

Abstract
Groups of pathogenic bacteria employ diffusible signals to regulate their virulence in a concerted manner. Pseudomonas aeruginosa uses 4-hydroxy-2-alkylquinolines (HAQs), including HHQ and PQS, as unique signals. We demonstrate that octanoic acid is directly incorporated into HHQ. This finding rules out the long-standing hypothesis that 3-ketofatty acids are the precursors of HAQs. We found that HAQ biosynthesis, which requires the PqsABCD enzymes, proceeds by a two-step pathway: [1] PqsD mediates the synthesis of 2-aminobenzoylacetate (2-ABA) from anthraniloyl-CoA and malonyl-CoA, then [2] the decarboxylating coupling of 2-ABA to an octanoate group linked to PqsC produces HHQ, the direct precursor of PQS (Pseudomonas Quinolone Signal). PqsB is tightly associated with PqsC and required for the second step. This finding uncovers promising targets for the development of specific antivirulence drugs to combat this opportunistic pathogen.
INTRODUCTION
The bacterium Pseudomonas aeruginosa is a Gram negative opportunistic pathogen frequently responsible for infections among immunocompromised individuals and is also often involved in hospital-acquired infections (Driscoll et al., 2007; Kerr and Snelling, 2009). Furthermore, it is the leading cause of morbidity and mortality in people affected with the genetic disease cystic fibrosis. Most virulence factors expressed by this bacterium are controlled in a cell density-dependent manner by a process called “quorum sensing” (QS), where cells communicate via small diffusible signalling molecules (Jimenez et al., 2012). There are three QS systems in P. aeruginosa, controlled by the transcriptional regulators LasR, RhlR and MvfR (PqsR) (Balasubramanian et al., 2013). In the two former systems, a cognate autoinducer synthase produces the signalling molecules N-(3-oxododecanoyl)-L-homoserine lactone (3-oxo-C12-HSL) and N-butanoyl-L-homoserine lactone (C4-HSL), respectively (Juhas et al., 2005; Smith and Iglewski, 2003). Each signal binds to its corresponding transcriptional regulator, thus activating the transcription of various downstream targets, including its synthase gene. Since production of many virulence determinants in pathogenic bacteria requires a fully functional quorum sensing circuitry, cell-to-cell communication represents an intensely investigated promising target as an alternative to antibiotics in virulence control (Bjarnsholt et al., 2010; Galloway et al., 2012).
In the P. aeruginosa MvfR system, the signalling molecules belong to a family of compounds that share a 4-hydroxy-2-alkylquinoline (HAQ) structure, such as 3,4-dihydroxy-2-heptylquinoline (PQS), its direct precursor 4-hydroxy-2-heptylquinoline (HHQ) and 4-hydroxy-2-heptylquinoline-N-oxide (HQNO) (Fig. 1) (Déziel et al., 2004; Lépine et al., 2004). HAQs are also called 2-alkyl-4(1H)-quinolones (Heeb et al., 2011). PQS and HHQ are both able to bind MvfR (Diggle et al., 2007; Wade et al., 2005; Xiao et al., 2006). Upon binding to its ligand, MvfR induces the expression of the pqsABCDE operon, which is responsible for the biosynthesis of HAQs (Déziel et al., 2004; Gallagher et al., 2002). In vivo, all of the enzymes encoded by this operon are essential for HAQ synthesis except for PqsE (Déziel et al., 2005; Diggle et al., 2003; Farrow et al., 2008). Two other metabolites, 2,4-dihydroxyquinoline (DHQ) (Lépine et al., 2007; Zhang et al., 2008) and 2-aminoacetophenone (2-AA) (Kesarwani et al., 2011) are also co-produced with HAQs (Fig. 1), but they only require the activity of PqsA and PqsD.
Figure 1. Proposed pathway for the biosynthesis of 2-ABA, 2-AA, HHQ and HQNO.

The * and + symbols refer to the source of the carbon as originating from the carbon 1 and 2 of acetate, respectively. Pathway in the box corresponds to the biosynthesis of DHQ as published by Zhang et al. (Zhang et al., 2008).
AA, anthranilic acid; 2-ABA, 2-aminobenzoylacetate; 2-AA, 2-aminoacetophenone; HHQ, 4-hydroxy-2-heptylquinoline; HQNO, 4-hydroxy-2-heptylquinoline N-oxide; DHQ, 2,4-dihydroxyquinoline; PQS (Pseudomonas Quinolone Signal), 3,4-dihydroxy-2-heptylquinoline.
Molecules produced from the activity of the pqsABCD gene products display additional biological activities besides MvfR activation. For instance, HHQ, PQS and 2-AA can modulate the innate immune response of mammalian hosts (Bandyopadhaya et al., 2012; Kim et al., 2010), while HQNO inhibits various cytochromes and decreases the antibacterial activity of aminoglycoside antibiotics towards Gram-positive bacteria (Hoffman et al., 2006; Lightbown, 1954). Of note, methylated analogs of HAQs are also produced in various Burkholderia species, and their production depends on an operon containing genes highly similar to those of P. aeruginosa (Vial et al., 2008).
Inhibition of the MvfR regulon by mutational inactivation of mvfR, pqsE or pqsA decreases P. aeruginosa virulence in a mouse acute infection model (Cao et al., 2001; Déziel et al., 2005). Furthermore, hindering HAQ synthesis protects mice from the infection (Lesic et al., 2007), which confirms that HAQ biosynthesis is a promising target to control virulence of this bacterium. However, the HAQ biosynthetic pathway is only partially deciphered.
We know that anthranilic acid is a precursor of HAQs (Calfee et al., 2001; Déziel et al., 2004), and that it is activated into anthraniloyl-CoA by PqsA. The generally accepted model states that the quinoline ring of HAQs originates from a one-step head-to-tail condensation of 3-ketofatty acids with anthranilic acid (Heeb et al., 2011). This hypothesis was initially proposed as early as in 1956 when the structure of HAQs was first uncovered (Cornforth and James, 1956). This was later given more credit by Luckner and Ritter (Luckner and Ritter, 1965; Ritter and Luckner, 1971) and by Bredenbruch et al. (Bredenbruch et al., 2005). The roles of PqsB, PqsC and PqsD in the biosynthesis of HAQs are still unknown.
In addition to the enzymes encoded pqsABCDE, the biosynthesis of HQNO and the other members of the N-oxides family requires the monoxygenase encoded by pqsL (Déziel et al., 2004; Lépine et al., 2004). The substrate of PqsL is unknown, but we have shown that HHQ is not the precursor of HQNO (Déziel et al., 2004). While the biosynthesis of 2-AA has not been solved, the biosynthesis of DHQ is better understood. In vitro studies revealed that PqsD binds to anthraniloyl-CoA and catalyses a reaction with malonyl-CoA to produce a hypothetical CoA-activated 2-aminobenzoylacetate (2-ABA-CoA) intermediate, which would spontaneously form DHQ (Fig. 1) (Zhang et al., 2008).
We have elucidated the function for PqsB, PqsC and PqsD in the biosynthesis of HAQs, and determined that the current model involving 3-ketofatty acids as precursors is incorrect. We present evidence that the actual precursors are fatty acids that can be produced through β-oxidation of longer chain fatty acids and that the other precursor is 2-aminobenzoylacetate produced by the action of anthraniloyl-PqsD and malonyl-CoA.
RESULTS
3-keto fatty acids are not precursors of HAQs
We were intrigued by a recent paper showing that adding 1 mM dodecanoic acid to a P. aeruginosa culture leads to increased production of pyocyanin, a virulence factor controlled by the MvfR regulon (Kwan et al., 2011). They hypothesized that this increased pyocyanin production was due to an increase in the production of 3-oxo-C12-HSL because of an increased production of its precursor 3-ketododecanoic acid through β-oxidation of dodecanoic acid (Kwan et al., 2011). We repeated this experiment using our P. aeruginosa PA14 strain to which was added the same concentration of dodecanoic acid and we similarly obtained an increase in pyocyanin (Fig. S1a). While the level of 3-oxo-C12-HSL was not significantly increased by the fatty acid, HAQ production was consistently elevated (Figs. S1b and S1c), an indication that the fatty acid increased the production of an HAQ precursor. Thus, to verify if a fatty acid could be directly incorporated into the structure of HHQ, 1 mM decanoic-9,9,10,10,10-d5 acid was provided to a PA14 culture. Under these conditions, HHQ was labelled at 75% with five deuterium (Fig. 2a, confirming Fig. S1c), showing that this fatty acid was incorporated within this HAQ.
Figure 2. Positive electrospray mass spectra of HHQ obtained after feeding P. aeruginosa with isotope-labelled precursors.
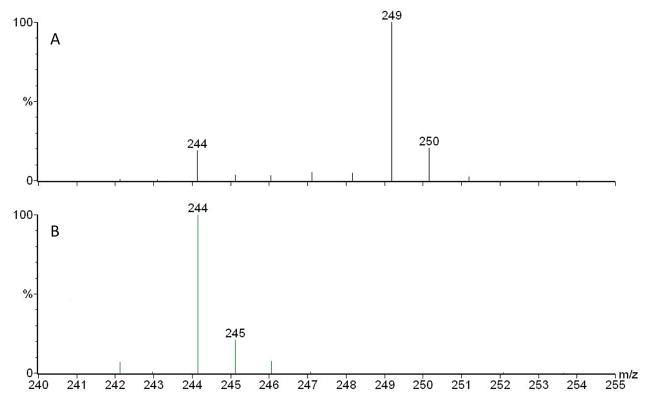
Addition of (a) 1 mM decanoic-9,9,10,10,10-d5 acid, or b) 1 mM decanoic-1,2-13C2 acid to a P. aeruginosa PA14 culture in TSB. See also Fig. S1, where we are showing that feeding dodecanoic acid is similarly increasing HAQ production.
As the generally accepted precursors of HAQs are 3-ketofatty acids, this labelled decanoic acid should have undergone first β-oxidation into 3-ketodecanoic acid before reacting with anthranilic acid, followed by concurrent loss of the acid function (as CO2) to produce HHQ. We tested the recognized model by adding 1 mM decanoic-1, 2-13C2 acid to a PA14 culture. β-oxidation of this compound should produce 3-oxodecanoic-1,2-13C2 acid which, taking into account the loss of the acid function (as CO2) should produce HHQ labelled with only one 13C. The extent of this 13C labelling should match the 75% level of labelling obtained when decanoic-9,9,10,10,10-d5 acid was provided to the cultures. However, the relative proportion of the m/z 245 over 244 was only 21% (Fig. 2b), very close to the theoretical value of 19% (due to the natural abundance of 13C), clearly indicating that 3-ketodecanoic acid cannot be a direct precursor of HHQ.
2-ABA is a precursor of HAQs
To test the hypothesis that the quinoline ring of HAQs might be the result of a two-step process involving an intermediate that can be isolated, we performed a series of cross-feeding experiments with various nonpolar mutants of the pqsABCDE operon involved in HAQ synthesis. In these mutants, the genes in the operon downstream from the inactivated gene are still transcribed. To maintain a WT level of expression of the other enzymes in the operon, we always included in all the cultures the MvfR-inducing ligand PQS, in its 2,3,4,5-tetradeuterated form (PQS-d4) to prevent interference with LC/MS quantification(Lépine et al., 2003). As a first step, we confirmed that none of the pqsA, pqsB, pqsC and pqsD mutant produces measurable amounts of HAQs (Fig. 3, on the right). Then, we fed the filter-sterilized supernatant of these pqs mutants to another pqs mutant and looked for the production of HAQs. Figure 3 shows that only the supernatant of a pqsB- or pqsC- mutant, when provided to a pqsA- or pqsD- mutant, results in HAQ production. This indicates that the culture supernatant of a nonpolar pqsB- or pqsC- mutant must contain an intermediate in the biosynthesis of HAQs.
Figure 3. Supernatant cross-feeding experiments.

Total HAQ production in the WT and in the supernatant (S) of nonpolar mutants of genes from pqsABCDE operon involved in HAQ synthesis. In a second step (B) the supernatants of each mutant were added to cultures of all the other nonpolar mutants. As shown in Fig. S2, the active intermediate present in the culture supernatant of a nonpolar pqsB- or pqsC- mutant was ultimately identified as 2-ABA.
The active intermediate was purified by activity-guided fractionation using preparative HPLC and thick layer chromatography, and ultimately identified by MS and MS/MS as 2-ABA. Negative electrospray ionization LC/MS/MS presents a pseudomolecular ion at m/z 178 and fragments at m/z 134 (loss of CO2) and 92 (loss of C2H2O), as expected (Fig. S2). Once purified, this compound is relatively unstable and decomposes in a matter of hours on standing at room temperature in water. 2-AA and DHQ are among the degradation products, the latter being predominantly produced under acidic conditions.
To conclusively prove the structure of the intermediate, 2-ABA was chemically synthesized (see METHODS). Like the compound obtained from purification, synthetic 2-ABA is relatively unstable in solution, but in a dry form can be stored for extended period of time at −20°C. When the synthetic product was fed to a double nonpolar pqsA- pqsH- mutant (unable to produce 2-ABA and to convert HHQ into PQS), 2-AA, HHQ and HQNO were produced. This mutant was used rather than a pqsA- mutant to avoid further transformation of HHQ into PQS, in order to simplify the analysis and increase HHQ concentration.
The synthesis of 2-aminobenzoylacetate-1,2-13C2 was also achieved. When it was fed to the nonpolar pqsA- pqsH- mutant, the 2-AA, HHQ and HQNO produced were totally labelled with only one 13C, as expected.
Biosynthesis of 2-ABA
To further investigate the biosynthesis of 2-ABA, the double nonpolar pqsB− pqsL− mutant, an overproducer of 2-ABA, was cultivated in a mineral medium with acetate 13C-labelled at position 1 or 2 as sole carbon source, supplemented with anthranilic acid to avoid incorporation of 13C abelling in the aromatic ring and PQS-d4 as inducer. The resulting supernatant was then added to a culture of the pqsA- pqsH- mutant in TSB. When the sole carbon source was acetate labelled at position 1, the pseudomolecular ion of the HHQ produced was predominantly not labelled (Fig. 4a). But when acetate was labelled at position 2, the HHQ produced appeared exclusively at m/z 245 showing that it had incorporated one 13C (Fig. 4b).
Figure 4. Positive electrospray spectra of HHQ produced by a double pqsA- pqsH- mutant fed with various culture supernatants providing 2-ABA.

When a double pqsB−pqsL− mutant was cultivated in M9 mineral medium with (a) acetate labelled at position 1 or (b) at position 2 with 13C as carbon source and the supernatant added to a nonpolar pqsA- pqsH- mutant culture in TSB. (c) pqsB− pqsL− mutant cultivated in M9 medium with unlabelled acetate and the supernatant fed to a double pqsA- pqsH- mutant mutant grown in fully 13C acetate, (d) supernatant of a pqsC- mutant cultivated in TSB and fed to a pqsA- pqsH- mutant grown in TSB and supplemented with fully 13C-labelled octanoic acid.
Malonyl-CoA is a precursor of HHQ
Because acetyl-CoA is a precursor of malonyl-CoA in vivo, and considering the published role of malonyl-CoA in the biosynthesis of DHQ, we wondered whether malonyl-CoA could also be involved in the biosynthesis of HHQ. To test this hypothesis, a PA14 cytoplasmic extract was combined with 1,2,3,4-tetradeuteroanthranilic acid (anthranilic-d4 acid), CoA, octanoyl-CoA (see below) and increasing levels of malonyl-CoA; the HHQ-d4 produced was monitored by LC/MS. Figure 5a shows that no HHQ-d4 is produced in absence of malonyl-CoA, while increasing the amount of malonyl-CoA leads to a corresponding augmentation in HHQ-d4, thus validating the hypothesis.
Figure 5. Malonyl-CoA and octanoyl-CoA are two precursors of HHQ.

(a) Dose-response relationship between added malonyl-CoA and HHQ-d4 production of a cytoplasmic extract of PA14 fed anthranilic acid-d4, CoA, and octanoyl-CoA. (b) Dose-response relationship between added octanoyl-CoA and in vitro HHQ production when copurified His-tagged PqsC and PqsB are provided with 2-ABA. Data are represented as mean +/− SD.
Octanoic acid and PqsC are involved in the coupling of the aliphatic tail of HHQ
To identify the precursor incorporated in the aliphatic tail of HAQs, the nonpolar pqsB− pqsL− mutant was cultured in TSB and the supernatant fed to the nonpolar pqsA- pqsH- mutant growing in mineral medium containing fully 13C-labelled acetate. The pseudomolecular ion of the HHQ produced showed an increase of 8 Da (Fig. 4c). This suggested that these eight carbons originating from acetate would come from octanoic acid produced from de novo fatty acid synthesis. To confirm this hypothesis, the supernatant of a nonpolar pqsC- mutant was provided to the nonpolar pqsA- pqsH- mutant fed with fully 13C-labelled octanoic acid instead of acetate, and the HHQ produced showed the same mass increase of 8 Da (Fig. 4d). These results identify octanoic acid as a direct precursor of HHQ.
Because of the amino acid sequence homologies between PqsC and PqsD, which performs the condensation of anthraniloyl-CoA with malonyl-CoA for the biosynthesis of DHQ (Zhang et al., 2008), we then decided to purify PqsC in order to study its potential role in the biosynthesis of HHQ. When trying to overexpress an His-tagged PqsC construct in E. coli strains BL21 or Origami under various temperature or IPTG concentrations, we always observed the protein inactive in inclusion bodies. Attempts to denature/renature the protein failed. However when co-expressed with a His-tagged PqsB construct, PqsC was found in the cytoplasmic fraction. After gel electrophoresis, the band corresponding to the molecular weight of PqsC was excised and analysed by MS after treatment with trypsin. MASCOT analysis confirmed that the protein was indeed PqsC, and also showed a tryptic peptide identified as ALPLDSQMECASFLLNLR (residues 120–137) carrying an additional mass of 126 Da, corresponding to an octanoyl moiety. This tryptic peptide contains many residues conserved in enzymes involved in condensation reactions, including the cysteine residue bearing the anthraniloyl group in PqsD (Zhang et al., 2008). The exact residue bearing the octanoyl group could not be identified by MS because it seemed too labile to survive collision-induced fragmentation of the peptide backbone. Nevertheless, we were able to determine that the octanoyl group was linked to the conserved cysteine residue, since a plasmid carrying a C129A mutation in pqsC could not complement the ability to produce HAQs of a pqsC- mutant, in contrast with the plasmid expressing the His-tagged pqsC. When overexpressing PqsC alone in E. coli, the same tryptic peptide was detected by MASCOT but without its octanoate adduct, an indication that the folding of PqsC and the binding of octanoate are linked, and promoted by PqsB.
Overexpressing an His-tagged PqsB construct in E. coli yielded a protein with the expected molecular weight (observed value: 31826 Da versus the theoretical value: 31830 Da), showing that there was no octanoyl group associated with this protein and that PqsB is not involved in vivo in providing octanoate to PqsC.
Interestingly, the co-purified His-tagged PqsB and PqsC when migrated together on a native polyacrylamide gel produce only one band, although their molecular weight differ considerably (Fig. 6). An attempt to purify the active form of PqsC without PqsB by coexpressing an His-tagged PqsC and a native PqsB using nickel-sepharose column failed, as PqsB was found to coelute with PqsC, an indication of the tight association between these two enzymes.
Figure 6. Polyacrylamide gels showing purified PqsB and PqsC.

(a) Native gel of copurified His-tagged PqsC and PqsB showing comigration (lane 1). (b) Denaturing gel showing purified PqsB and PqsC proteins (lane 1) and reanalysis of the complex from the native gel (a) after that the band was excised, showing two bands corresponding to PqsC and PqsB (lane 2). M= molecular weight ladder.
In order to conclusively prove that octanoate is the immediate intermediate in the biosynthesis of HHQ, we added increasing amounts of octanoyl-CoA to a synthetic 2-ABA solution to which were added the co-purified His-tagged PqsB and PqsC. This resulted in increasing production of HHQ (Fig. 5b), demonstrating that octanoyl-CoA is the direct precursor of HHQ and that PqsC is the enzyme coupling octanoic acid to 2-ABA.
DISCUSSION
Our experiment with decanoic-9,9,10,10,10 -d5 acid clearly shows that this fatty acid can be directly incorporated into HAQs, a departure from the current hypothesis that the fatty acid part of HAQs is produced from de novo fatty acid synthesis. It is noteworthy to mention the extensive labeling of the HAQs (75%) eventhough this experiment was conducted in TSB rich medium in which de novo biosynthesis of fatty acids is likely to be activated. The fact that no label was retained when decanoic-1,2 -13C2 acid was fed to the bacteria is in total contradiction with the current accepted model that the quinoline ring of HAQs originates from a one-step head-to-tail condensation of 3-ketofatty acids with anthranilic acid (Heeb et al., 2011).
Looking for an alternative to the one-step head-to-tail reaction, we found that an intermediate of HAQ biosynthesis is present in culture supernatants of nonpolar pqsB- and pqsC- mutants. Only the culture supernatants from these two nonpolar mutants provide the intermediate to nonpolar pqsA- or pqsD- mutants to produce HAQs. The results also show that PqsB and PqsC have to be present simultaneously to transform the intermediate into HHQ as the supernatant of a pqsC- mutant does not complement a pqsB- mutant, nor vice versa. The conclusion is that this extracellular intermediate is first synthesized by PqsA and PqsD, and is then further modified by PqsB and PqsC to produce HAQs. The fact that the supernatant of a double pqsB- pqsL- mutant enables the production of more HHQ than the supernatant of a single pqsB- mutant when fed to a pqsA- mutant also suggests that this intermediate is the substrate of PqsL in the production of HQNO.
This intermediate was identified as 2-ABA. Not unexpectedly, this compound once purified is rather unstable and tends to decompose into DHQ through attack of the carboxyl group of the acid function by the neighbouring amino group to produce the more stable quinoline ring of DHQ or, through decarboxylation of the rather unstable 3-ketofatty acid, into 2-AA. Interestingly the MS/MS analysis of 2-ABA also shows the loss of CO2 as the main decomposition pathway, even at very low collision energies.
2-AA is a P. aeruginosa volatile metabolite found in liquid culture and in the lungs of people affected by cystic fibrosis (Cox and Parker, 1979; Scott-Thomas et al., 2010). 2-AA modulates the virulence of the bacteria and promotes a chronic infection phenotype (Kesarwani et al., 2011), and can also modulate the immune response of the host (Bandyopadhaya et al., 2012). We knew that anthranilic acid is a precursor of 2-AA (Kesarwani et al., 2011), but the pathway leading to the addition of a methyl group adjacent to the carbonyl was unknown. Feeding of a pqsA- pqsH- mutant with 2-ABA-1,2-13C2 lead to quantitative labelling with one 13C, a demonstration that 2-AA is produced in vivo by the decarboxylation of 2-ABA, a reaction also spontaneously observed during the chemical synthesis of 2-ABA.
The feeding experiments with 13C-labelled acetate showed that the HHQ produced only incorporated one 13C and only if the label was at position 2 of acetate. The fact that only the carbon 2 of acetate was retained in HHQ implied that some form of malonate is involved in 2-ABA synthesis. That malonyl-CoA is the direct precursor of 2-ABA was demonstrated by the linearity of the dose-response production of HHQ-d4 upon feeding incremental concentrations of malonyl-CoA to a cytoplasmic extract of a nonpolar pqsB− pqsL− mutant supplemented with anthranilic-d4 acid, CoA and octanoyl-CoA. When synthetic 2-ABA-1,2-13C2 was provided to the pqsA- pqsH-mutant, the HHQ produced was labeled with only one 13C. This proves that 2-ABA has to undergo decarboxylation upon coupling with octanoate, and this also explains the absence of labelling of HHQ when acetate labelled at position 1 is fed to the pqsA- pqsH-mutant, as the carbon of the carbonyl group of 2-ABA also originates from the carbon 1 of acetate (Fig. 1).
The experiment involving feeding the supernatant of a nonpolar pqsB- pqsL- mutant to a nonpolar pqsA- pqsH- mutant grown only with fully 13C-labelled acetate as carbon source led to the incorporation of eight labelled carbons, an indication that octanoate, in this case produced through de novo synthesis, is the immediate precursor of HHQ. When this experiment was repeated with a pqsC- mutant supernatant given to a nonpolar pqsA- pqsH- mutant fed with fully 13C-labelled octanoic acid instead of acetate, the same eight 13C labelling was obtained, confirming that octanoic acid can be directly incorporated into HHQ without having to originate from de novo synthesis. The dose-response in vitro production of HHQ starting with synthetic 2-ABA and purified PqsB and PqsC with increasing amounts of octanoyl-CoA proves that octanoyl-CoA is the other direct precursor of HHQ.
Taking these results together, we propose a two-step pathway for the biosynthesis of HAQs in P. aeruginosa (Fig. 1). Initially, PqsA activates anthranilic acid into anthraniloyl-CoA (Coleman et al., 2008). Then anthraniloyl-CoA reacts with PqsD to produce anthraniloyl-PqsD, as described in the biosynthesis of DHQ (Zhang et al., 2008). The next step involves the reaction of anthraniloyl-PqsD with malonyl-CoA to form 2-ABA-CoA, as previously hypothesized for the synthesis of DHQ (Zhang et al., 2008). Then free 2-ABA undergoes decarboxylation and reacts with octanoate. This reaction is most likely performed by PqsC, as we have identified it as the carrier of the octanoyl group. Further supporting a model where PqsC is responsible for the coupling of the octanoate group and the closing of the quinoline ring, this group is located on the same conserved cysteine that carries anthranilate in PqsD (Zhang et al., 2008). Indeed, as noted by Zhang et al. (Zhang et al., 2008), PqsD and PqsC are structurally related as they both share the same cysteine 112 and histidine 244 found in the active site of E. coli FabH, although PqsC lacks the asparagine 274 shared by PqsD and FabH. As PqsD and FabH are both invoved in Claisen-type of condensations it is not unlikely that PqsC could perform the same type of reaction as depicted in Figure 1, with 2-ABA undergoing the loss of CO2 in the same way malonyl-CoA does in the biosynthesis of DHQ. Although PqsC lacks asparagine 244, 2-ABA is much likely to easily undergo decarboxylation than malonyl-CoA which could explain the absence of this residue in the active site of PqsC. While malonyl-CoA plays an essential role in the synthesis of DHQ and HAQ, we have previously shown that DHQ is not a precursor of HAQs (Lépine et al., 2007).
What remains to be determined is how free 2-ABA is produced from 2-ABA-CoA. Although it is possible that this is achieved by PqsB, adding PqsB to anthraniloyl-CoA, malonyl-CoA and PqsD did not alter the production of DHQ. However, under the tested conditions, the free 2-ABA produced spontaneously decomposes into DHQ, as we found this compound as a degradation product of 2-ABA. PqsB could not either directly hydrolyse anthraniloyl-CoA, which is somewhat structurally related to 2-ABA-CoA. In order to explain how free 2-ABA is found in the supernatant of a pqsB- mutant, we propose that in this mutant, unspecific thioesterases degrade the accumulating 2-ABA-CoA and that accumulated free 2-ABA is either excreted or diffused outside the cells. It is also possible that a 2-ABA-CoA-specific thioesterase exists. Thus, the role of PqsB remains to be confirmed but it is clear from the co-purification of the co-expressed PqsB-PqsC that these two enzymes are closely associated and that PqsB is required for PqsC to be active. When PqsC is expressed alone in E. coli, it is found mostly in the insoluble fraction of the cell lysate while when it is co-expressed with PqsB it is mostly found in the cytoplasmic fraction. Also, the cysteine of the active site of PqsC was not substituted with octanoate when PqsC was expressed alone, while it was substituted when coexpressed in presence of PqsB. These two facts suggest that PqsB is mostly involved in the proper folding of PqsC rather than having a direct enzymatic role in the process.
Another interesting finding is that synthetic 2-ABA, when provided to a pqsA- pqsH- mutant, also acts as a precursor of HQNO, along with HHQ. We have previously reported that pqsL, which codes for a monooxygenase, is required for the biosynthesis of HQNO (Lépine et al., 2004), and also, somewhat surprisingly, that HHQ is not the direct precursor of HQNO (Déziel et al., 2004). The present work indicates that 2-ABA is the likely elusive substrate of PqsL. Although we did not isolate the oxidized intermediate, we speculate that it is probably an hydroxylamino derivative of 2-ABA. The hydroxylamino function is strongly nucleophilic and capable to attack a neighbouring carbonyl group. Hydroxylamino groups are often obtained from the reduction of a nitro group with SnCl2 and this approach is used for the one-step chemical synthesis of HQNO by reducing the 2-nitro group of 1-(2-nitrophenyl)tetradecanoate-1,3-dione with SnCl2 (Taylor and Lynn, 1995). By analogy, the PqsL-mediated oxidation of the amino group of 2-ABA could produce a hydroxylamino group that would spontaneously attack the carbonyl function to produce HQNO (Fig. 1).
How do we reconcile our findings with prior claims that 3-ketofatty acids are the immediate precursors of HAQs ? (Bredenbruch et al., 2005; Luckner and Ritter, 1965; Pistorius et al., 2011; Ritter and Luckner, 1971). In fact, what was really demonstrated previously is that the carbons of the aliphatic chain alternatively originate from carbon 2 and carbon 1 of acetate, as expected from the de novo synthesis of fatty acids, and that the carbons 3 and 2 of the quinoline ring of HHQ also follow the same alternating origin, with these carbons originating from carbon 2 and 1 of acetate, respectively (Fig. 1). This is also true in the mechanism we propose, in which the carbon 2 of 2-ABA, which eventually ends up as carbon 3 of HHQ, also originates from carbon 2 of acetate, and for the carbon 1 of octanoate, which ends up as carbon 2 of HHQ, originates from carbon 1 of acetate, in the de novo biosynthesis of fatty acids. It remains to be determined whether the octanoic acid originates directly from de novo fatty acid synthesis or if it is rather a product of the β-oxidation of longer fatty acids. That octanoyl-CoA was found as a substrate of PqsC favours the second hypothesis because fatty acid-ACP, not fatty acid-CoAs, are the intermediate of de novo biosynthesis. Because labelling was predominant when labelled decanoic acid was provided exogenously, even when a rich culture medium was used, also supports the second hypothesis.
Pistorius et al. recently reported that the CoA and ACP esters of 3-ketodecanoic acid in presence of anthraniloyl-CoA and purified PqsD did not produce HHQ, as predicted by Bera et al. (2009) who described the active site of PqsD as being too small to accommodate such large substrates. But when Pistorius et al. fed free 3-ketodecanoic acid and anthraniloyl-CoA to PqsD, they reported the production of HHQ (Pistorius et al., 2011). However, as the authors themselves pointed out, their experiment cannot explain the essential role of PqsB and PqsC in vivo. Indeed, PqsD does not seem to be very substrate-specific as it can also accept benzoyl-CoA in addition to anthraniloyl-CoA (Pistorius et al., 2011). Moreover, their observed Kcat for the reaction of HHQ with 3-ketodecanoic acid was three orders of magnitude lower than the one for producing DHQ, while the relative proportion of DHQ and HHQ is much closer to a one to one ratio in cultures (Lépine et al., 2007). From this, we must conclude that 3-ketodecanoic can be transformed into HHQ in vitro with low efficiency by PqsD, but that 3-ketofatty acids are not the biologically relevant precursors of HAQs.
The discovery of the two-step mechanism responsible for the synthesis of HAQs is likely to foster the search for new and specific inhibitors of P. aeruginosa quorum sensing based on the 2-ABA structure, and provides a renewed perspective on the biosynthesis of 2-AA and HQNO, molecules with promising biological properties.
SIGNIFICANCE
Groups of pathogenic bacteria employ diffusible signaling molecules to regulate their virulence in a concerted manner (quorum sensing). In Pseudomonas aeruginosa, one family of signalling molecules share a 4-hydroxy-2-alkylquinoline (HAQ) structure, such as Pseudomonas Quinolone Signal (PQS), and its direct precursor 4-hydroxy-2-heptylquinoline (HHQ). PQS and HHQ activate the MvfR regulator, which then induces the expression of the pqsABCDE operon, responsible for the biosynthesis of HAQs.
Molecules produced from the activity of the pqsABCD gene products display additional biological activities besides MvfR activation. For instance, HHQ, PQS and 2-AA can modulate the innate immune response of mammalian hosts.
Inhibition of the MvfR regulon by mutational inactivation decreases P. aeruginosa virulence. Furthermore, hindering HAQ synthesis protects mice from the infection, which confirms that HAQ biosynthesis is a promising target to control virulence of this bacterium.
We know that anthranilic acid is a precursor of HAQs. It is widely accepted that the other precursors of HAQs are 3-ketofatty acids that condense with anthranilic acid in a head-to-head fashion.
The objective of the present work was to decipher the poorly understood HAQ biosynthetic pathway
We have elucidated the function for PqsB, PqsC and PqsD in the biosynthesis of HAQs, and determined that the current model involving 3-ketofatty acids as precursors is incorrect. We have found that HAQ biosynthesis proceeds by a two-step pathway: (1) the PqsD enzyme mediates the synthesis of 2-aminobenzoylacetate (2-ABA) from anthraniloyl-CoA and malonyl-CoA, then (2) the decarboxylating coupling of 2-ABA to an octanoate group linked to PqsC produces HHQ. PqsB is tightly associated with PqsC and required for the second step.
Our findings rule out the long standing hypothesis that 3-ketofatty acids are the precursors of HAQs, and uncover promising targets for the development of specific antivirulence drugs to combat this opportunistic pathogen, for which we need alternatives to traditional antibiotics.
METHODS
Bacteria and plasmids
The wildtype bacterium was P. aeruginosa strain PA14 (Lee et al., 2006). Bacteria and plasmids are presented in table 1.
TABLE 1.
Bacterial strains and plasmids used in this study.
| Strains | Characteristics | References |
|---|---|---|
| P. aeruginosa/Lab # | ||
| PA14/ED14 | Clinical isolate UCBPP-PA14 | (Rahme et al., 1995) |
| PA14 pqsA−np/ED83 | Unmarked pqsA deletion | L.G. Rahme |
| PA14 pqsB−/ED117 | pqsB::TnphoA | (Mahajan-Miklos et al., 1999) |
| PA14 pqsC−np/ED218 | Nonpolar pqsC::Kan | (Lesic and Rahme, 2008) |
| PA14 pqsD−np/ED690 | Nonpolar pqsD::Kan | L.G. Rahme |
| PA14 pqsA−pqsH−/ED170 | pqsA−np, pqsH::aacC1 | L.G. Rahme |
| PA14 pqsB−pqsL−/ED1156 | ΔpqsL, pqsB::ISlacZ | This study |
| E. coli DH5α | supE44 ΔlacU169 (φ80lacZΔM15) hsdR17 recA1 endA1 gyrA96 thi-1 relA1 | Invitrogen |
| E. coli Origami 2 (DE3) | D(ara-leu)7697 DlacX74 DphoA PvuII phoR araD139 ahpC galE galK rpsL F′[lac+ lacIq pro] (DE3) gor522::Tn10 trxB (StrR, TetR) | Novagen |
| E. coli BL21 (DE3) | fhuA2 [lon] ompT gal (λ DE3) [dcm] hsdS λ DE3 = λ sBamHIo ΔEcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 Δnin5 | Novagen |
| Vectors | Characteristics | Source |
| pET22b(+) | PT7, lacO, MCS, 6xHis·Tag, bla, lacI | Novagen |
| pET28a | PT7, lacO, MCS, 6xHis·Tag, lacI, KanR | Novagen |
| pGEM®-T easy | PSP6 RNA polymerase, lacO, bla, PT7, phage f1 region | Promega |
| pDN19 | Plac, PT7, lacZα′, tetA, oriT | (Nunn et al., 1990) |
| pMSR1 | PT7, lacO, pqsB-6xHis·Tag, bla, lacI, (from pET303) | L.G. Rahme |
| pCD1* | pqsB cloned in pET22b(+) | This study |
| pCD2* | pqsB-6xHis·Tag from pMRS1 cloned in pDN19 | This study |
| pCD3* | 6xHis·Tag-pqsC built into pET28a | This study |
| pCD4* | 6xHis·Tag-pqsC from pCD3 cloned in pDN19 | This study |
PCR primers used to construct these expression vectors are shown in Table S1.
Bacterial cultures
Tryptic soy broth (TSB; BD) was used for all cultures, unless otherwise stated. Bacteria were grown at 37°C in a TC-7 roller drum (New Brunswick) or on TSB agar plates. Cultures are typically performed at least in triplicates.
Construction of pqsC-6xHis expression vector pCD3
The pqsC coding sequence was amplified from PA14 genomic DNA as template with a Fast-Pfu DNA polymerase (Feldan) using primers F-pqsC-NdeI and R-pqsC-EcoRI (see Table S1). The PCR product was purified using the QIAquick PCR Purification Kit (QIAGEN), and sequentially digested with NdeI and EcoRI (NEB). In parallel, pET28a was similarly digested in similar conditions. Ligation reaction was carried out with the T4 DNA ligase (NEB), and transformation performed on chemically competent E. coli DH5α cells. Transformed cells were selected on LB agar plates containing 30 μg/mL kanamycin. After incubation at 37°C overnight, recombinant transformed clones were confirmed by colony PCR.
Construction of pqsB expression vector (pCD1)
In order to produce a not-tagged PqsB expression vector, reverse primer R-pqsB-HindII was designed with a stop codon. Forward primer was F-pqsB-NdeI. Construction of pCD1 was performed according to the method described above for pCD3.
Construction of a pqsC-6xHis vector for P. aeruginosa expression (pCD4)
pCD3 was digested using both XbaI and EcoRI restriction enzymes. The 6xHis-tag-pqsC fragment was separated after agarose gel electrophoresis and purified. Plasmid pDN19 was linearized using XbaI and EcoRI, and purified. The fragment was ligated in linearized pDN19 as above. Transformation in chemically competent E. coli DH5α was carried out and transformed cells were selected on LB agar plates containing 20 μg/mL tetracycline and after incubation at 37°C positive clones were verified by colony PCR. Functional expression of PqsC was confirmed by complementation of a nonpolar pqsC- mutant and HAQ detection by LC/MS (see below).
Construction of pqsB-6xHis vector for P. aeruginosa expression (pCD2)
pMSR1 was used as a template to PCR amplify the pqsB-6xHis fragment, using primers F-pqsB-XbaI and R-pqsBHisTag-SacI. After purification, the PCR product was cloned into the linearized plasmid pGEM®-T Easy according to the method described by Promega, and then transformed in E. coli DH5α. This subclone was purified using the Wizard Plus SV Minipreps (Promega) and digested with XbaI and SacI (NEB). The pqsB-6xHis-tag fragment was separated on agarose gel electrophoresis and purified. In parallel, pDN19 was digested with the same enzymes and purified. Subsequently, ligation was performed with T4 DNA ligase. Transformants in E. coli DH5α were selected on LB agar plates containing 20 μg/mL tetracycline. Functional expression of PqsB was confirmed by complementation of a nonpolar pqsB- mutant and HAQ detection by LC/MS.
Site-directed mutagenesis of Cys129 in pqsC
To examine if the Cys 129 residue of PqsC is required for binding octanoyl, this residue was changed to an alanine by QuikChange Site-Directed Mutagenesis (Wang and Malcolm, 1999). We used plasmid pCD3 as template primers PqsC_C129A and PqsC_C129A-Antisense. The PCR was carried out using the Fast-Pfu DNA polymerase. After purification of the product, this methylated template was digested with DpnI (NEB®) and the mutated construction was transformed in E. coli BL21 (DE3) chemical competent cells. Site-directed mutagenesis was verified by sequencing the pqsC gene on the mutated plasmid.
Mass spectrometry
The mass spectrometer was a triple quadrupole Quattro Premier XE (Waters, Missisissauga, Can.) interfaced to a Waters 2795 HPLC. The column was a 4.6 × 250 mm Agilent XDB-C8 column (particle size, 5 μm). The mobile phase was composed of water and acetonitrile containing 1% acetic acid. Quantification of the various HAQ was performed using HHQ-d4 as internal standard, as described previously (Lépine and Déziel, 2011; Lépine et al., 2003).
The mass spectrometer was operated in positive electrospray ionization mode with a capillary voltage of 3 kV and a cone voltage of 20 V. Nitrogen was the nebulising gas and the source was held at 120°C. Scanning was performed in full scan mode with a mass range of m/z 130 to 400. In negative mode, 2 mM ammonium acetate was included in the water and acetonitrile mobile phases. MS/MS analysis was performed with Argon as collision gas.
Isotope-labelled fatty acid feeding experiments
Decanoic-9,9,10,10,10-d5 acid (CDN isotopes, Pointe-Claire, Canada) and decanoic-1,2-13C2 acid (Sigma) stocks were prepared in MeOH at a final concentration of 100 mM. Overnight cultures of P. aeruginosa PA14 were diluted in fresh TSB to an OD600 = 0.5. Cultures were supplemented with 1 mM of the desired labeled fatty acid and 50 mg/L anthranilic acid and grown at 37°C with rotation. Control cultures were also prepared by replacing the labeled molecules with MeOH or with unlabeled decanoic acid (Sigma). Samples were collected at OD600 = 2.5 and analyzed for HHQ production.
Supernatant cross-feeding experiments
Supernatant from overnight cultures of various P. aeruginosa mutants supplemented with PQS-d4 (20 mg/l 2,3,4,5-tetradeuterated PQS) were collected by centrifugation and filtration (0,22 um). Supernatant-based medium was then prepared as follows: 2 mL of filtered supernatant, 0.5 mL of sterile water and 0.5 mL of 6X TSB broth. An overnight culture of a second mutant (see Fig. 3) was diluted in prepared supernatant-based medium to an OD600 = 0.5 and cultures were grown at 37°C. Samples were treated as usual for LC/MS quantification of HAQs, except that HHQ-d4 was used as internal standard.
Isotope-labelled acetate feeding experiments
To further investigate the biosynthesis of 2-ABA, the nonpolar pqsB− pqsL− double mutant, was cultivated in an M9 mineral medium with 20 mg/L PQS as inducer, 36 mM sodium acetate-13C-labelled at position 1 or 2, (Sigma-Aldrich), or unlabelled acetate, as sole carbon source, and 50 mg/L anthranilic acid to avoid incorporation of 13C labelling in the aromatic ring. Sterile supernatants were recovered by centrifugation and filtration and provided to the nonpolar pqsA- pqsH- double mutant growing in TSB, as described above for the cross-feeding experiments. For experiments shown in figures 4c and 4d, the 2-ABA present in the precultures was not labelled, and therefore the sodium acetate-1,2-13C or the fully 13C-labelled octanoic acid was provided to the pqsA- pqsH- mutant cultures along with supernatants.
Isolation of 2-ABA
Twenty 250 ml Erlenmeyer flasks each containing 50 mL of TSB supplemented with 20 mg/l of PQS were inoculated with a nonpolar pqsB− pqsL− mutant and incubated at 34°C and 240 rpm in a rotary shaker for 18h. The supernatant of a nonpolar pqsB− pqsL− double mutant produces more HHQ when fed to a nonpolar pqsA- mutant than the single non polar pqsB mutant (result not shown) and is thus ideal to maximize the production of the intermediate. The cultures were then pooled and centrifuged at 14,000 × g for 15 min. The supernatant was extracted twice with 600 ml ethyl acetate and once with 600 ml dichloromethane. The aqueous phase, which still contained the active intermediate, was concentrated to 50 ml in a rotary evaporator and 50 ml of methanol was added. After leaving the mixture at 4°C for 1 hr, it was filtrated and the filtrate evaporated to a volume of 30 ml. A volume of 50 ml of ethanol was then added, the precipitate removed by filtration and the filtrate evaporated to dryness. The residue was then fractionated with a Waters DeltaPrep 4000 preparative HPLC, equipped with a 50 × 21 mm Gemini NX-C18 column (particle size, 10 μm) (Phenomenex). Water (A) and acetonitrile (B), each containing 3 mM NH4OH, were used as eluents, with a flow rate of 3.5 ml·min−1. The initial gradient was 100% A for 9 min, then going to 20% B in 2 min, then to 100% B in 1 min, which was then maintained for 7 min. The absorbance was monitored at 364 nm and the active fraction, identified by feeding cultures of a nonpolar pqsA− mutant and looking for HHQ production, eluted between 7 and 9.7 min. The solvent was evaporated and the residue further purified by thick layer chromatography on a 20 × 20 cm Partisil PK6F silica plates (Whatman) using methanol/ethyl acetate (1:1) as eluent. The plates were revealed with UV light and the band with a Rf = 0.55 was collected.
Chemical synthesis of 2-ABA
2-ABA was chemically synthesized by hydrolysing ethyl 2-nitrobenzoylacetate with sulphuric acid and by hydrogenation of the acid with palladium, as described (Sicker and Mann, 1988). Exactly 240 mg ethyl-2-nitrobenzoylacetate were dissolved in 1 mL water to which was added 500 μL concentrated sulphuric acid and left at room temperature overnight. The mixture was poured on ice and extracted with ethyl acetate. The organic phase was then extracted with a 1% Na2CO3 aqueous solution which was acidified to pH 4–5. This solution was extracted with ethyl acetate to provide 167 mg of pure 2-nitrobenzoylacetic acid (mp = 118–119°C, litt. Mp = 117°C) (Overmyer, 1926). 2-nitrobenzoylacetic acid (200 mg) were dissolved in 10 mL isopropanol, and 50 μL of concentrated ammonium hydroxide and 10 mg of 5% palladium on charcoal were added. The mixture was hydrogenated at 25 psi for 1.5 hr. After filtration the solvent was evaporated and the residue purified par thick layer chromatography to produce 140 mg 2-ABA.
The synthesis of 2-aminobenzoylacetate-1,2-13C2 was also performed starting with ethyl acetoacetate-1,2-13C2 and 2-nitrobenzoyl chloride to produce ethyl 2-nitrobenzoylacetoacetate which upon treatment with pure sulphuric acid produced 2-nitrobenzoylacetic acid which was hydrogenated as above.
HHQ production from malonyl-CoA and PA14 cell extract
The PA14 cell lysate was prepared as follows: Bacteria grown at OD600 = 4 were recovered by centrifugation at 4,000 × g for 30 min at 4°C and washed twice in PBS. The cells were concentrated 10x in 20 mM Tris-HCl pH 7.6 buffer, and lyzed by three cycles of 30 sec sonication on ice with 1 min intervals (Branson sonifier 450, Duty cycle: 30%, Output control: 10). Finally, the insoluble fraction was removed by centrifugation at 15,000 × g for 30 min at 4°C and the cytoplasmic extract retrieved by filtration of the supernatant on 0.2 μm.
Increasing concentrations of malonyl-CoA (0–10 μM), were mixed with 100 μM anthranilic-d4 acid, 100 μM Coenzyme A, 650 μM ATP, and 100 μM octanoyl-CoA, in a volume of 90 μL completed with 50 μL of the soluble PA14 cytoplasmic extract. The reaction was incubated for 1 hrs at 37°C, and the production of HHQ-d4 was measured by LC/MS, using 4-hydroxy-3-methyl-2-heptenylquinoline as internal standard (Vial et al., 2008). The assay was performed in duplicate.
Coexpression and copurification of His·Tag-PqsB and PqsC-His·Tag
Plasmids pMSR1 and pCD3 were transformed in E. coli Origami 2 (DE3) and possible double transformants selected on LB agar plates containing 30 μg/ml kanamycin, and 50 μg/ml carbencilline. Presence of both plasmids was confirmed by colony PCR. A positive clone was grown O/N in LB with 30 μg/ml kanamycin, and 50 μg/ml carbencilline and diluted 1/100 in 500 mL LB. When the OD600 reached 0.8, expression was induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 2 hrs and the cells were then collected by centrifugation at 4,000 × g for 30 min at 4°C. The pelleted cells were then resuspended in 35 mL cold binding buffer (0.5 M NaCl, 20 mM Tris-HCl, 20 mM NaH2PO4, 20 mM Imidazole, pH 7.8) and lyzed by five cycles of 1 min sonication on ice with 1 min intervals (Branson sonifier 450, Duty cycle: 30%, Output control: 10). Finally, the insoluble fraction was removed by centrifugation at 15,000 × g for 30 min at 4°C and the soluble extract retrieved by filtration of the supernatant on 0.2 μm.
Co-purification of the tagged PqsB and PqsC proteins was performed using a 5 mL HisTrap FF affinity column (GE Healthcare) prepared according to the manufacturer’s instructions, and a ÄKTApurifier FPL (GE Healthcare). A flow of 1 ml/min was used throughout the purification steps. After the samples were loaded, the column was washed with five column volumes of binding buffer. For elution, buffer A was the same as binding buffer without imidazole, while buffer B contained 0.5 M imidazole. An increasing imidazole gradient was achieved by going from 4% buffer B to 60% buffer B over 20 column volumes. Two ml samples of flow-through and fractions were collected and analyzed on a 12% SDS-PAGE gel. Fractions containing the co-purified proteins were pooled, and the elution buffer was changed by dialysis with a 10 kDa cut-off dialysis bag over 80 volumes of 20 mM Tris-HCl, pH 8.0 at 4°C. Dialyzed proteins were then concentrated about 100x using an Amicon Ultra-15 centrifugal filter with a 10 kDa cutoff (Millipore). When the volume was reduced to about 500 μl, an equal volume of 2X conservation buffer (20 mM HEPES, 60% glycerol, pH 8.0) was added and aliquots were conserved at −80°C. We used the Bradford protein assay (BioRad) to measure the concentration of the co-purified PqsB/PqsC proteins.
Proteomic analysis
Co-purified PqsB and PqsC was separated from the protein mixture by running on SDS-PAGE 12% acrylamide gel. Both PqsB and PqsC bands were cut from the gel. Proteins were reduced with DTT and alkylated with iodoacetamide prior to in-gel digestion with trypsin. The tryptic peptides were eluted from the gel with acetonitrile containing 0.1% trifluoroacetic acid. The tryptic peptides were then separated on a Agilent Nanopump using a C18 ZORBAX trap and a SB-C18 ZORBAX 300 reversed phase column (150 mm × 75 μm, 3.5 μm particle size) (Agilent Technologies, Inc.). All mass spectra were recorded on a hybrid linear ion trap-triple quadrupole mass spectrometer (Q-Trap, AB Applied Biosystems, MDS SCIEX Instruments, California, USA) equipped with a nano-electrospray ionization source. The accumulation of MS/MS data was performed with the Analyst Software, version 1.4 (AB Applied Biosystems / MDS SCIEX Instruments, California, USA). MASCOT (Matrix Science, London, UK) was used to create peak lists from MS and MS/MS raw data.
In vitro synthesis of HHQ
For the enzymatic assay, 0 to 27.5 μM octanoyl-CoA, 100 μM 2-ABA, and 20 ng/μL of co-purified PqsB/PqsC were combined and completed to 100 μL with 20 mM Tris-HCl pH 7.6. The reaction was incubated for 1 hrs at 37°C, and the production of HHQ was measured by LC/MS, using 4-hydroxy-3-methyl-2-heptenylquinoline as internal standard (Vial et al., 2008). The assay was performed in duplicate.
Supplementary Material
Highlights.
We have deciphered the 4-hydroxy-2-alkylquinolines (HAQs) biosynthetic pathway
Contrary to the current model, fatty acids are directly incorporated into HAQs
PqsD produces 2-aminobenzoylacetate (2-ABA), which is then coupled to a fatty acid by PqsC
2-ABA is also likely the precursor of HQNO and 2-aminoacetophenone
Acknowledgments
Thanks to Melissa Starkey for providing plasmid pMSR1. This study was supported by Canadian Institutes of Health Research (CIHR) Operating grant MOP-97888 to ED, a NSERC Discovery grant to FL, and National Institute of Health (NIH) grant 2R56AI063433- 06A1 to LGR. CED and VD were respectively recipients of a M.Sc. and a Ph.D. scholarship from the Fondation Armand-Frappier. ED was a Chercheur-boursier Junior 2 of the Fonds de la recherche en santé du Québec (FRSQ) and holds a Canada Research Chair in Sociomicrobiology.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Balasubramanian D, Schneper L, Kumari H, Mathee K. A dynamic and intricate regulatory network determines Pseudomonas aeruginosa virulence. Nucleic Acids Res. 2013;41:1–20. doi: 10.1093/nar/gks1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandyopadhaya A, Kesarwani M, Que YA, He J, Padfield K, Tompkins R, Rahme LG. The quorum sensing volatile molecule 2-amino acetophenon modulates host immune responses in a manner that promotes life with unwanted guests. Plos Pathogens. 2012;8:e1003024. doi: 10.1371/journal.ppat.1003024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bera AK, Atanasova V, Robinson H, Eisenstein E, Coleman JP, Pesci EC, Parsons JF. Structure of PqsD, a Pseudomonas Quinolone Signal Biosynthetic Enzyme, in Complex with Anthranilate. Biochemistry-Us. 2009;48:8644–8655. doi: 10.1021/bi9009055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjarnsholt T, Tolker-Nielsen T, Hoiby N, Givskov M. Interference of Pseudomonas aeruginosa signalling and biofilm formation for infection control. Expert Rev Mol Med. 2010;12:e11. doi: 10.1017/S1462399410001420. [DOI] [PubMed] [Google Scholar]
- Bredenbruch F, Nimtz M, Wray V, Morr M, Müller R, Häussler S. Biosynthetic pathway of Pseudomonas aeruginosa 4-hydroxy-2-alkylquinolines. J Bacteriol. 2005;187:3630–3635. doi: 10.1128/JB.187.11.3630-3635.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calfee MW, Coleman JP, Pesci EC. Interference with Pseudomonas quinolone signal synthesis inhibits virulence factor expression by Pseudomonas aeruginosa. PNAS. 2001;98:11633–11637. doi: 10.1073/pnas.201328498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H, Krishnan G, Goumnerov B, Tsongalis J, Tompkins R, Rahme LG. A quorum sensing-associated virulence gene of Pseudomonas aeruginosa encodes a LysR-like transcription regulator with a unique self-regulatory mechanism. Proc Natl Acad Sci U S A. 2001;98:14613–14618. doi: 10.1073/pnas.251465298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman JP, Hudson LL, McKnight SL, Farrow JM, 3rd, Calfee MW, Lindsey CA, Pesci EC. Pseudomonas aeruginosa PqsA is an anthranilate-coenzyme A ligase. J Bacteriol. 2008;190:1247–1255. doi: 10.1128/JB.01140-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornforth JW, James AT. Structure of a naturally occurring antagonist of dihydrostreptomycin. Biochem J. 1956;63:124–130. doi: 10.1042/bj0630124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CD, Parker J. Use of 2-aminoacetophenone production in identification of Pseudomonas aeruginosa. J Clin Microbiol. 1979;9:479–484. doi: 10.1128/jcm.9.4.479-484.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Déziel E, Gopalan S, Tampakaki AP, Lépine F, Padfield KE, Saucier M, Xiao G, Rahme LG. The contribution of MvfR to Pseudomonas aeruginosa pathogenesis and quorum sensing circuitry regulation: multiple quorum sensing-regulated genes are modulated without affecting lasRI, rhlRI or the production of N-acyl-L-homoserine lactones. Mol Microbiol. 2005;55:998–1014. doi: 10.1111/j.1365-2958.2004.04448.x. [DOI] [PubMed] [Google Scholar]
- Déziel E, Lépine F, Milot S, He J, Mindrinos MN, Tompkins RG, Rahme LG. Analysis of Pseudomonas aeruginosa 4-hydroxy-2- alkylquinolines (HAQs) reveals a role for 4-hydroxy-2- heptylquinoline in cell-to-cell communication. Proc Natl Acad Sci U S A. 2004;101:1339–1344. doi: 10.1073/pnas.0307694100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggle SP, Matthijs S, Wright VJ, Fletcher MP, Chhabra SR, Lamont IL, Kong X, Hider RC, Cornelis P, Camara M, et al. The Pseudomonas aeruginosa 4-quinolone signal molecules HHQ and PQS play multifunctional roles in quorum sensing and iron entrapment. Chem Biol. 2007;14:87–96. doi: 10.1016/j.chembiol.2006.11.014. [DOI] [PubMed] [Google Scholar]
- Diggle SP, Winzer K, Chhabra SR, Worrall KE, Camara M, Williams P. The Pseudomonas aeruginosa quinolone signal molecule overcomes the cell density-dependency of the quorum sensing hierarchy, regulates rhl-dependent genes at the onset of stationary phase and can be produced in the absence of LasR. Mol Microbiol. 2003;50:29–43. doi: 10.1046/j.1365-2958.2003.03672.x. [DOI] [PubMed] [Google Scholar]
- Driscoll JA, Brody SL, Kollef MH. The epidemiology, pathogenesis and treatment of Pseudomonas aeruginosa infections. Drugs. 2007;67:351–368. doi: 10.2165/00003495-200767030-00003. [DOI] [PubMed] [Google Scholar]
- Farrow JM, 3rd, Sund ZM, Ellison ML, Wade DS, Coleman JP, Pesci EC. PqsE functions independently of PqsR-Pseudomonas quinolone signal and enhances the rhl quorum-sensing system. J Bacteriol. 2008;190:7043–7051. doi: 10.1128/JB.00753-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher LA, McKnight SL, Kuznetsova MS, Pesci EC, Manoil C. Functions required for extracellular quinolone signaling by Pseudomonas aeruginosa. J Bacteriol. 2002;184:6472–6480. doi: 10.1128/JB.184.23.6472-6480.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galloway WR, Hodgkinson JT, Bowden S, Welch M, Spring DR. Applications of small molecule activators and inhibitors of quorum sensing in Gram-negative bacteria. Trends Microbiol. 2012;20:449–458. doi: 10.1016/j.tim.2012.06.003. [DOI] [PubMed] [Google Scholar]
- Heeb S, Fletcher MP, Chhabra SR, Diggle SP, Williams P, Camara M. Quinolones: from antibiotics to autoinducers. FEMS microbiology reviews. 2011;35:247–274. doi: 10.1111/j.1574-6976.2010.00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman LR, Déziel E, D’Argenio DA, Lépine F, Emerson J, McNamara S, Gibson RL, Ramsey BW, Miller SI. Selection for Staphylococcus aureus small-colony variants due to growth in the presence of Pseudomonas aeruginosa. Proc Natl Acad Sci U S A. 2006;103:19890–19895. doi: 10.1073/pnas.0606756104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez PN, Koch G, Thompson JA, Xavier KB, Cool RH, Quax WJ. The multiple signaling systems regulating virulence in Pseudomonas aeruginosa. Microbiol Mol Biol Rev. 2012;76:46–65. doi: 10.1128/MMBR.05007-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhas M, Eberl L, Tummler B. Quorum sensing: the power of cooperation in the world of Pseudomonas. Environ Microbiol. 2005;7:459–471. doi: 10.1111/j.1462-2920.2005.00769.x. [DOI] [PubMed] [Google Scholar]
- Kerr KG, Snelling AM. Pseudomonas aeruginosa: a formidable and ever-present adversary. J Hosp Infect. 2009;73:338–344. doi: 10.1016/j.jhin.2009.04.020. [DOI] [PubMed] [Google Scholar]
- Kesarwani M, Hazan R, He J, Que Y, Apidianakis Y, Lesic B, Xiao G, Dekimpe V, Milot S, Déziel E, et al. A quorum sensing regulated small volatile molecule reduces acute virulence and promotes chronic infection phenotypes. PLoS pathogens. 2011;7:e1002192. doi: 10.1371/journal.ppat.1002192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Kim YU, Koh BH, Hwang SS, Kim SH, Lepine F, Cho YH, Lee GR. HHQ and PQS, two Pseudomonas aeruginosa quorum-sensing molecules, down-regulate the innate immune responses through the nuclear factor-kappaB pathway. Immunology. 2010;129:578–588. doi: 10.1111/j.1365-2567.2009.03160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan JC, Meickle T, Ladwa D, Teplitski M, Paul V, Luesch H. Lyngbyoic acid, a “tagged” fatty acid from a marine cyanobacterium, disrupts quorum sensing in Pseudomonas aeruginosa. Mol Biosyst. 2011;7:1205–1216. doi: 10.1039/c0mb00180e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DG, Urbach JM, Wu G, Liberati NT, Feinbaum RL, Miyata S, Diggins LT, He J, Saucier M, Déziel E, et al. Genomic analysis reveals that Pseudomonas aeruginosa virulence is combinatorial. Genome Biol. 2006;7:R90. doi: 10.1186/gb-2006-7-10-r90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lépine F, Dekimpe V, Lesic B, Milot S, Lesimple A, Mamer OA, Rahme LG, Déziel E. PqsA is required for the biosynthesis of 2,4-dihydroxyquinoline (DHQ), a newly identified metabolite produced by Pseudomonas aeruginosa and Burkholderia thailandensis. Biol Chem. 2007;388:839–845. doi: 10.1515/BC.2007.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lépine F, Déziel E. Liquid chromatography/mass spectrometry for the detection and quantification of N-acyl-L-homoserine lactones and 4-hydroxy-2-alkylquinolines. Methods Mol Biol. 2011;692:61–69. doi: 10.1007/978-1-60761-971-0_5. [DOI] [PubMed] [Google Scholar]
- Lépine F, Déziel E, Milot S, Rahme LG. A stable isotope dilution assay for the quantification of the Pseudomonas quinolone signal in Pseudomonas aeruginosa cultures. Biochim Biophys Acta. 2003;1622:36–41. doi: 10.1016/s0304-4165(03)00103-x. [DOI] [PubMed] [Google Scholar]
- Lépine F, Milot S, Déziel E, He J, Rahme LG. Electrospray/mass spectrometric identification and analysis of 4-hydroxy-2-alkylquinolines (HAQs) produced by Pseudomonas aeruginosa. J Am Soc Mass Spectrom. 2004;15:862–869. doi: 10.1016/j.jasms.2004.02.012. [DOI] [PubMed] [Google Scholar]
- Lesic B, Lépine F, Déziel E, Zhang J, Zhang Q, Padfield K, Castonguay MH, Milot S, Stachel S, Tzika AA, et al. Inhibitors of pathogen intercellular signals as selective anti-infective compounds. PLoS Pathog. 2007;3:1229–1239. doi: 10.1371/journal.ppat.0030126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesic B, Rahme LG. Use of the lambda Red recombinase system to rapidly generate mutants in Pseudomonas aeruginosa. BMC Mol Biol. 2008;9:20. doi: 10.1186/1471-2199-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lightbown JW. An antagonist of streptomycin and dihydrostreptomycin produced by Pseudomonas aeruginosa. J Gen Microbiol. 1954;11:477–492. doi: 10.1099/00221287-11-3-477. [DOI] [PubMed] [Google Scholar]
- Luckner M, Ritter C. On the biosynthesis of the 2-n-alkyl-4-hydroxyquinolines Pseudomonas aeruginosa (schroet.) migula. Tetrahedron Letters. 1965;12:741–744. doi: 10.1016/s0040-4039(01)83977-0. [DOI] [PubMed] [Google Scholar]
- Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa - Caenorhabditis elegans pathogenesis model. Cell. 1999;96:47–56. doi: 10.1016/s0092-8674(00)80958-7. [DOI] [PubMed] [Google Scholar]
- Nunn D, Bergman S, Lory S. Products of three accessory genes, pilB, pilC, and pilD, are required for biogenesis of Pseudomonas aeruginosa pili. J Bacteriol. 1990;172:2911–2919. doi: 10.1128/jb.172.6.2911-2919.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overmyer CJ. Synthesis of substitution derivatives of indigo. I. o-Nitrobenzoylacetic acid and related compounds. J Am Chem Soc. 1926;48:454–460. [Google Scholar]
- Pistorius D, Ullrich A, Lucas S, Hartmann RW, Kazmaier U, Muller R. Biosynthesis of 2-Alkyl-4(1H)-quinolones in Pseudomonas aeruginosa: potential for therapeutic interference with pathogenicity. Chembiochem. 2011;12:850–853. doi: 10.1002/cbic.201100014. [DOI] [PubMed] [Google Scholar]
- Rahme LG, Stevens EJ, Wolfort SF, Shao J, Tompkins RG, Ausubel FM. Common virulence factors for bacterial pathogenicity in plants and animals. Science. 1995;268:1899–1902. doi: 10.1126/science.7604262. [DOI] [PubMed] [Google Scholar]
- Ritter C, Luckner M. Biosynthesis of 2-n-alkyl-4-hydroxyquinoline derivatives (pseudane) in Pseudomonas aeruginosa. Eur J Biochem. 1971;18:391–400. doi: 10.1111/j.1432-1033.1971.tb01255.x. [DOI] [PubMed] [Google Scholar]
- Scott-Thomas AJ, Syhre M, Pattemore PK, Epton M, Laing R, Pearson J, Chambers ST. 2-Aminoacetophenone as a potential breath biomarker for Pseudomonas aeruginosa in the cystic fibrosis lung. Bmc Pulm Med. 2010:10. doi: 10.1186/1471-2466-10-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sicker D, Mann G. Synthesis of Ethyl Ortho-Substituted Benzoylacetates and Investigation of the Influence of Ortho-Substituents on Keto Enol Tautomerism and Ms Fragmentation Behavior. Collect Czech Chem C. 1988;53:839–850. [Google Scholar]
- Smith RS, Iglewski B. P. aeruginosa quorum-sensing systems and virulence. Curr Opin Microbiol. 2003;6:56–60. doi: 10.1016/s1369-5274(03)00008-0. [DOI] [PubMed] [Google Scholar]
- Taylor GW, Lynn SL. Quinoline-type antimicrobials for use against Helicobacter pylori infections. 1995. [Google Scholar]
- Vial L, Lépine F, Milot S, Groleau MC, Dekimpe V, Woods DE, Déziel E. Burkholderia pseudomallei, B. thailandensis, and B. ambifaria produce 4-hydroxy-2-alkylquinoline analogues with a methyl group at the 3 position that is required for quorum-sensing regulation. J Bacteriol. 2008;190:5339–5352. doi: 10.1128/JB.00400-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade DS, Calfee MW, Rocha ER, Ling EA, Engstrom E, Coleman JP, Pesci EC. Regulation of Pseudomonas quinolone signal synthesis in Pseudomonas aeruginosa. J Bacteriol. 2005;187:4372–4380. doi: 10.1128/JB.187.13.4372-4380.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Malcolm BA. Two-stage PCR protocol allowing introduction of multiple mutations, deletions and insertions using QuikChange (TM) site-directed mutagenesis. Biotechniques. 1999;26:680–682. doi: 10.2144/99264st03. [DOI] [PubMed] [Google Scholar]
- Xiao G, Déziel E, He J, Lépine F, Lesic B, Castonguay MH, Milot S, Tampakaki AP, Stachel SE, Rahme LG. MvfR, a key Pseudomonas aeruginosa pathogenicity LTTR-class regulatory protein, has dual ligands. Mol Microbiol. 2006;62:1689–1699. doi: 10.1111/j.1365-2958.2006.05462.x. [DOI] [PubMed] [Google Scholar]
- Zhang YM, Frank MW, Zhu K, Mayasundari A, Rock CO. PqsD is responsible for the synthesis of 2,4-dihydroxyquinoline, an extracellular metabolite produced by Pseudomonas aeruginosa. J Biol Chem. 2008;283:28788–28794. doi: 10.1074/jbc.M804555200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.