attributed to photoblinking, a phenomenon in which the acceptor dye transiently shifts to a non-emitting state [41]. Nevertheless, the uncertainty engendered by the single molecule FRET studies highlighted the value of using an entirely different method to measure nucleosomal DNA unwrapping and rewrapping rates.
For the study presented here, we assembled nucleosomes containing a single Tg lesion, located at sites where its helical orientation relative to the underlying histone octamer made it sterically inaccessible, except when exposed through nucleosome unwrapping. The first of these nucleosomes contained “601” DNA, which assembles into a highly stable and homogenously positioned nucleosome [23,36]. In reactions containing saturating amounts of the DNA glycosylase hNTH1, the resulting reaction progress curves fit Eq. (1). The rate extracted from these curves (0.0014 ± 0.0004 s−1) is similar to the exposure rate measured in a FRET-based assay, of a LexA binding site located 28–47 NT from the edge of a 601-based nucleosome (0.0017 ± 0.0010 s−1; [28]; Table 1). Since the Tg lesion in the Tg-601 nucleosome is ~27 NT from the nucleosome edge, one might have expected a significantly higher exposure rate, similar to that measured for LexA binding to a site located 18–37 NT from the nucleosome edge (0.016 ± 0.0003 s−1; [28]; Table 1). Clearly, however, the observed rate constants are influenced by the size and binding characteristics of the protein and target site used to measure unwrapping rates. Members of the hNTH1 glycosylase family bind the minor groove of DNA, inducing it to bend (on the order of ~60°) away from the active site; glycosylase activity also entails the insertion of several amino acid residues into the DNA helix, which helps stabilize the damaged base in its extra-helical configuration. Lesion processing may thus require the unwrapping of more DNA between the lesion and the histone octamer than is required for the binding of LexA at an equivalently positioned site.
Table 1.
Nucleosome unwrapping rates.
| Nucleosome | 601a | 601a | 601a | 5S (+1)b | 5S (0)b | 601b |
| Protein probe | LexA | LexA | LexA | hNTH1 | hNTH1 | hNTH1 |
|
| ||||||
| Distance of target sequence from the nucleosome edge (bp) | 17.5 | 27.5 | 37.5 | 16 | 26 | 27 |
| κ (s−1) | 4.1±0.2 | 0.016±0.003 | 0.0017±0.0010 | 0.13±0.03 | 0.003±0.0009 | 0.0014±0.0004 |
| Average lifetime of occluded state(s): τ (s) | 0.24 | 63 | 590 | 7.6 | 330 | 710 |
This manuscript.
As evident in Fig. 3, hNTH1 excised only ~1/3 of the Tg lesions in Tg-601 nucleosomes, even though reactions were continued for up to 60 min, time enough for ~5 lesion exposure events. Gel mobility shift experiments did not reveal any changes in nucleosome integrity during the 60 min reaction time; other control experiments ruled out loss of hNTH1 activity. This result is reminiscent of a previous study of nucleosome unwrapping [28], where researchers were unable to determine a Kd for binding of LexA to a similarly positioned target site in 601 nucleosomes, despite use of exceedingly high concentrations of LexA. Thus, the unwrapping rates measured in that study must also be attributed to a small fraction of the nucleosomes present. These findings together with ours suggest that, while 601 nucleosomes are homogenous with respect to their translational position, there may be conformational variants of some other kind that suppress or alter the timescale of DNA unwrapping. The basis for such heterogeneity might, for example, reside in a DNA stretching-associated kink that is evident in the 601 nucleosome crystal structure close to superhelix location 4.5 [42], immediately adjacent to the Tg in Tg-601 nucleosomes (located 47 nucleotides from the dyad axis). An unwrapping event of sufficient magnitude to permit hNTH1 binding might be expected to quickly ‘erase’ any unusual, local DNA configuration imposed by nucleosome formation. However, the anomalous electrophoretic migration of naked 601 DNA, which suggests the presence of intrinsic DNA bends [36], makes it possible that a portion of the lesion-exposed 601 DNA exists in a relatively stable configuration that reduces the probability of hNTH1 binding or processing within the lifetime of the exposed state. Intrinsic DNA bending might, for example, increase the energetic cost of flipping the Tg lesion into the active site of hNTH1. DNA bending may also help explain why the observed rate constant for the excision of Tg from naked 601 DNA (~0.03 s−1) is far lower than observed for both the 5S rDNA and the 35 bp DNA fragment used in Fig. 2 (0.21 s−1 and 0.19 s−1, respectively). Although the slower base excision rate measured with 601 DNA reduced the upper limit of detection for unwrapping rates, this had no impact in the present study, as the observed rates of lesion excision from the 601 nucleosomes proved to be 20-fold below this upper limit. Thus, we are confident that our measurements reflect a conformational change required for lesion exposure.
To measure unwrapping dynamics in a more biologically meaningful context, we used 5S rDNA-containing nucleosomes. 5S rDNA nucleosomes share a single common helical orientation but occupy a number of translational positions that differ from one another by multiples of 10 bp. Restriction enzyme analyses indicated that approximately half of the 5S rDNA nucleosomes had a translational position in which the Tg residue was located ~46 NT from the dyad axis [20]. The Tg residue was 10 NT closer to the dyad axis in another approximately one-third of the nucleosomes, and 10 NT further from the dyad axis in the remaining approximately one-sixth of the nucleosomes (Fig. 4A). One might expect different lesion exposure rates for the different translational variants and, indeed, the resulting hNTH1 reaction progress curves were biphasic. The faster of the observed rates was nearly as high as that measured for hNTH1 acting on naked DNA (Step 3 in Fig. 1) and thus is close the limit of detection for our experimental system. Nevertheless, one can be reasonably confident that the observed rate is due to a conformational change that is distinct from the base excision step, as the naked DNA and nucleosomal reactions were conducted as “side-by-side” replicates, using the same DNA and enzyme preparations to control for potential variation from one experiment to the next. At each of the early time points, reactions conducted with naked DNA contained more product than those conducted with nucleosomes, indicating a distinct, faster rate. It is noteworthy as well that the amplitude of this fast phase was equivalent to the proportion of the 5S rDNA nucleosomes predicted to have the Tg lesion 16–17 NT from the nucleosome edge. Thus, the fast phase of the hNTH1 reactions with 5S rDNA can be said to reflect a conformational change necessary for base excision step.
The second of the two kinetic constants measured for 5S rDNA nucleosomes reflects a slower conformational change that most likely reflects the unwrapping required to expose lesions that are ~46 NT from the dyad axis. This rate is slightly (~2-fold) faster than the rate of unwrapping that we measured for 601 nucleosomes containing a Tg at approximately the same location (i.e. 47 NT from the dyad axis). This rate difference is much smaller then the 40-fold difference in lesion exposure rates for the 5S translational variants +1 and 0 (Table 1), and could reflect either intrinsic (e.g. DNA sequence) or experimental differences. For example, the ~2-fold rate difference could be due to the post-translational modification of histones used to assemble 5S rDNA-containing nucleosomes (c.f. Section 2), as acetylation of K56 in histone H3 has been reported to increase unwrapping rates [43,44]. This is unlikely, however, not only because the influence of histone H3 K56 on unwrapping rates appears to be limited to DNA near the nucleosome edge, but also because we observed similar differences in the efficiency of lesion excision in studies where 5S rDNA and 601 nucleosomes were both assembled with recombinant histones [21]. Thus, it is most likely that DNA sequence differences account for the twofold difference in lesion exposure rates for the 601 and 5S rDNA nucleosomes.
The timescale for lesion exposure events detected in this study is defined by the base excision step of hNTH1. One would need to use a glycosylase with a faster catalytic rate to detect other more rapid conformational changes. A recent study by Stivers and colleagues provides evidence that more rapid conformational changes occur and also influence BER of DNA lesions in nucleosomes [45]. Those authors studied the excision of uracil residues from selected sites in nucleosomes by hUNG, a human uracil DNA glycosylase. Unlike hNTH1, hUNG is a monofunctional DNA glycosylase but shares with hNTH1 a similar glycosylase reaction mechanism. Under single-turnover reaction conditions, Stivers and colleagues observed several, relatively fast conformational changes associated with excision of residues from sites where there were no predicted steric clashes between the histone octamer and the incoming DNA glycosylase. Although the physical events behind these very rapid changes are not yet understood, their existence is consistent with our earlier studies, which indicated that favorably oriented oxidative lesions can be processed directly, without the involvement of DNA unwrapping [20].
Molecular tweezer studies indicated that DNA in nucleosomes can be “unzipped” up to the dyad, without irreversibly disrupting the nucleosome, whereas unzipping past the dyad resulted in removal of the octamer [46]. These observations suggested that spontaneous, transient DNA unwrapping events could, at least in principle, expose virtually all oxidative lesions in nucleosomes, enabling lesion capture and early steps in BER to occur without significant nucleosome disruption. (The nucleosome destabilizing activity of DNA ligase IIIα, in association with XRCC1, likely facilitates the final two steps in BER [21].) If the in vivo repair of the nucleosome lesions examined in the present study were limited by rates of spontaneous DNA unwrapping, it would require ~20 min in the case of 5S nucleosomes, and ~40 min in the case of 601 nucleosomes. These times are within the ~1 h time that cells require to completely repair ‘simple’ oxidative damages that result from exposure of cells to oxidizing agents [26,27]. However, the frequency of DNA unwrapping events that expose lesions closer to the dyad axis than those measured in the present study are likely to be far lower, resulting in repair times much longer than have been measured in cells. These considerations suggest that spontaneous unwrapping of nucleosomal DNA may contribute to but is not sufficient to account for the efficient repair of all oxidative lesions in vivo. Lesions that are not rendered accessible by unwrapping within a biologically meaningful time-frame might simply remain unrepaired until exposed through DNA replication- or transcription-mediated disruption of nucleosomes. Indeed, certain members of the Nei/fpg family of glycosylases may act principally during replication or transcription. Human NEIL1, for example, is expressed at high levels during S-phase and interacts with several components of the replisome [47–49], while human NEIL2 has been reported to associate with RPA and RNA polymerase II [50]. By contrast, hNTH1 and hOGG1 are present throughout the cell cycle and are more likely to function independently of replication and transcription; they may thus benefit most from unwrapping-driven exposure of nucleosome lesions.
Given the difficulty in measuring with precision the in vivo rates of BER, we cannot rule out the possibility that, together, replication-, transcription-, and unwrapping-mediated exposure of oxidative lesions is sufficient to account for BER in cells. Alternatively, there may be as yet undiscovered mechanisms that recruit chromatin remodeling agents to sites of oxidative damage. Some of these agents are thought to act by inducing or trapping a DNA loop within the nucleosome. The migration of such a loop around the histone octamer, followed by its release on the ‘other side’, would move the octamer by a loop-sized increment [51]. Conceivably, the spontaneous partial unwrapping of DNA, such as that investigated here, may help initiate or enhance formation of such loops, through out-of-register re-wrapping events. In this manner, chromatin remodeling factors may capitalize on dynamic behaviors that are intrinsic to nucleosomes.
Acknowledgements
We thank the UVM DNA analysis facility for sequencing and phosphorimagery support, April Averill for the isolation of DNA glycosylases, Scott Kathe for enzyme activity determinations, Joy-El Barbour and Ian Odell for isolation of histones, and Jeff Bond and members of the Pederson lab for helpful conversations, and Joyce Heckman for comments on the manuscript.
Funding This research was supported by NSF grant MCB-0821941 and NCI grant P01-CA098993 to D.S. Pederson. The authors of this study have no financial conflicts of interest.
Footnotes
Conflict of interest statement None.
References
- [1].David SS, O’Shea VL, Kundu S. Base-excision repair of oxidative DNA damage. Nature. 2007;447:941–950. doi: 10.1038/nature05978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wallace SS, Murphy DL, Sweasy JB. Base excision repair and cancer. Cancer Letters. 2012;327:73–89. doi: 10.1016/j.canlet.2011.12.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Dizdaroglu M. Oxidatively induced DNA damage: mechanisms, repair and disease. Cancer Letters. 2012;327:26–47. doi: 10.1016/j.canlet.2012.01.016. [DOI] [PubMed] [Google Scholar]
- [4].Hazra TK, Das A, Das S, Choudhury S, Kow YW, Roy R. Oxidative DNA damage repair in mammalian cells: a new perspective. DNA Repair. 2007;6:470–480. doi: 10.1016/j.dnarep.2006.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Hegde ML, Hazra TK, Mitra S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Research. 2008;18:27–47. doi: 10.1038/cr.2008.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Odell ID, Wallace SS, Pederson DS. Rules of engagement for base excision repair in chromatin. Journal of Cellular Physiology. 2013;228:258–266. doi: 10.1002/jcp.24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8Å resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- [8].Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annual Review of Biochemistry. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223. [DOI] [PubMed] [Google Scholar]
- [9].Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Flaus A, Owen-Hughes T. Mechanisms for ATP-dependent chromatin remodelling: the means to the end. FEBS Journal. 2011;278:3579–3595. doi: 10.1111/j.1742-4658.2011.08281.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Avvakumov N, Nourani A, Cote J. Histone chaperones: modulators of chromatin marks. Molecular Cell. 2011;41:502–514. doi: 10.1016/j.molcel.2011.02.013. [DOI] [PubMed] [Google Scholar]
- [12].Burgess RJ, Zhang Z. Histone chaperones in nucleosome assembly and human disease. Nature Structural & Molecular Biology. 2013;20:14–22. doi: 10.1038/nsmb.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Osley MA, Tsukuda T, Nickoloff JA. ATP-dependent chromatin remodeling factors and DNA damage repair. Mutation Research. 2007;618:65–80. doi: 10.1016/j.mrfmmm.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Seeber A, Hauer M, Gasser SM. Nucleosome remodelers in double-strand break repair. Current Opinion in Genetics & Development. 2013;23:174–184. doi: 10.1016/j.gde.2012.12.008. [DOI] [PubMed] [Google Scholar]
- [15].Smolle M, Workman JL. Transcription-associated histone modifications and cryptic transcription. Biochimica et Biophysica Acta. 2013;1829:84–97. doi: 10.1016/j.bbagrm.2012.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Menoni H, Gasparutto D, Hamiche A, Cadet J, Dimitrov S, Bouvet P, Angelov D. ATP-dependent chromatin remodeling is required for base excision repair in conventional but not in variant H2A.Bbd nucleosomes. Molecular and Cellular Biology. 2007;27:5949–5956. doi: 10.1128/MCB.00376-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Menoni H, Shukla MS, Gerson V, Dimitrov S, Angelov D. Base excision repair of 8-oxoG in dinucleosomes. Nucleic Acids Research. 2012;40:692–700. doi: 10.1093/nar/gkr761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Nakanishi S, Prasad R, Wilson SH, Smerdon M. Different structural states in oligonucleosomes are required for early versus late steps of base excision repair. Nucleic Acids Research. 2007;35:4313–4321. doi: 10.1093/nar/gkm436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Berquist BR, Wilson DM., 3rd Pathways for repairing and tolerating the spectrum of oxidative DNA lesions. Cancer Letters. 2012;327:61–72. doi: 10.1016/j.canlet.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Prasad A, Wallace SS, Pederson DS. Initiation of base excision repair of oxidative lesions in nucleosomes by the human, bifunctional DNA glycosylase NTH1. Molecular and Cellular Biology. 2007;27:8442–8453. doi: 10.1128/MCB.00791-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Odell ID, Barbour JE, Murphy DL, Della-Maria JA, Sweasy JB, Tomkinson AE, Wallace SS, Pederson DS. Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Molecular and Cellular Biology. 2011;31:4623–4632. doi: 10.1128/MCB.05715-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Polach KJ, Widom J. Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. Journal of Molecular Biology. 1995;254:130–149. doi: 10.1006/jmbi.1995.0606. [DOI] [PubMed] [Google Scholar]
- [23].Anderson JD, Widom J. Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. Journal of Molecular Biology. 2000;296:979–987. doi: 10.1006/jmbi.2000.3531. [DOI] [PubMed] [Google Scholar]
- [24].Geraghty DS, Sucic HB, Chen J, Pederson DS. Evidence that partial unwrapping of DNA from nucleosomes facilitates the binding of heat shock factor following DNA replication in yeast. Journal of Biological Chemistry. 1998;273:20463–20472. doi: 10.1074/jbc.273.32.20463. [DOI] [PubMed] [Google Scholar]
- [25].Odell ID, Newick K, Heintz NH, Wallace SS, Pederson DS. Non-specific DNA binding interferes with the efficient excision of oxidative lesions from chromatin by the human DNA glycosylase. NEIL1, DNA Repair. 2010;9:134–143. doi: 10.1016/j.dnarep.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Nyaga SG, Jaruga P, Lohani A, Dizdaroglu M, Evans MK. Accumulation of oxidatively induced DNA damage in human breast cancer cell lines following treatment with hydrogen peroxide. Cell Cycle. 2007;6:1472–1478. [PubMed] [Google Scholar]
- [27].Atamna H, Cheung I, Ames BN. A method for detecting abasic sites in living cells: age-dependent changes in base excision repair. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:686–691. doi: 10.1073/pnas.97.2.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tims HS, Gurunathan K, Levitus M, Widom J. Dynamics of nucleosome invasion by DNA binding proteins. Journal of Molecular Biology. 2011;411(2):430–448. doi: 10.1016/j.jmb.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ikeda S, Biswas T, Roy R, Izumi T, Boldogh I, Kurosky A, Sarker AH, Seki S, Mitra S. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III. Direct identification of Lys-212 as the active nucleophilic residue. Journal of Biological Chemistry. 1998;273:21585–21593. doi: 10.1074/jbc.273.34.21585. [DOI] [PubMed] [Google Scholar]
- [30].Marenstein DR, Chan MK, Altamirano A, Basu AK, Boorstein RJ, Cunningham RP, Teebor GW. Substrate specificity of human endonuclease III (hNTH1). effect of human APE1 on hNTH1 activity. Journal of Biological Chemistry. 2003;278:9005–9012. doi: 10.1074/jbc.M212168200. [DOI] [PubMed] [Google Scholar]
- [31].Liu X, Choudhury S, Roy R. In vitro and in vivo dimerization of human endonuclease III stimulates its activity. Journal of Biological Chemistry. 2003;278:50061–50069. doi: 10.1074/jbc.M309997200. [DOI] [PubMed] [Google Scholar]
- [32].Blaisdell JO, Wallace SS. Rapid determination of the active fraction of DNA repair glycosylases: a novel fluorescence assay for trapped intermediates. Nucleic Acids Research. 2007;35:1601–1611. doi: 10.1093/nar/gkm021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li G, Levitus M, Bustamante C, Widom J. Rapid spontaneous accessibility of nucleosomal DNA. Nature Structural & Molecular Biology. 2005;12:46–53. doi: 10.1038/nsmb869. [DOI] [PubMed] [Google Scholar]
- [34].Johnson KA. Transient-state kinetic analysis of enzyme reaction pathways. Enzymes. 1992;20:1–61. [Google Scholar]
- [35].Gutfreund H. Transients and relaxation kinetics of enzyme reactions. Annual Review of Biochemistry. 1971;40:315–344. doi: 10.1146/annurev.bi.40.070171.001531. [DOI] [PubMed] [Google Scholar]
- [36].Lowary PT, Widom J. New DNA sequence rules for high affinity binding to histone octamer and sequence-directed nucleosome positioning. Journal of Molecular Biology. 1998;276:19–42. doi: 10.1006/jmbi.1997.1494. [DOI] [PubMed] [Google Scholar]
- [37].Flaus A, Luger K, Tan S, Richmond TJ. Mapping nucleosome position at single base-pair resolution by using site-directed hydroxyl radicals. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:1370–1375. doi: 10.1073/pnas.93.4.1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dong F, Hansen JC, van Holde KE. DNA and protein determinants of nucleo-some positioning on sea urchin 5S rRNA gene sequences in vitro. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:5724–5728. doi: 10.1073/pnas.87.15.5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Li G, Widom J. Nucleosomes facilitate their own invasion. Nature Structural & Molecular Biology. 2004;11:763–769. doi: 10.1038/nsmb801. [DOI] [PubMed] [Google Scholar]
- [40].Tomschik M, Zheng H, van Holde K, Zlatanova J, Leuba SH. Fast, long-range, reversible conformational fluctuations in nucleosomes revealed by single-pair fluorescence resonance energy transfer. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3278–3283. doi: 10.1073/pnas.0500189102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tomschik M, van Holde K, Zlatanova J. Nucleosome dynamics as studied by single-pair fluorescence resonance energy transfer: a reevaluation. Journal of Fluorescence. 2009;19:53–62. doi: 10.1007/s10895-008-0379-1. [DOI] [PubMed] [Google Scholar]
- [42].Vasudevan D, Chua EY, Davey CA. Crystal structures of nucleosome core particles containing the ‘601’ strong positioning sequence. Journal of Molecular Biology. 2010;403:1–10. doi: 10.1016/j.jmb.2010.08.039. [DOI] [PubMed] [Google Scholar]
- [43].Neumann H, Hancock SM, Buning R, Routh A, Chapman L, Somers J, Owen-Hughes T, van Noort J, Rhodes D, Chin JW. A method for genetically installing site-specific acetylation in recombinant histones defines the effects of H3 K56 acetylation. Molecular Cell. 2009;36:153–163. doi: 10.1016/j.molcel.2009.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].North JA, Shimko JC, Javaid S, Mooney AM, Shoffner MA, Rose SD, Bundschuh R, Fishel R, Ottesen JJ, Poirier MG. Regulation of the nucleosome unwrapping rate controls DNA accessibility. Nucleic Acids Research. 2012;40:10215–10227. doi: 10.1093/nar/gks747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ye Y, Stahley MR, Xu J, Friedman JI, Sun Y, McKnight JN, Gray JJ, Bowman GD, Stivers JT. Enzymatic excision of uracil residues in nucleosomes depends on the local DNA structure and dynamics. Biochemistry. 2012;51:6028–6038. doi: 10.1021/bi3006412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hall MA, Shundrovsky A, Bai L, Fulbright RM, Lis JT, Wang MD. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nature Structural & Molecular Biology. 2009;16:124–129. doi: 10.1038/nsmb.1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Hazra TK, Izumi T, Boldogh I, Imhoff B, Kow YW, Jaruga P, Dizdaroglu M, Mitra S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:3523–3528. doi: 10.1073/pnas.062053799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Theriot CA, Hegde ML, Hazra TK, Mitra S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer–template structures. DNA Repair. 2010;9:643–652. doi: 10.1016/j.dnarep.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Dou H, Theriot CA, Das A, Hegde ML, Matsumoto Y, Boldogh I, Hazra TK, Bhakat KK, Mitra S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. Journal of Biological Chemistry. 2008;283:3130–3140. doi: 10.1074/jbc.M709186200. [DOI] [PubMed] [Google Scholar]
- [50].Banerjee D, Mandal SM, Das A, Hegde ML, Das S, Bhakat KK, Boldogh I, Sarkar PS, Mitra S, Hazra TK. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. Journal of Biological Chemistry. 2011;286:6006–6016. doi: 10.1074/jbc.M110.198796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Cairns BR. Chromatin remodeling: insights and intrigue from single-molecule studies. Nature Structural & Molecular Biology. 2007;14:989–996. doi: 10.1038/nsmb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]