

Abstract
Alzheimer disease (AD) is the most common form of dementia among the elderly and is characterized by progressive loss of memory and cognition. These clinical features are due in part to the increase of reactive oxygen and nitrogen species that mediate neurotoxic effects. The up-regulation of the heme oxygenase-1/biliverdin reductase-A (HO-1/BVR-A) system is one of the earlier events in the adaptive response to stress. HO-1/BVR-A reduces the intracellular levels of pro-oxidant heme and generates equimolar amounts of the free radical scavengers biliverdin-IX alpha (BV)/bilirubin-IX alpha (BR) as well as the pleiotropic gaseous neuromodulator carbon monoxide (CO) and ferrous iron. Two main and opposite hypotheses for a role of the HO-1/BVR-A system in AD propose that this system mediates neurotoxic and neuroprotective effects, respectively. This apparent controversy was mainly due to the fact that for over about 20 years HO-1 was the only player on which all the analyses were focused, excluding the other important and essential component of the entire system, BVR. Following studies from the Butterfield laboratory that reported alterations in BVR activity along with decreased phosphorylation and increased oxidative/nitrosative post-translational modifications in the brain of subjects with AD and amnestic mild cognitive impairment (MCI) subjects, a debate was opened on the real pathophysiological and clinical significance of BVR-A. In this paper we provide a review of the main discoveries about the HO/BVR system in AD and MCI, and propose a mechanism that reconciles these two hypotheses noted above of neurotoxic and the neuroprotective aspects of this important stress responsive system.
Keywords: Alzheimer disease, aging, biliverdin reductase, heme oxygenase, mild cogninitve impairment, oxidative stress
The aim of this review is to provide a comprehensive analysis about recent results involving the heme oxygenase/bileverdin reductase system with respect to Alzheimer disease (AD) and its arguably earliest form, amnestic mild cognitive impairment (MCI). In particular, a novel point of view regarding the existing paradigm about the HO/BVR-A system is proposed.
The heme oxygenase/biliverdin reductase system: an overview
Under physiological conditions, cell homeostasis is finely regulated by a balance between pro-oxidant and anti-oxidant stimuli; however, certain environmental factors, stressors, or diseases may affect this equilibrium and increase the production of reactive oxygen species (ROS) and reactive nitrogen species (RNS). Both ROS and RNS may react with biomolecules including proteins, lipids, carbohydrates, DNA and RNA (Halliwell, 2006) leading to their oxidative damage resulting in cellular dysfunction (Butterfield et al., 2001) (Lovell et al., 2001; Mark et al., 1997; Markesbery, 1997; Smith et al., 1994b).
The heme oxygenase/biliverdin reductase (HO/BVR) system is one of the main and evolutionarily conserved cellular cytoprotectants, whose up-regulation represents an early event in the adaptive response to stress (Poon et al., 2004). Despite that the initial attention by the scientific community was focused primarily on the ability of this system to degrade heme to be the main, if not the only, function, quite recently numerous different functions have been elucidated.
HO regulation and distribution
Humans and rodents have two HO isozymes, namely HO-1 (about 32 kDa, enzyme) and HO-2 (36 kDa) encoded by the HMOX1 and HMOX2 genes, respectively (Gozzelino et al., 2010). The third member of the family (HMOX3) has also been described, but it is generally believed that HO-3 is only represented by a pseudogene, with no coding function (Hayashi et al., 2004) (McCoubrey et al., 1997). Nevertheless, in light of the recent discoveries of the possible regulatory functions of pseudogenes (Pink et al., 2011), it is possible that HO-3 might also have biological effects by contributing to gene regulation (Vitek, 2012). Heme oxygenase-1, also known as heat shock protein (Hsp)-32, is induced by various stimuli, including oxidative and nitrosative stress, ischemia, heat shock, bacterial lipopolysaccharide (LPS), hemin, the neuroprotective agent leteprinim potassium (Neotrofin) (Maines, 1997; Maines, 2000) and several drugs currently used in the clinic, such as statins, non-steroidal antinflammatory drugs, antagonists to the adrenergic β receptor, cyclosporine A etc. (Butterfield et al., 2012a; Mancuso and Barone, 2009). Heme oxygenase-2, the constitutive isoform, is responsive to developmental factors, adrenal glucocorticoids and nitric oxide (NO) (Maines, 1997). Very interestingly, our group found that despite its constitutive nature, HO-2 protein levels can be up-regulated by atorvastatin in the cerebellum of aged beagles opening new frontiers in the comprehension of its features (Butterfield et al., 2012a). Similarly, HO-2 over-expression was detected as a consequence of the administration of drugs acting on the nervous system, such as morphine and glucocorticoids (Mancuso and Barone, 2009).
Under basal conditions, the expression of HO-1 is finely regulated at the HMOX1 gene level through the transcriptional repressor Bach-1, (Figure 1). Under pro-oxidant conditions, HO-1 undergoes up-regulation at both gene and protein levels. HMOX1 possesses two upstream enhancer regions, E1 and E2 (Sun et al., 2002), which contain multiple stress-responsive elements (SREs), also known as antioxidant-responsive elements (AREs), thus supporting the evidence of an oxidative-inducible nature of this protein (Figure 1). AREs share a consensus sequence (GCnnnGTA) with the Maf recognition element (MARE) (Stewart et al., 2003). The interactions between MAREs and heterodimers formed by a Maf protein (MafK, MafF, or MafG) and an NF-E2-related factor 2 (Nrf2), or activator protein 1 (AP-1) play a direct role in HO-1 induction (Maines, 2005a; Sun et al., 2002) (Figure 1). In contrast, HO-1 expression can be repressed by hypoxia, β- carotene, cigarette smoke, interferon-γ or desferrioxamine (Palozza et al., 2006; Shibahara et al., 2003). The inhibition is mediated by Bach1, which binds to AREs in the HMOX1 promoter, thus inhibiting the transcription by Nrf-2 or AP-1 (Kitamuro et al., 2003; Sun et al., 2004; Sun et al., 2002) (Figure 1).
Figure 1. The heme oxygenase/biliverdin reductase (HO/BVR) pathways.

Hemoprotein-derived heme is rapidly transformed by the activity of membrane-bound HO into equimolar amounts of carbon monoxide (CO), iron (II) (Fe2+) and biliverdin. Through the activity of cytosolic biliverdin reductase (BVR), HO-derived biliverdin is immediately reduced to bilirubin. Either heme and iron could be responsible for an increase of the oxidative stress levels in the cells. Thus, by the degradation of heme and through the antioxidant activity of both biliverdin and bilirubin, the HO/BVR system contributes to the maintenance of low oxidative stress levels in the cell. Furthermore, both heme and increased oxidative stress levels represent two main factors regulating HO-1 protein synthesis. Indeed, the HMOX1 promoter contains an ARE sequence recognized by specific transcription factors activated in response to oxidative stress. Under basal conditions, Bach1/small Maf dimers bind constitutively to ARE and inhibit HMOX1 transcription. However, in response to oxidative stress, heme binds Bach1, which is then exported from the nucleus, ubiquitinated and degraded, releasing transcriptional repression. Oxidative stress also induces Keap1 ubiquitination-degradation, allowing the transcription factor NF-E2-related factor-2 (Nrf2) to translocate into the nucleus. Nrf2/small Maf protein heterodimers bind to ARE and promote HMOX1 transcription. Most probably the Bach1/Nrf2 transcriptional system interacts functionally with other transcription factors to regulate HMOX1 transcription. In addition, BVR can function as a shuttle to vehicle heme to the nucleus. Transport of heme to the nucleus by BVR would enable its delivery to the transcriptional repressor Bach1, which, on binding heme dissociates from the DNA and is replaced by the Nrf2 transcription factor (Dhakshinamoorthy et al., 2005; Ogawa et al., 2001), thus allowing HMOX1 transcription. Arrows, stimulation; dotted line, inhibition.
Oxidative stress promotes HO-1 up-regulation through a double mechanism: (i) by inducing conformational modification of Bach1 structure, which leads to its translocation from the nucleus to the cytoplasm where Bach1 is ubiquitinated and degraded thereby, releasing transcriptional repression; and (ii) by promoting the ubiquitination and consequent degradation of Keap1, which under normal conditions sequesters Nrf-2 into the cytoplasm, avoiding its transcriptional activity (He et al., 2007; Zenke-Kawasaki et al., 2007) (Figure 1). Substances and drugs that differently affect HO-1 expression and activity have been extensively reviewed (Mancuso and Barone, 2009). As regard to HO-2 gene regulation, only limited evidence is available to implicate the glucocorticoid responsive elements (GRE) in the gene encoding for HO-2 as the main site involved in the modulation of HO-2 protein levels (see above) (Liu et al., 2000).
In the central nervous system (CNS), HO-2 is expressed in neuronal populations in almost all brain areas (Maines, 1997), whereas HO-1 is present at low levels in sparse groups of neurons, including the ventromedial and paraventricular nuclei of the hypothalamus (Maines, 1997). Heme oxygenase-1 is also found in cells of glial lineage, where its expression can be induced by oxidative stress (Dwyer et al., 1995).
HO-1 and HO-2 catalyze the same reaction, namely the transformation of iron-protoporphyrin-IX-alpha (heme) into equimolar amount of ferrous iron [Fe(II)], carbon monoxide (CO), and biliverdin-IX-alpha (BV-alpha) (Maines, 1997; Maines, 2000) (Figure 1). The activity of both enzymes can be regulated by post-translational modifications such as phosphorylation of specific serine or tyrosine residues (Figure 2). In particular, HO-1 activity might be regulated through Akt-mediated phosphorylation of Ser188 (Salinas et al., 2004). This kind of phosphorylation may change the strength of binding/interaction between HO-1 and BVR. However, considering the large number of residues involved in the interaction, a large change in binding affinity is not expected for a single phosphorylation event (Salinas et al., 2004); this explains why, after the phosphorylation of Ser188, a 1.6-fold increase in HO activity was measured in HEK293T cells with respect to controls (Salinas et al., 2004). HO-2 is activated during neuronal and odorant stimulation by phosphorylation of serine 79 by casein kinase 2 (CK-2) via participation of protein kinase C (PKC) and calmodulin (Boehning et al., 2003; Boehning et al., 2004; Dore et al., 1999) (Figure 2). In cerebral endothelial cells, stimulation of HO-2 activity by glutamate, via ionotropic glutamate receptors, involves tyrosine kinase-mediated but no protein kinase C- or CK-2-mediated phosphorylation (Leffler et al., 2003a; Leffler et al., 2003b).
Figure 2. Phosphorylation changes modulate HO and BVR activities.
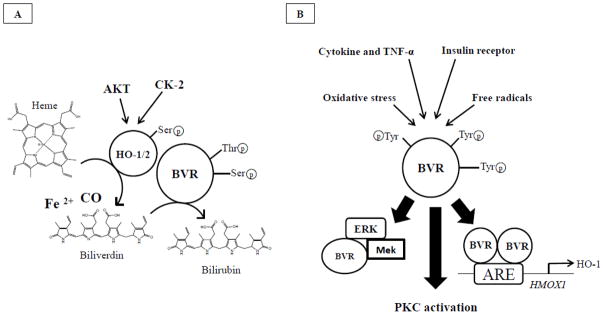
Panel A, HO-1 activity might be regulated through Akt-mediated phosphorylation of Ser188 (Salinas et al., 2004). This kind of phosphorylation may change the strength of binding/interaction between HO-1 and BVR. However, considering the large number of residues involved in the interaction, a large change in binding affinity is not expected for a single phosphorylation event (Salinas et al., 2004). HO-2 is activated during neuronal and odorant stimulation by phosphorylation of serine 79 by casein kinase 2 (CK-2) via participation of protein kinase C (PKC) and calmodulin (Boehning et al., 2003; Boehning et al., 2004; Dore et al., 1999). Similarly, BVR is able to phosphorylate itself on specific serine and threonine residues, and this step is essential for the activation of its reductase activity, named the ability to reduce BV to BR (Kapitulnik and Maines, 2009). Panel B, in order to function as Ser/Thr/Tyr kinase, BVR must be phosphorylated on specific Tyr residues by other kinases such as insulin receptor or other kinases induced under conditions of elevated oxidative stress levels. Following activation, BVR (i) modulates the activity of members of conventional and atypical groups of PKC isozymes (PKC-βII and PKC-ζ, respectively) (Kapitulnik and Maines, 2009; Lerner-Marmarosh et al., 2007; Maines et al., 2007); (ii) functions as a scaffold protein for the formation a ternary complex with MEK1 and ERK1/2, placing ERK in a position that enables its activation by MEK (Kapitulnik and Maines, 2009; Lerner-Marmarosh et al., 2008); and (iii) by forming a dimeric complex translocates into the nucleus where BVR regulates the expression of stress-responsive genes such as HO-1 (Tudor et al., 2008). Arrows, stimulation.
Although HO-1 and HO-2 share the same activity, they play different roles in protecting tissues against injuries (Maines, 2005a; Maines and Panahian, 2001). The most convincing hypothesis suggests that controlled HO-1 induction plays a pivotal role in the earliest stages of cellular responses to tissue damage, whereas HO-2 is constitutively expressed and is primarily involved in maintaining cell heme homeostasis and in sensing the intracellular levels of gaseous compounds including oxygen, nitric oxide (NO), and CO (Maines, 2005a).
HO by-products
The members of the HO family are pleiotropic enzymes playing an important role in the regulation of cell proliferation, differentiation, oxidative status and apoptosis, thereby influencing immune response, inflammatory reaction or angiogenesis. Thus, their significance is much wider than only heme elimination (Gozzelino et al., 2010; Maines, 2000; Mancuso and Barone, 2009). Indeed, the above-cited effects are mainly due to the products of HO activity.
Carbon monoxide
Carbon monoxide regulates long-term hippocampal potentiation, neuropeptide release, non-adrenergic non-cholinergic gastrointestinal relaxation, vessel tone, renal function (Kaide et al., 2001; Kooli et al., 2008; Piantadosi, 2008; Rodriguez et al., 2003; Wu and Wang, 2005; Zhang et al., 2001; Zhang et al., 2004), and inflammation processes (Piantadosi, 2008; Wu and Wang, 2005). Furthermore, low concentration of CO exposure by itself is antiapoptotic and cytoprotective against oxidative stress (Fujita et al., 2001; Liu et al., 2003; Song et al., 2003). Although CO has been shown to be cytoprotective in some experimental models, it also produces noxious effects in certain organs, such as the brain (Mancuso and Barone, 2009). The dual nature of CO’s effects depend on several factors, including cell type, the amount of CO formed or delivered to cell, and the tissue-specific signaling transduction pathway(s) involved in its biological activity. Carbon monoxide produced in rat hypothalamus by HO activity has displayed anti-inflammatory activity consisting in the attenuation of KCl-induced interleukin-1β release from interleukinergic neurons (Mancuso et al., 1998). However, hypothalamic CO also reduces stimulated increases in the in vitro and in vivo release of corticotropin releasing hormone (CRH) and arginine vasopressin (AVP) (Mancuso et al., 1997; Mancuso et al., 2010; Mancuso et al., 1999; Pozzoli et al., 1994), effects that are clearly pro-inflammatory. Indeed, their end result is decreased pituitary release of adrenocorticotropin hormone (ACTH), which, in turn, stimulates glucocorticoid production and release by the adrenal cortex. Additional evidence of CO’s dual role in the CNS has been provided by Luiz Branco’s group. In their studies on CO’s involvement in the pyrogenic response to stress, intracerebroventricular administration of HO inhibitors decreased LPS-induced fever in rats, while heme overload caused a rise in body temperature (Steiner and Branco, 2000; Steiner and Branco, 2001; Steiner et al., 1999; Steiner et al., 2003). In contrast, if the increased CO formation was confined to the locus coeruleus, the febrile response to LPS decreased (Ravanelli et al., 2007). Although it is only indirectly related to inflammation, CO’s effect on the release of gonadotropin-releasing-hormone (GnRH) is worth mentioning. Carbon monoxide was shown to up-regulate GnRH release in the hypothalamic GT1–7 cell line, and this effect seems to be dependent on the CO-mediated production of prostaglandin E2 (Errico et al., 2010).
Iron
Iron is a cofactor for several enzymes and is able to modulate specific brain functions by increasing the release and turnover of dopamine and other neurotransmitters (Chiueh, 2001). On the other hand, Fe(II) produced by HO can catalyze the production of free radicals through the Fenton chemistry and thus act as a cytotoxic pro-oxidant (Braughler et al., 1986; Bucher et al., 1983; Minotti and Aust, 1989). At the same time, Fe(II) released from heme is able to induce different metabolic pathways including the up-regulation of Fe-efflux pump and those of Ferritin-H with the final effect to neutralize the pro-oxidant activity of free Fe(II) (Gozzelino et al., 2010).
Biliverdin
Biliverdin (BV) is probably the least studied product of HO activity, mainly due to its high rate of catabolism once formed into the cell (Fakhrai and Maines, 1992). In fact, through the activity of BVR, BV is immediately reduced to bilirubin; therefore, its physiologic relevance remains to be established. That stated, several pathways involving BV were previously described. In particular, BV, by being a potent inhibitor of NF-kB (Gibbs and Maines, 2007), would offer a means for limiting the activity of this nuclear factor. Treatment with biliverdin enhances tolerance of cardiac allografts, and this tolerance is mediated by inhibition of the transcription factors NFAT and NF-kB (Yamashita et al., 2004). More recently it was reported that administration of BV markedly reduced mortality in experimental pancreatitis (Nuhn et al., 2013) Furthermore, BV administration protected against hemorrhagic shock and resuscitation induced lung injury through anti-inflammatory and anti-oxidant mechanisms involving reduction of TNF-α, iNOS and oxidative stress markers (Kosaka et al., 2013). Finally, BV exhibited a greater antioxidant activity than alpha-tocopherol in preventing oxidative stress damage in rat brain microsomes (Mancuso et al., 2012) (Figure 1).
BVR and bilirubin
Similar to HO, two isoforms of BVR were described and named BVR-A and BVR-B (Kapitulnik and Maines, 2009; Maines, 2005b; Pereira et al., 2001). Both these enzymes generate BR, but only BVR-A reduces BV-alpha into the powerful antioxidant and antinitrosative molecule BR-IX-alpha (thereafter BR) (Barone et al., 2009; Stocker, 2004), whereas BVR-B prefers the other BV isoforms, such as BV-β, BV-γ and BV-δ (Kapitulnik and Maines, 2009; Maines, 2005b; Pereira et al., 2001). Both BVR-A and BVR-B were identified in humans, with age-dependendent characteristic. Biliverdin reductase-A is the main form detected in the adult (95–97% BR is found in the bile), whereas BVR-B is predominant (~ 87%) in the fetus (Cunningham et al., 2000). A possible explanation for this ontogenesis-linked difference is that BVR-B-produced bilirubin-IX-beta does not undergo internal hydrogen bonding, unlike BR, and, therefore, has a much higher solubility (Cunningham et al., 2000). Indeed, it has been shown that bilirubin-IX-beta can be excreted directly into the bile without being conjugated with glucuronic acid (Cunningham et al., 2000).
Biliverdin reductase-A, is the product of a single transcript (McCoubrey et al., 1995) that encodes a soluble polypeptide that, in mammals, is in the range of about 300 amino acids. The human enzyme consists of 296 residues, whereas the rat enzyme is made of 295 amino acids (Fakhrai and Maines, 1992; Maines et al., 1996). Moreover, in the mammalian species, BVR shows a high degree of conservation of gene structure, which consists of seven coding exons and one noncoding exon at the 5′ end (McCoubrey et al., 1995). Because of extensive posttranscriptional modification, the mature protein displays a substantially larger apparent molecular weight than predicted, based on amino acid composition. The reported apparent molecular weight of BVR-A, as estimated by its electrophoretic mobility in SDS gels, ranges from 36 to 42 kDa. As described for the human and the rat, in the mature protein, the first methionine and the second residue, an asparagine, in the human, are deleted (Kapitulnik and Maines, 2009).
The promoter region of the human and rat genes contain consensus sequence elements associated with regulation of transcriptional activity and embryonic gene expression (McCoubrey et al., 1995). Furthermore, the gene encoding for BVR-A possesses a sequence upstream from the transcription start point, which matches that of the heat shock element (HSE) sequence, thus making BVR-A a heat shock-inducible protein similar to HO-1, despite the latter being induced more rapidly than the former (Maines, 2005b; McCoubrey et al., 1995). In addition, cytokines, LPS and atorvastatin induce BVR transcription (Barone et al., 2012b; Maines et al., 2001). Human BVR expression is downregulated by the zinc-finger hematopoietic transcription factor GATA1 and up-regulated by heme (Maines, 2005b).
Biliverdin reductase is co-expressed with HO-1 and/or HO-2 in cells of the rat brain that express these enzymes under normal conditions. BVR is also found in regions and cell types that can express heat shock-inducible HO-1 (Ewing et al., 1993).
BVR activity demonstrates a unique dual pH/cofactor-dependence nature: NADH at a pH of 6.8, and NADPH at pH 8.7 (Kapitulnik and Maines, 2009; Maines and Trakshel, 1993). Biliverdin reductase also requires free SH groups (Maines and Trakshel, 1993). BVR-A not only transforms BV into BR (by reducing the former’s C10 [γ bridge]), but it is also a serine/threonine/tyrosine kinase involved in various cellular functions (Kapitulnik and Maines, 2009; Maines, 2005b). In both cases, BVR activation through the phosphorylation of specific Ser/Thr/Tyr residues is required (Kapitulnik and Maines, 2009; Lerner-Marmarosh et al., 2005) (Figure 2B). Interestingly, two different ways through which BVR-A can be phosphorylated are known. Indeed, it was demonstrated that BVR-A is able to autophosphorylate, and this step is essential for the activation of its reductase activity, namely the ability to reduce BV to BR (Lerner-Marmarosh et al., 2005) (Figure 2A). Similarly, the phosphorylation by other kinases, including the insulin receptor kinase, activates BVR-A kinase activity (Kapitulnik and Maines, 2009; Lerner-Marmarosh et al., 2008; Maines, 2007; Tudor et al., 2008) Figure 2B). Once activated, BVR-A can: (i) catalyze the last step in the heme-degradation pathway by reducing the γ-meso (methylene) bridge of BV to BR (Kapitulnik and Maines, 2009); (ii) modulate the activity of members of conventional and atypical groups of PKC isozymes (PKC-βII and PKC-ζ, respectively) (Kapitulnik and Maines, 2009; Lerner-Marmarosh et al., 2007; Maines et al., 2007) (Gibbs et al., 2012b); (iii) function as a scaffold protein for the formation a ternary complex with MEK1 and ERK1/2, placing ERK in a position that enables its activation by MEK (Kapitulnik and Maines, 2009; Lerner-Marmarosh et al., 2008); and (iv) regulate the expression of stress-responsive genes such as HO-1 (Tudor et al., 2008) and iNOS (Di Domenico et al., 2013b; Gibbs et al., 2012a) (Figures 2A and 2B).
Once activated by the insulin receptor, BVR is able to modulate two of the most important arms of the insulin signaling pathway: MAPK and phosphatidylinositol-3-kinase (PI3K) (Kapitulnik and Maines, 2009). MAPK and PI3K pathways have essential roles in neuronal activity and development: (i) the PI3K pathway is involved in the maintenance of synaptic plasticity and memory consolidation (Horwood et al., 2006), Amyloid-β-peptide (Aβ)-induced memory loss (Chiang et al., 2010), synthesis of nitric oxide (NO), which in turn plays a role in learning and memory processes (Calabrese et al., 2007a); and (ii) the MAPK cascade is responsible both for the induction of several genes required for neuronal and synapse growth, maintenance and repair processes, as well as serving as a modulator of hippocampal synaptic plasticity that underlies learning and memory (Akter et al., 2011). Consequently, it is clear that the broad spectrum of pleiotropic actions mediated by BVR-A makes this enzyme an important interventional target for the development of new therapeutic strategies.
As with HO-1, also BVR at the beginning was considered relevant only for its ability to produce BR. However, in light of this pleiotropic nature of BVR, the discrimination between the effects directly mediated by BVR and those mediated by BR become essential in order to better understand and clarify the broad spectrum of actions to which we refer when we mention the HO/BVR system.
Bilirubin is a linear tetrapyrrole, characterized by high lipophylicity, and was extensively studied for its antioxidant and antinitrosative properties (Barone et al., 2009; Dore et al., 1999; Mancuso et al., 2003; Stocker et al., 1987a; Stocker et al., 1987b; Takahashi et al., 2000). Despite this important antioxidant behavior, if produced in excess, as during hemolytic anemia or sepsis, unconjugated BR becomes neurotoxic through multiple mechanisms involving the disruption of cell membrane structure, the reduction of mitochondrial transmembrane potential and the activation of the apoptotic cascade (Brito et al., 2004; Kapitulnik, 2004). Other than its antioxidant activity, BR increased neuronal NOS expression and nitric oxide formation in both primary cultures of cerebellar granule neurons and neurotrophin-sensitive PC12 cells (Mancuso et al., 2008), and it was shown that this gaseous neurotransmitter plays a key role in the long-term potentiation and synaptic plasticity (Calabrese et al., 2007a). In addition, in PC12 cells BR upregulated CREB (Mancuso et al., 2008), which is considered an important transcription factor regulating both short- and long- term memory (Suzuki et al., 2011).
Alzheimer disease pathology: the involvement of oxidative stress
AD is one of the most disabling neurodegenerative disorders that cause dementia and affect middle- to old-aged individuals, with a prevalence that increases markedly after age 65. AD is characterized pathologically by the presence of senile plaques (SPs), neurofibrillary tangles (NFTs), decreased synaptic density and brain atrophy particularly in the hippocampus, amygdala and frontal cortex, consistent with cognitive and memory deficits observed. The main component of SPs is amyloid β-peptide (Aβ), comprising 40–42 amino acids and generated by proteolytic cleavage of amyloid precursor protein (APP), a type I transmembrane protein, by β-secretase and γ-secretase. Aβ exists in various soluble and insoluble forms including aggregates, soluble monomers, oligomers, protofibrils, and fibrils (Haass and Selkoe, 2007; Walsh et al., 2002). Recent studies have suggested that soluble oligomers are the most toxic form of Aβ. NFT are formed by hyperphosphorylation of tau, a microtubule-associated protein, causing it to aggregate to an insoluble form and lose the affinity for microtubules (Querfurth and LaFerla, 2010). Aβ oligomeric and fibrillary forms and hyperphosphotylated tau are normally degraded by the unfolded protein response, however, when this system is dysfunctional, as in AD progression, contribution toward an aberrant deposition of Aβ occurs.
Sporadic AD that accounts for approximately 95% of AD cases results from a complex array of etiological factors in addition to age such as family history of dementia, head trauma, gender, education level, vascular disease, general lifestyles and other environmental factors. AD is often preceded by three stages of progression characterized by gradual increase of AD hallmarks starting from preclinical AD (PCAD), to mild cognitive impairment (MCI) and early AD (EAD) (Morris and Cummings, 2005; Price and Morris, 1999). Despite continued efforts, the development of an effective treatment for AD remains elusive. Current therapeutic strategies are limited to those that attenuate AD symptoms without modifying the progress of the disease itself, and thus only postpone the inevitable deterioration of the affected individual (Bonda et al., 2010).
The amended amyloid cascade hypothesis is one of the leading notions of underlying mechanisms of AD, stating that Aβ oligomer formation and deposition are the cause of this disorder. The importance of APP, and consequently Aβ, in AD pathogenesis has arisen from genetic evidence of patients with familial AD (FAD) and Down syndrome (DS) (Butterfield et al., 2013). Indeed, it has been reported that rare FAD forms are linked directly to highly penetrant autosomal dominant genetic mutations in the APP and presenilin 1 and 2 (PS1, PS2) genes. In addition, AD pathology is found prematurely in Down syndrome, given that the APP can be found on chromosome 21 and that Down syndrome subjects have an extra copy of chromosome 21 (Di Domenico et al., 2013a; Perluigi and Butterfield, 2012). Many studies reported that Aβ toxicity leads to AD development through the alteration of several neuronal mechanisms, which include formation of free radicals, oxidative stress, mitochondrial dysfunction, inflammatory processes, and apoptosis. These factors may interact and amplify each other in a vicious cycle of toxicity, leading to neuronal impairment, cell dysfunction, and finally cell death (Butterfield et al., 2001; Butterfield et al., 2013).
Oxidative stress undoubtedly plays a critical role, as evidence for its molecular impact exists very early in disease progression (Behl, 2012; Hensley et al., 1995; Markesbery, 1999; Zafrilla et al., 2006). Due to its elevated levels of peroxidizable fatty acids, high requirement for oxygen, relative paucity of antioxidant systems, and richness in iron content, the brain is extremely sensitive to oxidative stress (Butterfield, 2006; Markesbery, 1997). Normal metabolism generates oxygen free radicals and other reactive oxygen species (ROS) that are part of several physiologic processes including signal transduction pathways (e.g., related to some growth factors, cytokines and calcium signaling). When oxidative stress exceeds the capacity to terminate ROS, then oxidative damage ensues (Butterfield and Stadtman, 1997). ROS can damage cell or organelle membranes directly (e.g., through lipid peroxidation), and can react with metals, nitrogen or carbon to form intermediates that react with proteins (e.g., through nitration, carbonylation and nitrosylation). ROS (including superoxide anion radical (O2•−), hydrogen peroxide (H2O2), hydroxyl radical (•OH), singlet oxygen (O2), alkoxyl radicals (RO•), peroxyl radicals (ROO•) and peroxynitrite (ONOO−), contribute to pathogenesis of numerous human neurodegenerative diseases. Specific antioxidants, both endogenous and exogenous, such as glutathione, α-tocopherol (vitamin E), carotenoids, and ascorbic acid, and antioxidant enzymes, such as catalase, peroxiredoxins, and glutathione peroxidases, are able to detoxify H2O2 by converting it to O2 and H2O under physiological conditions (Feng and Wang, 2012). The brain in AD appears to sustain more oxidative damage than normal brain, exhibits an increased susceptibility to oxidative stress and has relatively low levels of naturally occurring antioxidants such as α-tocopherol. Aβ peptides, together with altered mitochondrial function, and the presence of trace metal ions such as iron and copper, have been identified as potential sources of oxidative stress (Butterfield et al., 2001; Cai et al., 2011; Clark et al., 2010). In accordance with the Aβ-induced oxidative stress hypothesis, oxidative stress is the result of Aβ insertion as oligomers into the bilayer causing ROS production and initiating lipid peroxidation and protein oxidation in AD pathology (Axelsen et al., 2011; Butterfield et al., 2001; Butterfield and Lauderback, 2002; Butterfield et al., 2007). Studies on AD transgenic animal models expressing Aβ peptide confirmed the association between Aβ and oxidative stress suggesting the involvement of methionine 35 of Aβ peptide in the mechanism of oxidative damage (Butterfield et al., 2010; Butterfield et al., 2013; Sultana et al., 2012).
However, despite an extensive understanding of each of the phenomena occurring within the cell during AD, an adequate explanation for AD development and progression is still lacking. Aβ has been shown to induce oxidative stress in vitro and in AD model systems in vivo, as evidenced by leading to protein oxidation, lipid oxidation, DNA oxidation, and glycoxidation (Butterfield et al., 2001; Nunomura et al., 2012). Oxidative stress and its effects have been found as early as MCI and EAD in the progression toward AD. Many studies conducted in our and other laboratories have found that oxidative stress markers for protein oxidation/nitration, such as protein carbonyls and 3-nitrotyrosine, are elevated in brains from subjects with MCI and EAD (Butterfield et al., 2006; Keller et al., 2005; Reed et al., 2009; Sultana and Butterfield, 2010). Regions of the brain rich in Aβ proteins have increased levels of protein oxidation, while Aβ-poor cerebellum does not (Hensley et al., 1995). In addition high levels of free and protein-bound HNE were found in AD brain (Lovell et al., 2001; Lauderback et al., 2001) as well as protein carbonyls an protein nitration in regions of the brain heavily associated with AD, including the hippocampus and parietal cortex, while leaving the cerebellum relatively untouched (Castegna et al., 2002a; Castegna et al., 2002b; Markesbery and Lovell, 1998; Perluigi et al., 2009; Sultana et al., 2006).
The use of redox proteomics (Butterfield et al., 2012; Butterfield et al., 2013) to identify oxidatively modified brain proteins in AD and MCI revealed a number of oxidatively modified brain proteins that are associated with the mitochondrial functionality, energy metabolism and antioxidant response suggesting that the alteration of these pathways by Aβ-induced oxidative stress is involved in AD progression or pathogenesis (Butterfield, 2002; Butterfield, 2006; Butterfield et al., 2012b; Butterfield and Stadman, 1997; Butterfield et al., 2013). Significant DNA and RNA oxidation has been shown to exist in AD, as have been found since the early stages of the disease. In AD brain, 8-hydroxy-2-deoxyguanosine (8OHdG) and 8-hydroxyguanosine (8OHG) were found to be elevated in AD hippocampus, frontal, and occipital neocortex, which correlated with the β-amyloid load (Mecocci et al., 1994; Nunomura et al., 2012). Elevated levels of protein-bound HNE, protein carbonyls, 3-NT, free HNE and MDA have been described not only in brain but also in cerebrospinal fluid (CSF), blood, and urine of AD patients when compared with healthy controls (Dildar et al., 2010; Irizarry et al., 2007; Pratico et al., 2000; Pratico et al., 1998).
On the other hand, increased oxidative stress has been proposed to contribute to Aβ generation and the formation of NFT (Butterfield and Boyd-Kimball, 2004). Indeed several reports stated that interaction between oxidative stress and neuroinflammation leads to Aβ production (Akama et al., 1998; Cai et al., 2011). AD is associated with an increase in blood–brain barrier (BBB) permeability due to disruption of tight junction. One of the closest links pertain to the BBB, where oxidative stress decreases the expression and oxidatively modifies low-density lipoprotein receptor-related protein 1 (LRP-1) (Owen et al., 2010), up-regulates the receptor for advanced glycation end products (RAGE) and increases BBB permeability, which could potentially lead to increased deposition of Aβ within the AD parenchyma (Deane et al., 2004; Srikanth et al., 2011). Interestingly, a recent study (Badia et al., 2013) suggested that persons at risk of AD suffer from reductive stress (indicators of oxidative stress being lower in healthy individuals at risk than in those with low risk of developing the disease) but during the persistent formation of radicals, the capacity of the cells to react and the antioxidant response collapse. Overwhelming evidence supports the notion that oxidative stress occurs in AD and its earlier forms, and oxidative stress and compensatory mechanisms to oxidative stress may contribute to development of AD pathological and clinical hallmarks.
The role of HO/BVR-A system in Alzheimer disease: a new perspective
The first evidence about the association between HO and AD hails from a paper by Smith and colleagues who demonstrated that in AD brain pronounced HO-1 immunoreactivity is seen localized with neurofibrillary tangles, senile plaque neurites, neuropil threads (i.e., the neurofibrillary pathology), and granulovacuolar degeneration (Smith et al., 1994a). For about twenty years since, during which the main emphases were focused on the role of HO-1/HO-2 (Calabrese et al., 2006; Dore et al., 1999; Mueller et al., 2010; Poon et al., 2004; Takahashi et al., 2000) or bilirubin (Dore et al., 1999; Kimpara et al., 2000), the story on the involvement of the HO/BVR system in AD is still open, in particular because of the novel findings of our group highlighting an oxidative/nitrosative-induced impairment of both HO-1 and BVR-A in AD and MCI brain (Barone et al., 2011a; Barone et al., 2011b; Barone et al., 2012a; Barone et al., 2012b; Butterfield et al., 2012a; Di Domenico et al., 2012). These findings, discussed in detail below, led to our proposed new paradigm for the HO/BVR system uniting the Janus nature of the literature with respect to this system in AD (Barone et al., 2011a; Barone et al., 2011b; Barone et al., 2012a; Barone et al., 2012b; Butterfield et al., 2012a; Di Domenico et al., 2012).
In order to untangle the complex role of the HO/BVR system in AD and to provide readers with a clear and easily understandable explanation about the numerous effects mediated by the system, it is helpful to review the main achievements obtained over the years.
Due to the inducible nature of HO-1 (HO-2 is constitutive), and due to its ability to be up-regulated following stress stimuli as outlined above, most of the prior observations about AD regard the role of HO-1. Binding of APP inhibits both HO-1 and HO-2 activity, and APP with mutations linked to the familial Alzheimer’s disease (FAD) provided substantially greater inhibition of HO activity than wild-type APP. These findings indicated that augmented neurotoxicity caused by APP–HO interactions may contribute to neuronal cell death in AD (Takahashi et al., 2000).
Given that up-regulation of HO-1 is widely accepted as a sensitive and fairly ubiquitous marker of oxidative stress, two main schools of thought exist with regard to the role of HO-1/BVR-A system in AD. One of these posits a detrimental activity of HO-1 suggesting that iron deposition and attendant neuronal dysfunction in AD may represent downstream effects of sustained HO-1 over-activity within the astrocyte compartment (Hascalovici et al., 2009; Schipper, 2011; Smith et al., 1997; Takeda et al., 2004); whereas the other, coming from our group, proposed the up-regulation of HO-1/BVR-A system as a neuroprotective mechanism aimed to counteract the rise of oxidative stress observed during the onset and the progression of AD (Butterfield et al., 2001; Butterfield and Lauderback, 2002; Di Domenico et al., 2010; Markesbery, 1997; Poon et al., 2004).
However, despite while at first glance it may appear that these two hypotheses appear completely different since they propose two opposite effects, an in-depth analysis based on our novel findings (Barone et al., 2011a; Barone et al., 2011b; Barone et al., 2012a; Di Domenico et al., 2012) suggest that they complement each other based on the pathophysiological conditions relevant to AD.
The role of HO-1 in AD and MCI: a brief history in time
A brief excursus on the progress obtained in the study of the HO/BVR-A system in AD will help to clarify the common points of these two different views noted above. In the first part of this section the main discoveries by others will be outlined. Then, we will discuss our novel findings, with an aim to reconcile the two main hypotheses.
After the discovery in 1994 of the co-localization of HO-1 and the pathological hallmarks of AD (Smith et al., 1994a), in 1995 the increased expression of HO-1 but not HO-2 mRNA transcripts in cerebral cortex and cerebral vessels from subjects with AD compared with age-matched non-AD controls was demonstrated (Table 1). These lines of evidence suggested the specific induction of HO-1 mRNA and protein in the cerebral cortex and cerebral vessels in association with pathological lesions of AD (Premkumar et al., 1995). Later, in 1997, Smith et al. proposed that redox-active iron is associated with the senile plaques and neurofibrillary tangles, indicating that iron accumulation could be an important contributor toward the oxidative damage of Alzheimer disease (Smith et al., 1997), thus providing a basis for the future involvement of HO-1 as one of the main source of iron deposition and accumulation.
Table 1.
Changes observed by other investigators with regard to HO-1 (Ishizuka et al., 2002; Premkumar et al., 1995; Schipper et al., 2006; Schipper et al., 2000).
| Plasma | Lymphocyte | Cerebral cortex | Cerebral vessels | CSF | Mononuclear cells | Temporal Lobe astrocytes | Hippocampal astrocytes | ||
|---|---|---|---|---|---|---|---|---|---|
| HO-1 protein levels | MCI | - | - | - | - | - | - |

|
|
| AD |
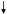
|
mRNA |
mRNA |
mRNA |
|
mRNA |
|
|
|
| BVR protein levels | MCI | - | - | - | - | - | - | - | - |
| AD |
BVR-B |
- | - | - | - | - | - | - | |
|
HO-1/BVR-A ratio |
In 2000, Schipper et al. found decreased plasma and CSF HO-1 protein and lymphocyte HO-1 mRNA levels in subjects with sporadic AD proposing for the first time, the quantitative assay for lymphocyte HO-1 mRNA expression as a useful biologic marker in early sporadic AD (Schipper et al., 2000) (Table 1). Similarly in 2002, Ishizuka et al. showed plasma HO-1 protein and mononuclear cell HO-1 mRNA levels significantly suppressed in AD subjects compared to controls (Ishizuka et al., 2002) (Table 1). However, the reports of HO-1 plasma levels in AD subjects are controversial as highlighted below in this review (Tables 1 and 2).
Table 2.
Changes from control observed by the Butterfield group with regard to the HO/BVR-A system in AD and MCI subjects (Barone et al., 2011a; Barone et al., 2011b; Barone et al., 2012a; Calabrese et al., 2006; Di Domenico et al., 2012; Di Domenico et al., 2010)
| Hippocampus | Cerebellum | IPL | Plasma | Lymphocyte | ||
|---|---|---|---|---|---|---|
| HO-1 protein levels | MCI | ≠ | ≠ | N/A |
70%, *
|
- |
| AD |
119%, **
|
38%, ns |
400%,**
|
130%, *
|
150%, **,%
|
|
| HO-1 oxidation | MCI |
HNE 85%, *
|
HNE 36%, *
|
- | - | - |
| AD |
PC 30%, **
|
PC 32%, *
|
- | - | - | |
|
HNE 52%, *
|
||||||
| HO-1 phosphorylation | MCI | ≠ |
pSer 24%, *
|
- | - | - |
| AD |
pSer 26%, *
|
pSer 21%, *
|
- | - | - | |
| HO-2 protein levels | MCI |
32%, *
|
≠ |
270%, **
|
- | - |
| AD |
32%, *
|
≠ | - | - | - | |
| BVR-A protein levels | MCI |
44%, *
|
≠ | - |
10%, ns |
- |
| AD |
45%, **
|
≠ | - |
36%, *
|
- | |
| BVR-A oxidation | MCI |
3-NT 30%, **
|
≠ | - |
3-NT 24%, *
|
- |
| AD |
3-NT 95%, **
|
≠ | - |
3-NT 44%, *
|
- | |
| BVR-A phosphorylation | MCI |
pTyr 20%, *
|
≠ |
pTyr 22%, *
|
||
|
pSer/Thr 40%, **
|
- | - | ||||
| AD |
pTyr 40%, **
|
≠ |
pTyr 57%, *
|
|||
|
pSer/Thr 28%, **
|
- | - | ||||
| BVR activity | MCI |
40%, **
|
≠ | - | ≠ | - |
| AD |
27%, **
|
≠ | - |
30%, *
|
- |
Increase,
decrease, ≠ no changes,
p<0.05,
p<0.01,
ns non significant, N/A not applicable, IPL Inferior Parietal Lobule,
Corresponding author: V. Calabrese.
The year 2000 was a rich one for of findings related to HO-1. First, the ability of APP to bind HO-1 and HO-2 and inhibit their activity was demonstrated (Takahashi et al., 2000). An increase of BR levels was demonstrated in the CSF of AD subjects. These findings imply that BR synthesis might be activated in the AD brain. Therefore, it is plausible that BR is locally produced in lesions of the AD brain to work as an efficient scavenger of ROS. (Kimpara et al., 2000). In the same 2000 year, up-regulation of HO-1 was demonstrated to be necessary and sufficient for subsequent induction of the MnSOD gene, consistent with the notion of a compensatory upregulation of MnSOD to protect against oxidative damage accruing from heme-derived free iron and CO liberated by the activity of HO-1 (Frankel et al., 2000).
In 2006, Calabrese et al. showed that HO-1 is overexpressed in the inferior parietal lobule of AD subjects (Calabrese et al., 2006). In the same paper, a significant down-regulation of HO-2 was found in this brain area, probably due to the massive neuronal death secondary to the disease (Calabrese et al., 2006). In addition, a marked increase in HO-1 expression and HO activity were found in the plasma and lymphocytes of these AD subjects (Calabrese et al., 2006). In the same year, the neurotoxic role of HO-1 was highlighted by a study showing that up-regulation of HO-1 engenders oxidative mitochondrial injury in cultured rat astroglia (Song et al., 2006). Heme-derived ferrous iron and CO may mediate the oxidative modification of mitochondrial lipids, proteins and nucleic acids in these cells. Glial HO-1 hyperactivity may contribute to cellular oxidative stress, pathological iron deposition, and bioenergetic failure characteristic of degenerating and inflamed neural tissues as observed in AD (Song et al., 2006). The same group demonstrated that immunoreactive HO-1 protein was significantly increased in temporal lobe and hippocampal astrocytes in subjects with MCI and AD (Table 1), and was associated with global measures of cognitive impairment and specific memory deficits in these individuals. The authors suggested a mechanism favoring early mobilization of free iron, mitochondrial insufficiency and corpora amylacea formation in this neurodegenerative disorder (Schipper et al., 2006). Later in the same year, decreased levels of antioxidants, including bilirubin, were found in the plasma of subjects with AD, strengthening the idea that antioxidant dysregulation might be associated with cognitive dysfunctions observed in AD (Kim et al., 2006).
In 2007, it was demonstrated that transient transfection of rat astroglia with human (h)ho-1 cDNA for 3 days significantly decreased intracellular cholesterol concentrations and increased levels of four well-known neurotoxic oxysterol species (Vaya et al., 2007). This study opened a new avenue with regard to the possible involvement of HO-1 in cholesterol homeostasis and provided new basis to understand cholesterol homeostasis dysregulation observed in AD (Dufouil et al., 2005; Hajjar et al., 2002; Jick et al., 2000; Rockwood et al., 2002; Rodriguez et al., 2002; Wolozin et al., 2000; Wolozin et al., 2007; Zamrini et al., 2004). An advance was made in 2009 when decreased cholesterol, increased oxysterol and increased cholesterol precursor concentrations were found significantly correlated with HO-1 levels in the cortex of MCI and AD subjects (Hascalovici et al., 2009).
The last discovery, which we want to mention in this brief discussion, was reported in 2009, when Kanninen et al. indicated that significant reductions in spatial learning deficits of aged APP/PS1 mice can be achieved by modulating levels of Nrf2 in the brain. This may represent a potential therapeutic strategy to pursue in AD, particularly in view of the multiple mechanisms by which Nrf2 can exert its protective effects including the increase of HO-1 protein levels (Kanninen et al., 2009).
In the same years, in parallel to these studies, our group started to look at HO-1 from another point of view.
Indeed, it is conceivable that the dramatic increase in HO-1 in AD may be a direct response to increased free heme associated with neurodegeneration and an attempt to convert highly damaging heme into BV, and then BR by BVR, with the latter a cytoprotective molecule. Heme oxygenase-1 is rapidly upregulated by oxidative and nitrosative stresses, as well as by glutathione depletion. All these findings have introduced new perspectives in medicine and pharmacology, as molecules activating this defense mechanism appear to be possible candidates for novel cytoprotective strategies (Butterfield et al., 2002a; Butterfield et al., 2002b; Butterfield and Lauderback, 2002; Calabrese et al., 2003). Furthermore, considerable attention has been focused on identifying dietary and medicinal phytochemicals that can inhibit, retard or reverse the multi-stage pathophysiological events underlying AD pathology (Butterfield et al., 2002b; Butterfield et al., 2001).
In 2004, it was reported that treating neurons with ferulic acid ethyl-ester (FAEE) resulted in an enhanced cellular resistance to glucose oxidase-mediated oxidative damage; this cytoprotective effect was considerably attenuated by zinc protoporphyrin IX, an inhibitor of HO activity. This study identified a novel modified natural compound that potentially could be used for therapeutic purposes as a potent inducer of HO-1 for the protection of brain cells against oxidative and neurodegenerative conditions (Scapagnini et al., 2004). One year later, in 2005, we demonstrated that cortical neurons treated with FAEE showed a marked increase of HO-1, which may strengthen the cellular defense mechanisms against Aβ-induced neurotoxicity (Sultana et al., 2005). In 2006, we extended our knowledge about the beneficial effects of FAEE by showing that FAEE can act as a potent antioxidant in vivo, thus providing neuroprotection against Aβ-induced oxidative stress, and that these effects can be mediated at least in part by the up-regulation of HO-1 (Perluigi et al., 2006).
Based on others and ours evidence, increased interest has been focused on identifying dietary compounds that can inhibit, retard, or reverse the multistage pathophysiologic events underlying AD pathology. Indeed, the stimulation of various repair pathways, such as HO-1, by mild stress has significant effects on delaying the onset of various age-associated alterations in cells, tissues, and organisms (Mancuso et al.,2012).
Due to the main role played by oxidative and nitrosative stress in the pathogenesis of AD, and the importance of heat shock proteins, including HO-1, as molecular chaperones involved in the protection of cells from various forms of stress, we then proposed for the first time the existence of a link between nitrosative stress and HO-1 in brain and plasma of AD subjects (Table 2). Indeed, (i) elevation of HO-1 protein levels in the inferior parietal lobule, and (ii) elevation of HO-1 protein levels and activity in plasma and lymphocytes from AD subjects (Calabrese et al., 2006) (Table 2), suggested that HO-1 is redox regulated, similar to other antioxidant enzymes (Alam and Cook, 2003; Balogun et al., 2003). This suggestion has merit because the HO-1 gene contains an ARE motif in its promoter region.
In 2008, the first evidence of a dietary antioxidant treatment-mediated beneficial effects on cognition and oxidative stress levels was provided in a well-characterized pre-clinical model of AD, the aged beagle (Cotman and Head, 2008; Johnstone et al., 1991; Opii et al., 2008; Torp, 2000a; Torp, 2000b; Torp et al., 2003). These effects were mediated at least in part through the up-regulation of the HO-1 protein (Opii et al., 2008). Very interestingly, the higher HO-1 protein levels after antioxidant treatment were associated with lower error scores on individual cognitive tasks. As a result HO-1 was one of the best predictors of error scores on black/white reversal learning, i.e., higher HO-1 protein levels were associated with improved cognitive function (Opii et al., 2008).
Oxidative/nitrosative stress-induced modifications of the HO-1/BVR-A system in AD and MCI
At this point it was quite clear that there was something missing in the comprehension of the role of the HO-1/BVR-A system in AD. Is HO-1 up-regulation neuroprotective or neurotoxic? Indeed, by looking at the previous studies, the neurotoxic effects were ascribed in part to its by-products, i.e., iron and CO, whereas the neuroprotective effects were explained by noting that the up-regulation of HO-1, together with increased HO activity, was associated with elevated levels of BR, which in turn, due to its antioxidant power, mediates beneficial effects. However, while iron and CO were generated directly by HO activity, BR was not, and as explained above, the enzyme responsible for the production of BR is BVR (Figure 1).
Thus, what about BVR? As noted above despite many studies over about 20 years to clarify the role of HO-1in AD, few addressed the role of BVR in AD.
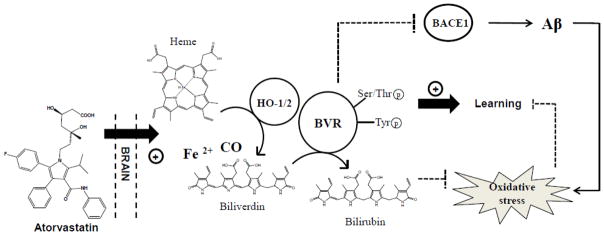
In 2011, we were the first to report alterations in BVR activity along with decreased phosphorylation and increased oxidative/nitrosative post-translational modifications in the brain of subjects with AD and amnestic MCI subjects (Barone et al., 2011a; Barone et al., 2011b) (Table 2). The first novel finding provided by these studies was the relationship between BVR-A protein levels and activity. Total BVR-A protein levels were increased in the hippocampi of both AD and MCI subjects, but its activity was reduced. No changes were observed in cerebellum (Barone et al., 2011a; Barone et al., 2011b) (Table 2). Hippocampus is broadly recognized as a main target of neurodegenerative damage during AD progression, presenting increased levels of oxidative stress, neuronal loss and marked atrophy in respect to whole brain (Aksenov et al., 1995; Keller et al., 2005; Markesbery, 2009). Conversely, cerebellum is largely devoid of pathology and oxidative stress (Hensley et al., 1995), consistent with the hypothesis of lack of AD pathology due to BVR-A levels, activity and phosphorylation state observed in this brain area (Barone et al., 2011a; Barone et al., 2011b). As mentioned above, BVR-A must be phosphorylated on specific Ser/Thr/Tyr residues in order to be activated. The significant reductions of both pSer/Thr-BVR-A and pTyr-BVR-A and BVR activity we discovered in AD brain (Table 2) agree with this paradigm. Interestingly, other than the reductase activity (the ability to produce BR) also its kinase activity was reduced as demonstrated by the decreased associations with ERK-2 in hippocampus (Barone et al., 2011a). This result lends support to the hypothesis that BVR could contribute to the ERK1/2 dysregulation detected in this brain area in AD subjects (Hyman et al., 1994), and confirmed the importance of BVR kinase activity even in the modulation of cell stress response (Barone et al., 2011a; Barone et al., 2011b).
In a following study, we demonstrated that BVR-A undergoes nitrosative stress-induced modifications. We showed increased 3-NT levels on BVR-A in the hippocampus of subjects with AD and MCI (Barone et al., 2011a; Barone et al., 2011b) (Table 2). Since it is well known that the formation of oxidative/nitrosative post-translational modifications alters protein structure (Subramaniam et al., 1997) and most often results in a marked decrease of their function (Butterfield and Lauderback, 2002; Lauderback et al., 2001; Owen et al., 2010), it is plausible to argue that the rise of 3-NT levels on BVR-A could be responsible at least in part for the observed reduced activity (Barone et al., 2011a; Barone et al., 2011b). Indeed, nitration and phosphorylation processes occur on the same residues, i.e., Tyr residues (Butterfield and Stadman, 1997). Currently it is not known if exactly the same Tyr residues are the substrate of these kinds of modifications, but it is conceivable that, due to the decreased Tyr phosphorylation and the increased Tyr nitration, a competition between nitration and phosphorylation processes occurs. Certainly, from a chemical point of view, steric hindrance of the NO2 group on the 3-position of Tyr could significantly modulate activity of Tyr kinases for the 4-OH group. This notion strengthens the hypothesis that nitrosative stress prevents/inhibits Tyr phosphorylation on BVR-A (Barone et al., 2011a; Barone et al., 2011b). The evidence that BVR-A nitration occurred also in the hippocampus of MCI subjects (Table 2), suggests that any modification in terms of cell stress response is an early event in the pathogenesis and progression of AD (Barone et al., 2011b).
These findings raised the question about the effective neuroprotective role of the HO-1/BVR-A system in AD. Why, despite the up-regulation of both HO-1 and BVR-A protein levels, are the oxidative stress markers levels in the hippocampus still higher and the pathological features of AD still present? Based on our results, the answer is quite simple: it is no longer correct to measure only total protein levels as an index to evaluate the involvement of these enzymes in cell stress response since post-translational modifications appear to play a main role in the regulation of the neuroprotective and/or metabolic activities of these proteins. In fact, the observed impairment of BVR-A activity blunts the effects that could be mediated by the up-regulation of this enzyme (Barone et al., 2011a; Barone et al., 2011b).
These observations were logically followed by the analysis of the post-translational modifications of HO-1. The question to be addressed at this stage was to know if only BVR-A was impaired or also HO-1.
We then extended the investigation on the neurobiological features of both HO-1 and HO-2 in the brain of AD and MCI subjects to include: (i) increase of HO-1 protein levels in another well-known brain area involved in AD pathology such as hippocampus; (ii) decrease of HO-2 protein levels in the same brain area; and (iii) the observation that no changes for HO-2 protein levels in cerebellum of MCI subjects were observed (Barone et al., 2012a) (Table 2). Furthermore, we showed a significant increase of Ser-residue phosphorylation along with oxidative post-translational (PC- and HNE-adducts) modifications in the hippocampus of only AD subjects (Barone et al., 2012a) (Table 2). In hippocampus of MCI subjects only a significant increase of HNE-adducts on HO-1 was observed without changes in phosphorylation (Barone et al., 2012a) (Table 2).
Since HO-1 is a stress-inducible protein, the increase of oxidative stress levels in the hippocampus of AD subjects could lead to an increase in HO-1 protein levels and phosphorylation in order to promote its activity and its interaction with BVR (Salinas et al., 2004). At the same time, the increased oxidative stress could be responsible for the observed rise of PC and HNE-adducts, already demonstrated for other proteins in AD (Sultana et al., 2009), including BVR-A (Barone et al., 2011a; Barone et al., 2011b), leading to altered protein structure and function impairment (Butterfield and Lauderback, 2002; Lauderback et al., 2001; Owen et al., 2010; Subramaniam et al., 1997). Based on our experimental model, it is difficult to state which post translational modification precedes the other between phosphorylation and oxidative modification and at least two interpretations could be conceivable: 1) oxidative stress promotes the increase of oxidative damage to HO-1 (e.g., increased PC and HNE-adducts on its structure). Consequently, the cell tries to restore the functionality of the protein by increasing Ser residue phosphorylation; 2) Oxidative stress promotes the increase of Ser-residue phosphorylation in order to activate protein functions, but HO-1 quickly becomes a target for oxidative post-translational modifications, that in turn could impair its function (Barone et al., 2012a) (Figure 3).
Figure 3. The neuroprotective vs neurotoxic hypothesis.

The apparent discrepancy between the neuroprotective and neurotoxic hypothesis, with regard to the role played by the HO/BVR system in the pathogenesis of AD, could be solved by considering different phases in the progression of the pathology. Panel A, neuroprotective. By considering oxidative stress as a central event in AD pathology, it is conceivable that during an initial phase, which could be represented by an early stage even preceding MCI, the elevation of oxidative stress levels promotes the increase of HO-1 and BVR-A protein levels which could still work properly in order to counteract, the noxious effects related to augmented oxidative and nitrosative stress levels through: (i) the production of antioxidant and antinitrosative bilirubin; and (ii) the pleiotropic functions of BVR regulating cell survival. With the progression of the pathology through the progression from MCI and AD, characterized by a continuous increase of oxidative stress levels, the neuroprotective activities mediated by the HO/BVR system would not be sufficiently efficacious anymore. Panel B, neurotoxic. Taking into account the observed impairment of BVR-A in both MCI and AD (Barone et al., 2011a; Barone et al., 2011b), the presence of both oxidative post-translational modifications and Ser phosphorylation on HO-1 in AD brain makes it difficult to state which post translational modification precedes the other and at least two interpretations could be conceivable: (1) oxidative stress promotes the increase of HO-1 oxidative damage (e.g., increased PC and HNE-adducts to key amino acids within HO-1). Consequently, the cell tries to restore the functionality of the protein by increasing Ser residue phosphorylation; (2) Oxidative stress promotes the increase of Ser-residue phosphorylation in order to activate protein functions, but HO-1 quickly becomes a target for oxidative post-translational modifications, that in turn could impair its function (Barone et al., 2012a). Arrows, stimulation; dotted lines, inhibition.
With regard to MCI, the results from hippocampus add new elements to the comprehension of the contribution of the HO-1/BVR-A system to AD pathogenesis. Unlike BVR-A, whose expression levels were found significantly increased even in the hippocampus of subjects with MCI (Barone et al., 2011a), HO-1 protein levels do not present any differences (Di Domenico et al., 2010) (Table 2). This result could mean that the induction of each member of the HO-1/BVR-A system is not correlated and probably the threshold levels of oxidative/nitrosative stress needed to induce HO-1 and BVR-A are different. Due to the pleiotropic functions of BVR-A in the maintenance of cellular homeostasis (Kapitulnik and Maines, 2009), we speculate that the induction of BVR-A precedes those of HO-1. On the contrary, the formation of HNE-adducts on HO-1(Barone et al., 2012a), along with BVR-A nitration (Barone et al., 2011b), are already evident in the hippocampus of subjects with MCI. In this light, despite the progressive increase of HO-1/BVR-A protein levels observed from MCI to AD (Barone et al., 2012a), the impairment of the system appears to be an early event in the pathogenesis and progression of the disease.
Furthermore, the increased Ser-residue phosphorylation along with increased protein levels conceivably could act as a compensatory mechanism to overcome the inactivation of HO-1 by oxidative damage (Barone et al., 2012a) (Figure 3). However, whether or not HO-1 functionality is in part restored after Ser-residue phosphorylation remains an unsolved question. In order to complete this intricate puzzle, the measure of HO-1 activity should be considered. However, in our experimental model it is not possible to single out the differential contribution of HO-1 and HO-2 to the generation of their products (i.e., CO, ferrous iron and biliverdin) due to lack of reliable selective inhibitors of the two isoforms (Mancuso and Barone, 2009).
Reconciliation of the two hypotheses
Based on the scenario described above, it is possible to reconcile the apparent contradictory roles of the HO-1/BVR-A system in AD brain [neuroprotective or neurotoxic] as follows: i) The failure to protect neurons against the deleterious effects of oxidative/nitrosative stress could be due to an impairment of HO-1, together with BVR-A, as suggested by our group (Barone et al., 2011a; Barone et al., 2011b); ii) Phosphorylation might be able to restore HO-1 functionality, and as a consequence the sustained activation of HO-1 could be responsible, at least in part, for the observed increased oxidative stress, as well as tau phosphorylation, in the hippocampus of AD subjects, as suggested by other groups (Hui et al., 2011; Schipper et al., 2009); iii) the reduced activation of BVR-A implies either a reduced production of the powerful antioxidant/antinitrosative molecule BR (Barone et al., 2009; Stocker, 2004) and/or a dysfunction of all the cellular pathways regulated by BVR-A that are essential for cellular homeostasis (Kapitulnik and Maines, 2009) (Figure 3).
Our studies demonstrated that HO-1/BVR are not very protective in AD/MCI brain due to the post-translational modifications which decrease both the reductase and kinase activities. This view in the end results in the same place as that proposed by the neurotoxic hypothesis associated with the HO/BVR system: damage to AD and MCI brain, including oxidative damage. Hence, it is time to come together and see that both notions lead to the same conclusion: oxidative damage in AD and MCI brain, produced in part either as a result of the products of HO-1 (ferrous iron for example) or as a result of a dysfunctional HO/BVR system as a consequence of oxidative and/or nitrosative modification.
The challenge in AD and MCI will be to find an effective pharmacological treatment that might conceivably be capable of overcoming or at least reducing these obstacles related to the neurotoxic effects.
With the aim to realize this ambitious goal, we evaluated the effect of atorvastatin treatment (80 mg/day for 14.5 months) on oxidative stress levels and the HO/BVR-A system in the parietal cortex, cerebellum and liver of a well characterized pre-clinical model of AD, the aged beagles (Cotman and Head, 2008; Johnstone et al., 1991). We found that atorvastatin, which only to a minute extent can cross the BBB, in brain significantly: (i) decreased HNE, PC and 3-NT total levels; (ii) increased GSH levels; (iii) increased HO-1 protein levels; (iv) increased BVR-A protein levels, phosphorylation and activity (Barone et al., 2012b; Butterfield et al., 2012a) (Table 3, Figure 4). Additionally, significant correlations were found among: (i) decreased levels of oxidative/nitrosative stress markers and decreased discriminate learning error score (DLES), reflecting improved cognition; (ii) HO-1 and BVR-A and decreased oxidative/nitrosative stress indices, as well as DLES (Barone et al., 2012b; Butterfield et al., 2012a). Furthermore, BVR-A up-regulation and post-translational modifications significantly correlated with β-secretase protein levels in the brain, suggesting a possible role for BVR-A in Aβ formation (Barone et al., 2012b) (Table 3, Figure 4).
Table 3.
Main effects observed during previous studies by the Butterfield group about (i) oxidative stress levels and (ii) HO-1/BVR-A system in the brain and liver of aged beagles following atorvastatin treatment. Each value is expressed with respect to the control group (Barone et al., 2012b; Butterfield et al., 2012a).
| Parietal Cortex | Cerebellum | Liver | |
|---|---|---|---|
| PC total levels |
11%, *
|
≠ | ≠ |
| HNE total levels |
32%, *
|
≠ | ≠ |
| 3-NT total levels |
26%, *
|
≠ | ≠ |
| HO-1protein levels |
75%, **
|
≠ | ≠ |
| BVR-A protein levels |
21%, *
|
≠ |
60%, **
|
| pTyr-BVR-A |
54%, *
|
≠ | ≠ |
| pSer/Thr-BVR-A |
17% |
≠ | ≠ |
| BVR activity |
35%, *
|
≠ | ≠ |
| PC-BVR-A | ≠ | ≠ |
60%, ns |
| HNE-BVR-A | ≠ | ≠ |
60%, **
|
| 3-NT-BVR-A |
18%, ns |
≠ | ≠ |
Increase,
decrease, ≠ no changes,
p<0.05,
p<0.01,
ns non significant
Figure 4. Schematic representation of atorvastatin-induced BVR-A neuroprotective effects in the parietal cortex of aged beagles.
Aged beagles are a good preclinical model of Alzheimer disease since they naturally develops learning and memory impairments in association with the accumulation of human-sequence Aβ and increased oxidative stress levels (Cotman and Head, 2008; Head et al., 2008) (right side). Atorvastatin increases (i) HO-1 protein levels and (ii) both BVR-A protein levels and phosphorylation on Tyr/Ser/Thr residues in parietal cortex of aged beagles. As a consequence, an increase of its reductase activity (increased bilirubin (BR) production) is observed. Either BVR-A and BR possesses antioxidant features responsible of the reduction of oxidative stress in the parietal cortex, as demonstrated by the negative correlations found between oxidative stress biomarkers levels and (i) BVR-A protein levels or (ii) BVR activity in the same brain area (Barone et al., 2012b). Furthermore BVR-A is associated with an improvement of cognitive functions (learning) following atorvastatin treatment (Barone et al., 2012b). Finally, BVR-A protein levels and pTyr-BVR-A were significantly associated with decreased BACE1 protein levels suggesting a role for BVR-A in Aβ production (Barone et al., 2012b). All these effects contribute to the neuroprotective role of BVR-A in the brain. Arrows, stimulation; dotted lines, inhibition.
We believe that an increase of a drug-related induction of BVR-A protein levels together with its improved functioning could trigger a cell stress response and thus improve cognitive behavior by the following mechanisms: (i) Interaction with members of the MAPK family, such as ERK1/2-Mek-Elk1, through which BVR-A regulates important metabolic pathway as well as the expression of oxidative-stress-responsive genes such as HO-1 or inducible nitric oxide synthase (iNOS) (Di Domenico et al., 2013b; Kapitulnik and Maines, 2009; Lerner-Marmarosh et al., 2008; Maines, 2007; Tudor et al., 2008); (ii) Production of the powerful antioxidant BR as result of BVR-A’s reductase activity; (iii) and speculatively, activation of both conventional and atypical protein kinase C isoforms (Kapitulnik and Maines, 2009), whose involvement in memory function is now well established (Sacktor, 2011) [but see Gibbs et al. (2012), who show that PKC activation does not always lead to protection]. In these scenarios, since the phosphorylation of BVR-A on Tyr residues is required to interact with ERK-Mek-Elk1 (Lerner-Marmarosh et al., 2008), the increase of pTyr-BVR-A in the parietal cortex following atorvastatin treatment, coupled with the negative correlation between pTyr-BVR-A and size discrimination error scores, could suggest an activation of the MAPK-related signal transduction pathways that in turn promote a robust cell stress response (Kapitulnik and Maines, 2009) (Figure 4). At the same time, the significant correlations found between BVR activity and decreased total PC and 3-NT levels suggest a main antioxidant role for BR, consistent with prior studies (Barone et al., 2009; Dore et al., 1999; Stocker et al., 1987a; Stocker et al., 1987b) (Figure 4).
Based on these observations, we propose a novel mechanism of action for atorvastatin which, through the activation of HO/BVR-A system, may contribute to the neuroprotective effects thus suggesting a potential therapeutic role in AD and potentially accounting for the observation of decreased AD incidence with persons on statin (Figure 4).
HO-1/BVR-A system as an AD diagnostic tool
A definitive diagnosis of AD requires post-mortem neuropathological examination for the presence of two hallmarks of AD brain lesions: extracellular amyloid plaques and intraneuronal neurofibrillary tangles (Fagan and Perrin, 2012). Clinical diagnosis of AD during the patient’s life is based on both novel techniques of brain imaging (e.g. positron-emission tomography, PET) (Chen and Zhong, 2013) and a battery of probabilistic neuropsychological, cognitive and functional tests that however, have low accuracy when applied at very early stages or used to observe the effects of disease-modifying drugs (Gustaw-Rothenberg et al., 2010). In this frame, particularly interesting is the study by Shokouhi et al. (Shokouhi et al., 2013), who demonstrated a longitudinal progression between the cognitive decline of MCI subjects and the brain pattern of 18F-fluordeoxyglucose detected by PET. A novel drug, florbetapir, was recently studied as an Aβ-tracer and a potential role for this agent as a tool to monitor Aβ formation/disappearance by PET has been proposed. (Saint-Aubert et al., 2013). Treatments approved for AD, often initiated only at the time dementia is recognized, are considered to have marginal efficacy if administered at late stages of the disease when irreversible brain damage has already occurred (Di Domenico et al., 2011). In this view, the diagnosis of AD at earlier stages represent a key step for the administration of preventive and disease-modifying therapies that conceivably could protect brain from neurodegeneration.
Therefore, there is an urgent need for objective diagnostic tests of AD onset and progression. AD biomarkers based on imaging and body fluid analytes have been proposed and the combined detection of three well recognized CSF biomarkers: Aβ1–42, total tau and phosphorylated tau (p-tau) reach high sensitivity and specificity for AD prediction (Grossman et al., 2005; Jack et al., 2010; Mulder et al., 2010; Petersen et al., 2010). However, a significant limitation to these methods is represented by their costs, availability and invasiveness that impedes their routine use especially for the diagnosis of asymptomatic early stages of AD. Current studies on biochemical, easy detectable, markers of AD and MCI in blood are based on the analyses of inflammatory proteins, markers of cholesterol homeostasis, oxidative stress, or related to characteristic pathological alterations in AD (Di Domenico et al., 2011; Galasko and Montine, 2010; Padurariu et al., 2010; Song et al., 2009). In this context, the analysis in peripheral fluids (serum/plasma) of the HO-1/BVR-A system, as outlined before, is closely related to oxidative status, one of the main features of AD, and might represent a promising strategy to predict AD onset, staging and progression.
As noted above, HO-1 has been the object of several studies on biomarker discovery regarding AD and other degenerative pathologies (Schipper, 2007). In 2000 Schipper and colleagues (Schipper et al., 2000) and in 2002 Ishizuka and colleagues (Ishizuka et al., 2002) showed that plasma HO-1 protein and mononuclear cell HO-1 mRNA levels were significantly suppressed in subjects with probable early sporadic AD compared to normal elderly controls and individuals with various neurological and medical disorders (Table 1). MCI subjects reportedly had HO-1 mRNA and protein levels that were intermediate between controls and AD values indicating a correlation between disease progression and peripheral decrease in HO-1. In addition to plasma levels, CSF HO-1 levels also were found suppressed in AD patients supporting their previous finding (Table 1). In 2006 Maes et al. (Maes et al., 2006) explained such HO-1 plasma and CSF reduction by the presence of a circulating suppressor of HO-1 expression, identified as alpha-1 anti-chymotrypsin (AAT) in patients with sporadic AD. The same inhibition does not occur in CNS of AD patients due to the high AAT exposure to disease- related protein oxidation and nitration. In contrast to the above-referenced studies, a recent research by Mateo et al. (Mateo et al., 2010) described unaltered HO-1 serum levels between control and AD subjects, and a study (Calabrese et al., 2006) demonstrated increased levels of HO-1 in AD lymphocytes compared with control (Table 2). Extending AD studies from brain to plasma, the Butterfield laboratory investigated the status of the peripheral HO-1/BVR system with the idea that it might reflect brain pathology. We showed, recently, that plasma levels of HO-1 are increased in AD and MCI subjects following disease severity. Our data on plasma HO-1 levels correlate with brain data previously discussed; however, none of the HO-1 aberrant modifications (protein bound-HNE or phosphorylation) seen in brain were found in plasma from AD subjects suggesting that HO-1 analysis may lack AD specificity as a disease biomarker (Di Domenico et al., 2012) (Barone et al., 2012a) (Table 2). The analysis of the literature shows the presence of a number of investigations that propose HO-1 altered expression levels as a biomarker of several different diseases, such as lung function decline in silicosis patients, secondary hemophagocytic syndrome (HPS) or adult-onset Still’s disease, type-2 diabetes mellitus and coronary atherosclerosis (Brydun et al., 2007; Miyazaki et al., 2010) (Calabrese et al., 2007b). Thus, altered HO-1 expression in a such number of heterogenic diseases suggests that HO-1 alteration may be specific of an event common to all of these diseases, such as oxidative stress or antioxidant response, but not exclusive of one particular disease.
In 2010 Mueller et al. (Mueller et al., 2010) identified the association of BVR-A and -B altered expression in AD and MCI pathology in plasma samples suggesting that the heme degradation pathway, with the focus on BVR, may represent a new avenue for biomarker search (Table 1). Subsequent studies from our laboratory showed that, in AD, data from BVR-A plasma are closely related to BVR-A results in hippocampus with regard to increased protein quantity, increased protein nitration, decreased tyrosine phosphorylation, and decreased protein reductase activity (Di Domenico et al., 2012) (Table 2). Interestingly, we showed that in plasma from probable AD patients (pAD), in which the pro-oxidant conditions are steadily higher than control, BVR-A protein levels and activity follow their relationship seen in hippocampus. Conversely, in MCI, the lack of significant increase of BVR-A levels is coupled with unchanged reductase activity levels (Di Domenico et al., 2012) (Table 2). The differences between pAD and MCI plasma data might be related to the severity of the disease that results in different degrees of protein induction, oxidative modification, phosphorylation, and finally protein activity. Correlation data among nitration, pTyr alterations, and BVR-A levels in pAD strengthen our findings. Moreover, correlations of HO-1 and BVR-A quantity and BVR-A post-translational modifications, with cognitive performance parameters such as Mini Mental State Examination (MMSE) and instrumental activities of daily living (IADL) in control, AD, and MCI confirm that the alteration of the HO-1/BVR-A system in plasma is dependent on disease stage, increasing with the severity or rate of progression of AD pathology (Di Domenico et al., 2012). Currently, the source of plasma BVR-A is not known.
The results noted above suggest that plasma BVR-A status, more than HO-1, might represent a reliable monitor of hippocampal BVR-A status and brain damage in pAD. Indeed, BVR-A post-translational modifications and protein activity add a further degree of complexity and consistency to a potential diagnostic test and provides deeper and more detailed information about BVR-A status during AD pathology (Figure 5).
Figure 5. Schematic view of plasma HO-1/BVR-A system reflecting CNS pathology in AD subjects.
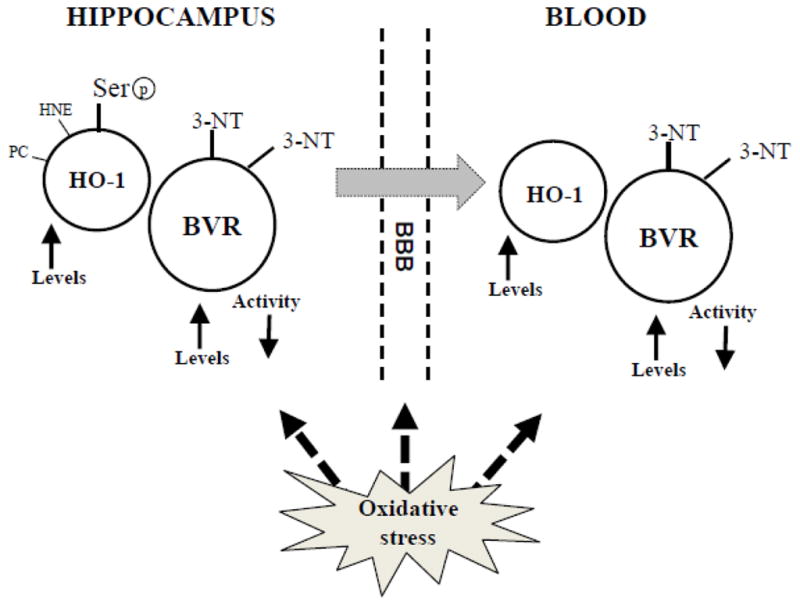
We reported in AD plasma that HO-1/BVR-A system status could represent a reliable tool to monitor CNS ongoing AD pathology. Indeed, we found altered expression levels for both the protein components of the system and aberrant BVR-A protein nitration and phosphorylation that mimic the hippocampal situation during AD pathology. We speculate that the increased OS levels at both CNS and blood levels might also account for BVR-A aberrant expression, oxidation and phosphorylation during AD.
In conclusion, the data reported above suggest that even if the blood proteome profile is relatively different from the brain protein profile, the HO-1/BVR-A system status in plasma could mimic the ongoing situation in the brain. Therefore, the analysis of HO- 1/BVR-A system in blood-related biofluids might represent a reasonable way to gain information on increased oxidative and nitrosative stress ongoing in the brain. In light of such considerations, the HO-1/BVR-A system could conceivably predict AD onset and advancement, expanding its significance as a potential AD biomarker, from the early stages of the disease, in combination with other AD diagnostic tools.
Future perspective
The role of the HO/BVR axis in the pathogenesis and therapy of AD and MCI is still a developing research area. From a pathogenetic viewpoint, our data confirm a limited neuroprotective role for the HO-1/BVR system in normal brain in terms of enhancement of the cell stress response. However, loss of activity of HO-1/BVR-A leads to loss of neuroprotective BR and the pleiotropic neuroprotective activities of BVR. This loss of function in both enzymes is due to the post-translational modifications on both, under pro-oxidant conditions. On the other hand, it is necessary to point out that the brain has many others enzymes that can be protective under conditions of oxidative/nitrosative stress, such as Hsp70, thioredoxin reductase, catalase, superoxide dismutase and Nrf-2-dependent phase II enzymes, etc. (Calabrese et al., 2008; Calabrese et al., 2007a; Di Domenico et al., 2010; Joshi et al., 2007; Mancuso et al., 2008). Therapeutic manipulation of the HO/BVR system is so far in the pre-clinical stage. Particularly interesting is the neuroprotective role of statins.
In conclusion, several efforts are still necessary for the full comprehension of the importance of both HO-1 and BVR in the pathophysiology and therapy of neurodegenerative diseases, in particular AD, and this goal can be achieved only on the basis of a strong collaboration between chemists, pharmacologists and clinicians. Such studies are in progress in the Butterfield laboratory.
Acknowledgments
This work was supported in part by a NIH grant to DAB [AG-05119] and Fondi Ateneo to CM.
Abbreviations
- BVR-A
biliverdin reductase isoform A
- HO-1/2
heme oxygenase isoform 1 or 2
- HNE
4-hydroxy-2-nonenal
- PC
protein carbonyls
- 3-NT
3-nitrotyrosine
- ARE
antioxidant responsive elements
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akama KT, et al. Amyloid beta-peptide stimulates nitric oxide production in astrocytes through an NFkappaB-dependent mechanism. Proc Natl Acad Sci U S A. 1998;95:5795–800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksenov M, et al. Enhancement of beta-amyloid peptide A beta(1–40)-mediated neurotoxicity by glutamine synthetase. J Neurochem. 1995;65:1899–902. doi: 10.1046/j.1471-4159.1995.65041899.x. [DOI] [PubMed] [Google Scholar]
- Akter K, et al. Diabetes mellitus and Alzheimer’s disease: shared pathology and treatment? Br J Clin Pharmacol. 2011;71:365–76. doi: 10.1111/j.1365-2125.2010.03830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alam J, Cook JL. Transcriptional regulation of the heme oxygenase-1 gene via the stress response element pathway. Curr Pharm Des. 2003;9:2499–511. doi: 10.2174/1381612033453730. [DOI] [PubMed] [Google Scholar]
- Axelsen PH, et al. Oxidative stress and cell membranes in the pathogenesis of Alzheimer’s disease. Physiology (Bethesda) 2011;26:54–69. doi: 10.1152/physiol.00024.2010. [DOI] [PubMed] [Google Scholar]
- Badia MC, et al. Reductive stress in young healthy individuals at risk of Alzheimer disease. Free Radic Biol Med. 2013;63C:274–279. doi: 10.1016/j.freeradbiomed.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Balogun E, et al. Curcumin activates the haem oxygenase-1 gene via regulation of Nrf2 and the antioxidant-responsive element. Biochem J. 2003;371:887–95. doi: 10.1042/BJ20021619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone E, et al. Biliverdin reductase--a protein levels and activity in the brains of subjects with Alzheimer disease and mild cognitive impairment. Biochim Biophys Acta. 2011a;1812:480–7. doi: 10.1016/j.bbadis.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone E, et al. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer’s disease and amnestic mild cognitive impairment. J Alzheimers Dis. 2011b;25:623–33. doi: 10.3233/JAD-2011-110092. [DOI] [PubMed] [Google Scholar]
- Barone E, et al. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med. 2012a;52:2292–301. doi: 10.1016/j.freeradbiomed.2012.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barone E, et al. Biliverdin reductase-A: a novel drug target for atorvastatin in a dog pre-clinical model of Alzheimer disease. J Neurochem. 2012b;120:135–46. doi: 10.1111/j.1471-4159.2011.07538.x. [DOI] [PubMed] [Google Scholar]
- Barone E, et al. Characterization of the S-denitrosylating activity of bilirubin. J Cell Mol Med. 2009;13:2365–75. doi: 10.1111/j.1582-4934.2008.00680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behl C. Brain aging and late-onset Alzheimer’s disease: many open questions. Int Psychogeriatr. 2012;24(Suppl 1):S3–9. doi: 10.1017/S104161021200052X. [DOI] [PubMed] [Google Scholar]
- Boehning D, et al. Carbon monoxide neurotransmission activated by CK2 phosphorylation of heme oxygenase-2. Neuron. 2003;40:129–37. doi: 10.1016/s0896-6273(03)00596-8. [DOI] [PubMed] [Google Scholar]
- Boehning D, et al. Heme oxygenase-2 is activated by calcium-calmodulin. J Biol Chem. 2004;279:30927–30. doi: 10.1074/jbc.C400222200. [DOI] [PubMed] [Google Scholar]
- Bonda DJ, et al. Oxidative stress in Alzheimer disease: a possibility for prevention. Neuropharmacology. 2010;59:290–4. doi: 10.1016/j.neuropharm.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Braughler JM, et al. The involvement of iron in lipid peroxidation. Importance of ferric to ferrous ratios in initiation. J Biol Chem. 1986;261:10282–9. [PubMed] [Google Scholar]
- Brito MA, et al. A link between hyperbilirubinemia, oxidative stress and injury to neocortical synaptosomes. Brain Res. 2004;1026:33–43. doi: 10.1016/j.brainres.2004.07.063. [DOI] [PubMed] [Google Scholar]
- Brydun A, et al. Reduced expression of heme oxygenase-1 in patients with coronary atherosclerosis. Hypertension Research. 2007;30:341–348. doi: 10.1291/hypres.30.341. [DOI] [PubMed] [Google Scholar]
- Bucher JR, et al. Redox cycling and lipid peroxidation: the central role of iron chelates. Fundam Appl Toxicol. 1983;3:222–6. doi: 10.1016/s0272-0590(83)80130-4. [DOI] [PubMed] [Google Scholar]
- Butterfield D, et al. Nutritional approaches to combat oxidative stress in Alzheimer’s disease. J Nutr Biochem. 2002a;13:444. doi: 10.1016/s0955-2863(02)00205-x. [DOI] [PubMed] [Google Scholar]
- Butterfield DA. Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer’s disease brain. A review. Free Radic Res. 2002;36:1307–13. doi: 10.1080/1071576021000049890. [DOI] [PubMed] [Google Scholar]
- Butterfield DA. Oxidative stress in neurodegenerative disorders. Antioxid Redox Signal. 2006;8:1971–3. doi: 10.1089/ars.2006.8.1971. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, et al. Atorvastatin treatment in a dog preclinical model of Alzheimer’s disease leads to up-regulation of haem oxygenase-1 and is associated with reduced oxidative stress in brain. Int J Neuropsychopharmacol. 2012a;15:981–7. doi: 10.1017/S1461145711001118. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Boyd-Kimball D. Amyloid beta-peptide(1–42) contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathol. 2004;14:426–32. doi: 10.1111/j.1750-3639.2004.tb00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, et al. Vitamin E and neurodegenerative disorders associated with oxidative stress. Nutr Neurosci. 2002b;5:229–39. doi: 10.1080/10284150290028954. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, et al. Evidence of oxidative damage in Alzheimer’s disease brain: central role for amyloid beta-peptide. Trends Mol Med. 2001;7:548–54. doi: 10.1016/s1471-4914(01)02173-6. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, et al. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid beta-peptide of APP. Free Radic Biol Med. 2010;48:136–44. doi: 10.1016/j.freeradbiomed.2009.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Lauderback CM. Lipid peroxidation and protein oxidation in Alzheimer’s disease brain: potential causes and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic Biol Med. 2002;32:1050–60. doi: 10.1016/s0891-5849(02)00794-3. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, et al. Redox proteomics in selected neurodegenerative disorders: from its infancy to future applications. Antioxid Redox Signal. 2012b;17:1610–55. doi: 10.1089/ars.2011.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiol Dis. 2006;22:223–32. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, et al. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic Biol Med. 2007;43:658–77. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterfield DA, Stadman ER. Protein oxidation processes in aging brain. In: Paula ST, Bittar EE, editors. Advances in Cell Aging and Gerontology. 1997. pp. 161–191. [Google Scholar]
- Butterfield DA, Stadtman ER. Protein Oxidation Processes in Aging. Brain 1997 [Google Scholar]
- Butterfield DA, et al. Amyloid beta-Peptide (1–42)-Induced Oxidative Stress in Alzheimer Disease: Importance in Disease Pathogenesis and Progression. Antioxid Redox Signal. 2013 doi: 10.1089/ars.2012.5027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, et al. Oxidative stress and beta-amyloid protein in Alzheimer’s disease. Neuromolecular Med. 2011;13:223–50. doi: 10.1007/s12017-011-8155-9. [DOI] [PubMed] [Google Scholar]
- Calabrese V, et al. Cellular stress response: a novel target for chemoprevention and nutritional neuroprotection in aging, neurodegenerative disorders and longevity. Neurochem Res. 2008;33:2444–71. doi: 10.1007/s11064-008-9775-9. [DOI] [PubMed] [Google Scholar]
- Calabrese V, et al. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007a;8:766–75. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- Calabrese V, et al. Oxidative stress and cellular stress response in diabetic nephropathy. Cell Stress Chaperones. 2007b;12:299–306. doi: 10.1379/CSC-270.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V, et al. Redox regulation of heat shock protein expression in aging and neurodegenerative disorders associated with oxidative stress: a nutritional approach. Amino Acids. 2003;25:437–44. doi: 10.1007/s00726-003-0048-2. [DOI] [PubMed] [Google Scholar]
- Calabrese V, et al. Nitrosative stress, cellular stress response, and thiol homeostasis in patients with Alzheimer’s disease. Antioxid Redox Signal. 2006;8:1975–86. doi: 10.1089/ars.2006.8.1975. [DOI] [PubMed] [Google Scholar]
- Castegna A, et al. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I: creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic Biol Med. 2002a;33:562–71. doi: 10.1016/s0891-5849(02)00914-0. [DOI] [PubMed] [Google Scholar]
- Castegna A, et al. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II: dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J Neurochem. 2002b;82:1524–32. doi: 10.1046/j.1471-4159.2002.01103.x. [DOI] [PubMed] [Google Scholar]
- Chen Z, Zhong C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog Neurobiol. 2013 doi: 10.1016/j.pneurobio.2013.06.004. [DOI] [PubMed] [Google Scholar]
- Chiang HC, et al. PI3 kinase signaling is involved in Abeta-induced memory loss in Drosophila. Proc Natl Acad Sci U S A. 2010;107:7060–5. doi: 10.1073/pnas.0909314107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiueh CC. Iron overload, oxidative stress, and axonal dystrophy in brain disorders. Pediatr Neurol. 2001;25:138–47. doi: 10.1016/s0887-8994(01)00266-1. [DOI] [PubMed] [Google Scholar]
- Clark TA, et al. Oxidative Stress and its Implications for Future Treatments and Management of Alzheimer Disease. Int J Biomed Sci. 2010;6:225–227. [PMC free article] [PubMed] [Google Scholar]
- Cotman CW, Head E. The canine (dog) model of human aging and disease: dietary, environmental and immunotherapy approaches. J Alzheimers Dis. 2008;15:685–707. doi: 10.3233/jad-2008-15413. [DOI] [PubMed] [Google Scholar]
- Cunningham O, et al. Initial-rate kinetics of the flavin reductase reaction catalysed by human biliverdin-IXbeta reductase (BVR-B) Biochem J. 2000;345(Pt 2):393–9. [PMC free article] [PubMed] [Google Scholar]
- Deane R, et al. RAGE (yin) versus LRP (yang) balance regulates alzheimer amyloid beta-peptide clearance through transport across the blood-brain barrier. Stroke. 2004;35:2628–31. doi: 10.1161/01.STR.0000143452.85382.d1. [DOI] [PubMed] [Google Scholar]
- Dhakshinamoorthy S, et al. Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem. 2005;280:16891–900. doi: 10.1074/jbc.M500166200. [DOI] [PubMed] [Google Scholar]
- Di Domenico F, et al. HO-1/BVR-a system analysis in plasma from probable Alzheimer’s disease and mild cognitive impairment subjects: a potential biochemical marker for the prediction of the disease. J Alzheimers Dis. 2012;32:277–89. doi: 10.3233/JAD-2012-121045. [DOI] [PubMed] [Google Scholar]
- Di Domenico F, et al. Circulating biomarkers of protein oxidation for Alzheimer disease: expectations within limits. Biochim Biophys Acta. 2011;1814:1785–95. doi: 10.1016/j.bbapap.2011.10.001. [DOI] [PubMed] [Google Scholar]
- Di Domenico F, et al. Impairment of proteostasis network in Down syndrome prior to the development of Alzheimer’s disease neuropathology: Redox proteomics analysis of human brain. Biochim Biophys Acta. 2013a;1832:1249–59. doi: 10.1016/j.bbadis.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F, et al. Biliverdin Reductase-A correlates with iNOS in atorvastatin treated aged canine brain. Neural Reg Res. 2013b doi: 10.3969/j.issn.1673-5374.2013.21.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domenico F, et al. Protein levels of heat shock proteins 27, 32, 60, 70, 90 and thioredoxin-1 in amnestic mild cognitive impairment: an investigation on the role of cellular stress response in the progression of Alzheimer disease. Brain Res. 2010;1333:72–81. doi: 10.1016/j.brainres.2010.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dildar K, et al. Serum nitrosative stress levels are increased in Alzheimer disease but not in vascular dementia. Alzheimer Dis Assoc Disord. 2010;24:194–7. doi: 10.1097/WAD.0b013e3181c53d0d. [DOI] [PubMed] [Google Scholar]
- Dore S, et al. Bilirubin, formed by activation of heme oxygenase-2, protects neurons against oxidative stress injury. Proc Natl Acad Sci U S A. 1999;96:2445–50. doi: 10.1073/pnas.96.5.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufouil C, et al. APOE genotype, cholesterol level, lipid-lowering treatment, and dementia: the Three-City Study. Neurology. 2005;64:1531–8. doi: 10.1212/01.WNL.0000160114.42643.31. [DOI] [PubMed] [Google Scholar]
- Dwyer BE, et al. Differential expression of heme oxygenase-1 in cultured cortical neurons and astrocytes determined by the aid of a new heme oxygenase antibody. Response to oxidative stress. Brain Res Mol Brain Res. 1995;30:37–47. doi: 10.1016/0169-328x(94)00273-h. [DOI] [PubMed] [Google Scholar]
- Errico S, et al. Heme oxygenase-derived carbon monoxide modulates gonadotropin-releasing hormone release in immortalized hypothalamic neurons. Neurosci Lett. 2010;471:175–8. doi: 10.1016/j.neulet.2010.01.036. [DOI] [PubMed] [Google Scholar]
- Ewing JF, et al. Biliverdin reductase is heat resistant and coexpressed with constitutive and heat shock forms of heme oxygenase in brain. J Neurochem. 1993;61:1015–23. doi: 10.1111/j.1471-4159.1993.tb03615.x. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Perrin RJ. Upcoming candidate cerebrospinal fluid biomarkers of Alzheimer’s disease. Biomark Med. 2012;6:455–76. doi: 10.2217/bmm.12.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhrai H, Maines MD. Expression and characterization of a cDNA for rat kidney biliverdin reductase. Evidence suggesting the liver and kidney enzymes are the same transcript product. J Biol Chem. 1992;267:4023–9. [PubMed] [Google Scholar]
- Feng Y, Wang X. Antioxidant therapies for Alzheimer’s disease. Oxid Med Cell Longev. 2012;2012:472932. doi: 10.1155/2012/472932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel D, et al. Role of heme oxygenase-1 in the regulation of manganese superoxide dismutase gene expression in oxidatively-challenged astroglia. J Cell Physiol. 2000;185:80–6. doi: 10.1002/1097-4652(200010)185:1<80::AID-JCP7>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Fujita T, et al. Paradoxical rescue from ischemic lung injury by inhaled carbon monoxide driven by derepression of fibrinolysis. Nat Med. 2001;7:598–604. doi: 10.1038/87929. [DOI] [PubMed] [Google Scholar]
- Galasko D, Montine TJ. Biomarkers of oxidative damage and inflammation in Alzheimer’s disease. Biomark Med. 2010;4:27–36. doi: 10.2217/bmm.09.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs PE, Maines MD. Biliverdin inhibits activation of NF-kappaB: reversal of inhibition by human biliverdin reductase. Int J Cancer. 2007;121:2567–74. doi: 10.1002/ijc.22978. [DOI] [PubMed] [Google Scholar]
- Gibbs PE, et al. Formation of ternary complex of human biliverdin reductase-protein kinase Cdelta-ERK2 protein is essential for ERK2-mediated activation of Elk1 protein, nuclear factor-kappaB, and inducible nitric-oxidase synthase (iNOS) J Biol Chem. 2012a;287:1066–79. doi: 10.1074/jbc.M111.279612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs PE, et al. Biliverdin reductase: more than a namesake - the reductase, its Peptide fragments, and biliverdin regulate activity of the three classes of protein kinase C. Front Pharmacol. 2012b;3:31. doi: 10.3389/fphar.2012.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozzelino R, et al. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–54. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- Grossman M, et al. Cerebrospinal fluid profile in frontotemporal dementia and Alzheimer’s disease. Ann Neurol. 2005;57:721–9. doi: 10.1002/ana.20477. [DOI] [PubMed] [Google Scholar]
- Gustaw-Rothenberg K, et al. Biomarkers in Alzheimer’s disease: past, present and future. Biomark Med. 2010;4:15–26. doi: 10.2217/bmm.09.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Selkoe DJ. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid beta-peptide. Nat Rev Mol Cell Biol. 2007;8:101–12. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- Hajjar I, et al. The impact of the use of statins on the prevalence of dementia and the progression of cognitive impairment. J Gerontol A Biol Sci Med Sci. 2002;57:M414–8. doi: 10.1093/gerona/57.7.m414. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 2006;97:1634–58. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- Hascalovici JR, et al. Brain sterol dysregulation in sporadic AD and MCI: relationship to heme oxygenase-1. J Neurochem. 2009;110:1241–53. doi: 10.1111/j.1471-4159.2009.06213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi S, et al. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene. 2004;336:241–50. doi: 10.1016/j.gene.2004.04.002. [DOI] [PubMed] [Google Scholar]
- He X, et al. Protection against chromium (VI)-induced oxidative stress and apoptosis by Nrf2. Recruiting Nrf2 into the nucleus and disrupting the nuclear Nrf2/Keap1 association. Toxicol Sci. 2007;98:298–309. doi: 10.1093/toxsci/kfm081. [DOI] [PubMed] [Google Scholar]
- Head E, et al. Oxidative stress, aging, and central nervous system disease in the canine model of human brain aging. Vet Clin North Am Small Anim Pract. 2008;38:167–78. vi. doi: 10.1016/j.cvsm.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K, et al. Brain regional correspondence between Alzheimer’s disease histopathology and biomarkers of protein oxidation. J Neurochem. 1995;65:2146–56. doi: 10.1046/j.1471-4159.1995.65052146.x. [DOI] [PubMed] [Google Scholar]
- Horwood JM, et al. Signalling mechanisms mediated by the phosphoinositide 3-kinase/Akt cascade in synaptic plasticity and memory in the rat. Eur J Neurosci. 2006;23:3375–84. doi: 10.1111/j.1460-9568.2006.04859.x. [DOI] [PubMed] [Google Scholar]
- Hui Y, et al. Long-term overexpression of heme oxygenase 1 promotes tau aggregation in mouse brain by inducing tau phosphorylation. J Alzheimers Dis. 2011;26:299–313. doi: 10.3233/JAD-2011-102061. [DOI] [PubMed] [Google Scholar]
- Hyman BT, et al. Extracellular signal regulated kinases. Localization of protein and mRNA in the human hippocampal formation in Alzheimer’s disease. Am J Pathol. 1994;144:565–72. [PMC free article] [PubMed] [Google Scholar]
- Irizarry MC, et al. Plasma F2A isoprostane levels in Alzheimer’s and Parkinson’s disease. Neurodegener Dis. 2007;4:403–5. doi: 10.1159/000107699. [DOI] [PubMed] [Google Scholar]
- Ishizuka K, et al. Possible assessment for antioxidant capacity in Alzheimer’s disease by measuring lymphocyte heme oxygenase-1 expression with real-time RT-PCR. Ann N Y Acad Sci. 2002;977:173–8. doi: 10.1111/j.1749-6632.2002.tb04814.x. [DOI] [PubMed] [Google Scholar]
- Jack CR, Jr, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jick H, et al. Statins and the risk of dementia. Lancet. 2000;356:1627–31. doi: 10.1016/s0140-6736(00)03155-x. [DOI] [PubMed] [Google Scholar]
- Johnstone EM, et al. Conservation of the sequence of the Alzheimer’s disease amyloid peptide in dog, polar bear and five other mammals by cross-species polymerase chain reaction analysis. Brain Res Mol Brain Res. 1991;10:299–305. doi: 10.1016/0169-328x(91)90088-f. [DOI] [PubMed] [Google Scholar]
- Joshi G, et al. Glutathione elevation by gamma-glutamyl cysteine ethyl ester as a potential therapeutic strategy for preventing oxidative stress in brain mediated by in vivo administration of adriamycin: Implication for chemobrain. J Neurosci Res. 2007;85:497–503. doi: 10.1002/jnr.21158. [DOI] [PubMed] [Google Scholar]
- Kaide JI, et al. Carbon monoxide of vascular origin attenuates the sensitivity of renal arterial vessels to vasoconstrictors. J Clin Invest. 2001;107:1163–71. doi: 10.1172/JCI11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanninen K, et al. Intrahippocampal injection of a lentiviral vector expressing Nrf2 improves spatial learning in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2009;106:16505–10. doi: 10.1073/pnas.0908397106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapitulnik J. Bilirubin: an endogenous product of heme degradation with both cytotoxic and cytoprotective properties. Mol Pharmacol. 2004;66:773–9. doi: 10.1124/mol.104.002832. [DOI] [PubMed] [Google Scholar]
- Kapitulnik J, Maines MD. Pleiotropic functions of biliverdin reductase: cellular signaling and generation of cytoprotective and cytotoxic bilirubin. Trends Pharmacol Sci. 2009;30:129–37. doi: 10.1016/j.tips.2008.12.003. [DOI] [PubMed] [Google Scholar]
- Keller JN, et al. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–6. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- Kim TS, et al. Decreased plasma antioxidants in patients with Alzheimer’s disease. Int J Geriatr Psychiatry. 2006;21:344–8. doi: 10.1002/gps.1469. [DOI] [PubMed] [Google Scholar]
- Kimpara T, et al. Increased bilirubins and their derivatives in cerebrospinal fluid in Alzheimer’s disease. Neurobiol Aging. 2000;21:551–4. doi: 10.1016/s0197-4580(00)00128-7. [DOI] [PubMed] [Google Scholar]
- Kitamuro T, et al. Bach1 functions as a hypoxia-inducible repressor for the heme oxygenase-1 gene in human cells. J Biol Chem. 2003;278:9125–33. doi: 10.1074/jbc.M209939200. [DOI] [PubMed] [Google Scholar]
- Kooli A, et al. trans-Arachidonic acids induce a heme oxygenase-dependent vasorelaxation of cerebral microvasculature. Free Radic Biol Med. 2008;44:815–25. doi: 10.1016/j.freeradbiomed.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Kosaka J, et al. Effects of biliverdin administration on acute lung injury induced by hemorrhagic shock and resuscitation in rats. PLoS One. 2013;8:e63606. doi: 10.1371/journal.pone.0063606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauderback CM, et al. The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1–42. J Neurochem. 2001;78:413–6. doi: 10.1046/j.1471-4159.2001.00451.x. [DOI] [PubMed] [Google Scholar]
- Leffler CW, et al. Mechanism of glutamate stimulation of CO production in cerebral microvessels. Am J Physiol Heart Circ Physiol. 2003a;285:H74–80. doi: 10.1152/ajpheart.01081.2002. [DOI] [PubMed] [Google Scholar]
- Leffler CW, et al. Regulation of CO production in cerebral microvessels of newborn pigs. Am J Physiol Heart Circ Physiol. 2003b;285:H292–7. doi: 10.1152/ajpheart.01059.2002. [DOI] [PubMed] [Google Scholar]
- Lerner-Marmarosh N, et al. Regulation of TNF-alpha-activated PKC-zeta signaling by the human biliverdin reductase: identification of activating and inhibitory domains of the reductase. FASEB J. 2007;21:3949–62. doi: 10.1096/fj.07-8544com. [DOI] [PubMed] [Google Scholar]
- Lerner-Marmarosh N, et al. Human biliverdin reductase is an ERK activator; hBVR is an ERK nuclear transporter and is required for MAPK signaling. Proc Natl Acad Sci U S A. 2008;105:6870–5. doi: 10.1073/pnas.0800750105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner-Marmarosh N, et al. Human biliverdin reductase: a member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc Natl Acad Sci U S A. 2005;102:7109–14. doi: 10.1073/pnas.0502173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N, et al. Developmentally regulated expression of two transcripts for heme oxygenase-2 with a first exon unique to rat testis: control by corticosterone of the oxygenase protein expression. Gene. 2000;241:175–83. doi: 10.1016/s0378-1119(99)00439-4. [DOI] [PubMed] [Google Scholar]
- Liu XM, et al. Antiapoptotic action of carbon monoxide on cultured vascular smooth muscle cells. Exp Biol Med (Maywood) 2003;228:572–5. doi: 10.1177/15353702-0322805-30. [DOI] [PubMed] [Google Scholar]
- Lovell MA, et al. Acrolein is increased in Alzheimer’s disease brain and is toxic to primary hippocampal cultures. Neurobiol Aging. 2001;22:187–94. doi: 10.1016/s0197-4580(00)00235-9. [DOI] [PubMed] [Google Scholar]
- Maes OC, et al. Characterization of alpha1-antitrypsin as a heme oxygenase-1 suppressor in Alzheimer plasma. Neurobiol Dis. 2006;24:89–100. doi: 10.1016/j.nbd.2006.06.009. [DOI] [PubMed] [Google Scholar]
- Maines MD. The heme oxygenase system: a regulator of second messenger gases. Annu Rev Pharmacol Toxicol. 1997;37:517–54. doi: 10.1146/annurev.pharmtox.37.1.517. [DOI] [PubMed] [Google Scholar]
- Maines MD. The heme oxygenase system and its functions in the brain. Cell Mol Biol (Noisy-le-grand) 2000;46:573–85. [PubMed] [Google Scholar]
- Maines MD. The heme oxygenase system: update 2005. Antioxid Redox Signal. 2005a;7:1761–6. doi: 10.1089/ars.2005.7.1761. [DOI] [PubMed] [Google Scholar]
- Maines MD. New insights into biliverdin reductase functions: linking heme metabolism to cell signaling. Physiology (Bethesda) 2005b;20:382–9. doi: 10.1152/physiol.00029.2005. [DOI] [PubMed] [Google Scholar]
- Maines MD. Biliverdin reductase: PKC interaction at the cross-talk of MAPK and PI3K signaling pathways. Antioxid Redox Signal. 2007;9:2187–95. doi: 10.1089/ars.2007.1805. [DOI] [PubMed] [Google Scholar]
- Maines MD, et al. Nuclear localization of biliverdin reductase in the rat kidney: response to nephrotoxins that induce heme oxygenase-1. J Pharmacol Exp Ther. 2001;296:1091–7. [PubMed] [Google Scholar]
- Maines MD, et al. Human biliverdin reductase, a previously unknown activator of protein kinase C betaII. J Biol Chem. 2007;282:8110–22. doi: 10.1074/jbc.M513427200. [DOI] [PubMed] [Google Scholar]
- Maines MD, Panahian N. The heme oxygenase system and cellular defense mechanisms. Do HO-1 and HO-2 have different functions? Adv Exp Med Biol. 2001;502:249–72. doi: 10.1007/978-1-4757-3401-0_17. [DOI] [PubMed] [Google Scholar]
- Maines MD, et al. Human biliverdin IXalpha reductase is a zinc-metalloprotein. Characterization of purified and Escherichia coli expressed enzymes. Eur J Biochem. 1996;235:372–81. doi: 10.1111/j.1432-1033.1996.00372.x. [DOI] [PubMed] [Google Scholar]
- Maines MD, Trakshel GM. Purification and characterization of human biliverdin reductase. Arch Biochem Biophys. 1993;300:320–6. doi: 10.1006/abbi.1993.1044. [DOI] [PubMed] [Google Scholar]
- Mancuso C, Barone E. The heme oxygenase/biliverdin reductase pathway in drug research and development. Curr Drug Metab. 2009;10:579–94. doi: 10.2174/138920009789375405. [DOI] [PubMed] [Google Scholar]
- Mancuso C, et al. Inhibition of lipid peroxidation and protein oxidation by endogenous and exogenous antioxidants in rat brain microsomes in vitro. Neurosci Lett. 2012;518:101–5. doi: 10.1016/j.neulet.2012.04.062. [DOI] [PubMed] [Google Scholar]
- Mancuso C, et al. Bilirubin and S-nitrosothiols interaction: evidence for a possible role of bilirubin as a scavenger of nitric oxide. Biochem Pharmacol. 2003;66:2355–63. doi: 10.1016/j.bcp.2003.08.022. [DOI] [PubMed] [Google Scholar]
- Mancuso C, et al. Bilirubin as an endogenous modulator of neurotrophin redox signaling. J Neurosci Res. 2008;86:2235–49. doi: 10.1002/jnr.21665. [DOI] [PubMed] [Google Scholar]
- Mancuso C, et al. Activation of heme oxygenase and consequent carbon monoxide formation inhibits the release of arginine vasopressin from rat hypothalamic explants. Molecular linkage between heme catabolism and neuroendocrine function. Brain Res Mol Brain Res. 1997;50:267–76. doi: 10.1016/s0169-328x(97)00197-6. [DOI] [PubMed] [Google Scholar]
- Mancuso C, et al. Roles of nitric oxide, carbon monoxide, and hydrogen sulfide in the regulation of the hypothalamic-pituitary-adrenal axis. J Neurochem. 2010;113:563–75. doi: 10.1111/j.1471-4159.2010.06606.x. [DOI] [PubMed] [Google Scholar]
- Mancuso C, et al. Inhibition of heme oxygenase in the central nervous system potentiates endotoxin-induced vasopressin release in the rat. J Neuroimmunol. 1999;99:189–94. doi: 10.1016/s0165-5728(99)00112-5. [DOI] [PubMed] [Google Scholar]
- Mancuso C, et al. The generation of nitric oxide and carbon monoxide produces opposite effects on the release of immunoreactive interleukin-1beta from the rat hypothalamus in vitro: evidence for the involvement of different signaling pathways. Endocrinology. 1998;139:1031–7. doi: 10.1210/endo.139.3.5822. [DOI] [PubMed] [Google Scholar]
- Mancuso C, Siciliano R, Barone E, Preziosi P. Natural substances and Alzheimer’s disease: from preclinical studies to evidence based medicine. Biochim Biophys Acta. 2012;1822:616–624. doi: 10.1016/j.bbadis.2011.09.004. [DOI] [PubMed] [Google Scholar]
- Mark RJ, et al. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997;68:255–64. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- Markesbery WR. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med. 1997;23:134–47. doi: 10.1016/s0891-5849(96)00629-6. [DOI] [PubMed] [Google Scholar]
- Markesbery WR. The role of oxidative stress in Alzheimer disease. Arch Neurol. 1999;56:1449–52. doi: 10.1001/archneur.56.12.1449. [DOI] [PubMed] [Google Scholar]
- Markesbery WR. Neuropathologic Alterations in Mild Cognitive Impairment: A Review. J Alzheimers Dis. 2009 doi: 10.3233/JAD-2010-1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery WR, Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol Aging. 1998;19:33–6. doi: 10.1016/s0197-4580(98)00009-8. [DOI] [PubMed] [Google Scholar]
- Mateo I, et al. Serum heme oxygenase-1 levels are increased in Parkinson’s disease but not in Alzheimer’s disease. Acta Neurol Scand. 2010;121:136–8. doi: 10.1111/j.1600-0404.2009.01261.x. [DOI] [PubMed] [Google Scholar]
- McCoubrey WK, Jr, et al. The structure, organization and differential expression of the rat gene encoding biliverdin reductase. Gene. 1995;160:235–40. doi: 10.1016/0378-1119(95)00112-j. [DOI] [PubMed] [Google Scholar]
- McCoubrey WK, Jr, et al. Isolation and characterization of a cDNA from the rat brain that encodes hemoprotein heme oxygenase-3. Eur J Biochem. 1997;247:725–32. doi: 10.1111/j.1432-1033.1997.00725.x. [DOI] [PubMed] [Google Scholar]
- Mecocci P, et al. Oxidative damage to mitochondrial DNA is increased in Alzheimer’s disease. Ann Neurol. 1994;36:747–51. doi: 10.1002/ana.410360510. [DOI] [PubMed] [Google Scholar]
- Minotti G, Aust SD. The role of iron in oxygen radical mediated lipid peroxidation. Chem Biol Interact. 1989;71:1–19. doi: 10.1016/0009-2797(89)90087-2. [DOI] [PubMed] [Google Scholar]
- Miyazaki T, et al. Serum HO-1 is useful to make differential diagnosis of secondary hemophagocytic syndrome from other similar hematological conditions. International Journal of Hematology. 2010;91:229–237. doi: 10.1007/s12185-010-0495-y. [DOI] [PubMed] [Google Scholar]
- Morris JC, Cummings J. Mild cognitive impairment (MCI) represents early-stage Alzheimer’s disease. J Alzheimers Dis. 2005;7:235–9. doi: 10.3233/jad-2005-7306. discussion 255–62. [DOI] [PubMed] [Google Scholar]
- Mueller C, et al. The heme degradation pathway is a promising serum biomarker source for the early detection of Alzheimer’s disease. J Alzheimers Dis. 2010;19:1081–91. doi: 10.3233/JAD-2010-1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder C, et al. Amyloid-beta(1–42), total tau, and phosphorylated tau as cerebrospinal fluid biomarkers for the diagnosis of Alzheimer disease. Clin Chem. 2010;56:248–53. doi: 10.1373/clinchem.2009.130518. [DOI] [PubMed] [Google Scholar]
- Nuhn P, et al. Heme oxygenase 1-generated carbon monoxide and biliverdin attenuate the course of experimental necrotizing pancreatitis. Pancreas. 2013;42:265–71. doi: 10.1097/MPA.0b013e318264cc8b. [DOI] [PubMed] [Google Scholar]
- Nunomura A, et al. Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox Res. 2012;22:231–48. doi: 10.1007/s12640-012-9331-x. [DOI] [PubMed] [Google Scholar]
- Ogawa K, et al. Heme mediates derepression of Maf recognition element through direct binding to transcription repressor Bach1. EMBO J. 2001;20:2835–43. doi: 10.1093/emboj/20.11.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opii WO, et al. Proteomic identification of brain proteins in the canine model of human aging following a long-term treatment with antioxidants and a program of behavioral enrichment: relevance to Alzheimer’s disease. Neurobiol Aging. 2008;29:51–70. doi: 10.1016/j.neurobiolaging.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen JB, et al. Oxidative modification to LDL receptor-related protein 1 in hippocampus from subjects with Alzheimer disease: implications for Abeta accumulation in AD brain. Free Radic Biol Med. 2010;49:1798–803. doi: 10.1016/j.freeradbiomed.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padurariu M, et al. Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer’s disease. Neurosci Lett. 2010;469:6–10. doi: 10.1016/j.neulet.2009.11.033. [DOI] [PubMed] [Google Scholar]
- Palozza P, et al. beta-Carotene and cigarette smoke condensate regulate heme oxygenase-1 and its repressor factor Bach1: relationship with cell growth. Antioxid Redox Signal. 2006;8:1069–80. doi: 10.1089/ars.2006.8.1069. [DOI] [PubMed] [Google Scholar]
- Pereira PJ, et al. Structure of human biliverdin IXbeta reductase, an early fetal bilirubin IXbeta producing enzyme. Nat Struct Biol. 2001;8:215–20. doi: 10.1038/84948. [DOI] [PubMed] [Google Scholar]
- Perluigi M, Butterfield DA. Oxidative Stress and Down Syndrome: A Route toward Alzheimer-Like Dementia. Curr Gerontol Geriatr Res. 2012;2012:724904. doi: 10.1155/2012/724904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perluigi M, et al. In vivo protective effects of ferulic acid ethyl ester against amyloid-beta peptide 1–42-induced oxidative stress. J Neurosci Res. 2006;84:418–26. doi: 10.1002/jnr.20879. [DOI] [PubMed] [Google Scholar]
- Perluigi M, et al. Redox proteomics identification of 4-hydroxynonenal-modified brain proteins in Alzheimer’s disease: Role of lipid peroxidation in Alzheimer’s disease pathogenesis. Proteomics Clin Appl. 2009;3:682–693. doi: 10.1002/prca.200800161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, et al. Alzheimer’s Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology. 2010;74:201–9. doi: 10.1212/WNL.0b013e3181cb3e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi CA. Carbon monoxide, reactive oxygen signaling, and oxidative stress. Free Radic Biol Med. 2008;45:562–9. doi: 10.1016/j.freeradbiomed.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pink RC, et al. Pseudogenes: pseudo-functional or key regulators in health and disease? RNA. 2011;17:792–8. doi: 10.1261/rna.2658311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon HF, et al. Free radicals: key to brain aging and heme oxygenase as a cellular response to oxidative stress. J Gerontol A Biol Sci Med Sci. 2004;59:478–93. doi: 10.1093/gerona/59.5.m478. [DOI] [PubMed] [Google Scholar]
- Pozzoli G, et al. Carbon monoxide as a novel neuroendocrine modulator: inhibition of stimulated corticotropin-releasing hormone release from acute rat hypothalamic explants. Endocrinology. 1994;135:2314–7. doi: 10.1210/endo.135.6.7988414. [DOI] [PubMed] [Google Scholar]
- Pratico D, et al. Increased 8,12-iso-iPF2alpha-VI in Alzheimer’s disease: correlation of a noninvasive index of lipid peroxidation with disease severity. Ann Neurol. 2000;48:809–12. [PubMed] [Google Scholar]
- Pratico D, et al. Increased F2-isoprostanes in Alzheimer’s disease: evidence for enhanced lipid peroxidation in vivo. FASEB J. 1998;12:1777–83. doi: 10.1096/fasebj.12.15.1777. [DOI] [PubMed] [Google Scholar]
- Premkumar DR, et al. Induction of heme oxygenase-1 mRNA and protein in neocortex and cerebral vessels in Alzheimer’s disease. J Neurochem. 1995;65:1399–402. doi: 10.1046/j.1471-4159.1995.65031399.x. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999;45:358–68. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Ravanelli MI, et al. Role of the locus coeruleus carbon monoxide pathway in endotoxin fever in rats. Pflugers Arch. 2007;453:471–6. doi: 10.1007/s00424-006-0136-8. [DOI] [PubMed] [Google Scholar]
- Reed TT, et al. Proteomic identification of HNE-bound proteins in early Alzheimer disease: Insights into the role of lipid peroxidation in the progression of AD. Brain Res. 2009;1274:66–76. doi: 10.1016/j.brainres.2009.04.009. [DOI] [PubMed] [Google Scholar]
- Rockwood K, et al. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol. 2002;59:223–7. doi: 10.1001/archneur.59.2.223. [DOI] [PubMed] [Google Scholar]
- Rodriguez EG, et al. Use of lipid-lowering drugs in older adults with and without dementia: a community-based epidemiological study. J Am Geriatr Soc. 2002;50:1852–6. doi: 10.1046/j.1532-5415.2002.50515.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez F, et al. Effects of exogenous heme on renal function: role of heme oxygenase and cyclooxygenase. Hypertension. 2003;42:680–4. doi: 10.1161/01.HYP.0000085785.40581.1A. [DOI] [PubMed] [Google Scholar]
- Saint-Aubert L, et al. Cortical florbetapir-PET amyloid load in prodromal Alzheimer’s disease patients. EJNMMI Res. 2013;3:43. doi: 10.1186/2191-219X-3-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salinas M, et al. Protein kinase Akt/PKB phosphorylates heme oxygenase-1 in vitro and in vivo. FEBS Lett. 2004;578:90–4. doi: 10.1016/j.febslet.2004.10.077. [DOI] [PubMed] [Google Scholar]
- Scapagnini G, et al. Ethyl ferulate, a lipophilic polyphenol, induces HO-1 and protects rat neurons against oxidative stress. Antioxid Redox Signal. 2004;6:811–8. doi: 10.1089/ars.2004.6.811. [DOI] [PubMed] [Google Scholar]
- Schipper HM. Biomarker potential of heme oxygenase-1 in Alzheimer’s disease and mild cognitive impairment. Biomark Med. 2007;1:375–85. doi: 10.2217/17520363.1.3.375. [DOI] [PubMed] [Google Scholar]
- Schipper HM. Heme oxygenase-1 in Alzheimer disease: a tribute to Moussa Youdim. J Neural Transm. 2011;118:381–7. doi: 10.1007/s00702-010-0436-1. [DOI] [PubMed] [Google Scholar]
- Schipper HM, et al. Glial heme oxygenase-1 expression in Alzheimer disease and mild cognitive impairment. Neurobiol Aging. 2006;27:252–61. doi: 10.1016/j.neurobiolaging.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Schipper HM, et al. Evaluation of heme oxygenase-1 as a systemic biological marker of sporadic AD. Neurology. 2000;54:1297–304. doi: 10.1212/wnl.54.6.1297. [DOI] [PubMed] [Google Scholar]
- Schipper HM, et al. Suppression of glial HO-1 activity as a potential neurotherapeutic intervention in AD. Curr Alzheimer Res. 2009;6:424–30. doi: 10.2174/156720509789207985. [DOI] [PubMed] [Google Scholar]
- Shibahara S, et al. Repression of heme oxygenase-1 expression as a defense strategy in humans. Exp Biol Med (Maywood) 2003;228:472–3. doi: 10.1177/15353702-0322805-08. [DOI] [PubMed] [Google Scholar]
- Shokouhi S, et al. Longitudinal Progression of Cognitive Decline Correlates with Changes in the Spatial Pattern of Brain 18F-FDG PET. J Nucl Med. 2013 doi: 10.2967/jnumed.112.116137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, et al. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A. 1997;94:9866–8. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, et al. Heme oxygenase-1 is associated with the neurofibrillary pathology of Alzheimer’s disease. Am J Pathol. 1994a;145:42–7. [PMC free article] [PubMed] [Google Scholar]
- Smith MA, et al. Advanced Maillard reaction end products, free radicals, and protein oxidation in Alzheimer’s disease. Ann N Y Acad Sci. 1994b;738:447–54. doi: 10.1111/j.1749-6632.1994.tb21836.x. [DOI] [PubMed] [Google Scholar]
- Song F, et al. Plasma biomarkers for mild cognitive impairment and Alzheimer’s disease. Brain Res Rev. 2009;61:69–80. doi: 10.1016/j.brainresrev.2009.05.003. [DOI] [PubMed] [Google Scholar]
- Song R, et al. Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am J Pathol. 2003;163:231–42. doi: 10.1016/S0002-9440(10)63646-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, et al. Over-expression of heme oxygenase-1 promotes oxidative mitochondrial damage in rat astroglia. J Cell Physiol. 2006;206:655–63. doi: 10.1002/jcp.20509. [DOI] [PubMed] [Google Scholar]
- Srikanth V, et al. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging. 2011;32:763–77. doi: 10.1016/j.neurobiolaging.2009.04.016. [DOI] [PubMed] [Google Scholar]
- Steiner AA, Branco LG. Central CO-heme oxygenase pathway raises body temperature by a prostaglandin-independent way. J Appl Physiol. 2000;88:1607–13. doi: 10.1152/jappl.2000.88.5.1607. [DOI] [PubMed] [Google Scholar]
- Steiner AA, Branco LG. Carbon monoxide is the heme oxygenase product with a pyretic action: evidence for a cGMP signaling pathway. Am J Physiol Regul Integr Comp Physiol. 2001;280:R448–57. doi: 10.1152/ajpregu.2001.280.2.R448. [DOI] [PubMed] [Google Scholar]
- Steiner AA, et al. Carbon monoxide as a novel mediator of the febrile response in the central nervous system. Am J Physiol. 1999;277:R499–507. doi: 10.1152/ajpregu.1999.277.2.R499. [DOI] [PubMed] [Google Scholar]
- Steiner AA, et al. Role of the brain heme oxygenase-carbon monoxide pathway in stress fever in rats. Neurosci Lett. 2003;341:193–6. doi: 10.1016/s0304-3940(03)00197-6. [DOI] [PubMed] [Google Scholar]
- Stewart D, et al. Degradation of transcription factor Nrf2 via the ubiquitin-proteasome pathway and stabilization by cadmium. J Biol Chem. 2003;278:2396–402. doi: 10.1074/jbc.M209195200. [DOI] [PubMed] [Google Scholar]
- Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal. 2004;6:841–9. doi: 10.1089/ars.2004.6.841. [DOI] [PubMed] [Google Scholar]
- Stocker R, et al. Antioxidant activity of albumin-bound bilirubin. Proc Natl Acad Sci U S A. 1987a;84:5918–22. doi: 10.1073/pnas.84.16.5918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocker R, et al. Bilirubin is an antioxidant of possible physiological importance. Science. 1987b;235:1043–6. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- Subramaniam R, et al. The lipid peroxidation product, 4-hydroxy-2-trans-nonenal, alters the conformation of cortical synaptosomal membrane proteins. J Neurochem. 1997;69:1161–9. doi: 10.1046/j.1471-4159.1997.69031161.x. [DOI] [PubMed] [Google Scholar]
- Sultana R, Butterfield DA. Role of oxidative stress in the progression of Alzheimer’s disease. J Alzheimers Dis. 2010;19:341–53. doi: 10.3233/JAD-2010-1222. [DOI] [PubMed] [Google Scholar]
- Sultana R, et al. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal. 2006;8:2021–37. doi: 10.1089/ars.2006.8.2021. [DOI] [PubMed] [Google Scholar]
- Sultana R, et al. Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Acta Neuropathol. 2009;118:131–50. doi: 10.1007/s00401-009-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, et al. Ferulic acid ethyl ester protects neurons against amyloid beta- peptide(1–42)-induced oxidative stress and neurotoxicity: relationship to antioxidant activity. J Neurochem. 2005;92:749–58. doi: 10.1111/j.1471-4159.2004.02899.x. [DOI] [PubMed] [Google Scholar]
- Sultana R, et al. Do proteomics analyses provide insights into reduced oxidative stress in the brain of an Alzheimer disease transgenic mouse model with an M631L amyloid precursor protein substitution and thereby the importance of amyloid-beta-resident methionine 35 in Alzheimer disease pathogenesis? Antioxid Redox Signal. 2012;17:1507–14. doi: 10.1089/ars.2011.4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, et al. Heme regulates the dynamic exchange of Bach1 and NF-E2-related factors in the Maf transcription factor network. Proc Natl Acad Sci U S A. 2004;101:1461–6. doi: 10.1073/pnas.0308083100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, et al. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase-1 gene. EMBO J. 2002;21:5216–24. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki A, et al. Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J Neurosci. 2011;31:8786–802. doi: 10.1523/JNEUROSCI.3257-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, et al. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron. 2000;28:461–73. doi: 10.1016/s0896-6273(00)00125-2. [DOI] [PubMed] [Google Scholar]
- Takeda A, et al. Heme catabolism and heme oxygenase in neurodegenerative disease. Antioxid Redox Signal. 2004;6:888–94. doi: 10.1089/ars.2004.6.888. [DOI] [PubMed] [Google Scholar]
- Torp R, Head E, Cotman CW. Ultrastructural analyses of beta-amyloid in the aged dog brain: Neuronal beta-amyloid is localized to the plasma membrane. Progress in Neuro-Psychopharmacology & Biological Psychiatry. 2000a;24:801–810. doi: 10.1016/s0278-5846(00)00107-x. [DOI] [PubMed] [Google Scholar]
- Torp R, Head E, Milgram NW, Hahn F, Ottersen OP, Cotman CW. Ultrastructural evidence of fibrillar β–amyloid associated with neuronal membranes in behaviorally characterized aged dog brains. Neuroscience. 2000b;93:495–506. doi: 10.1016/s0306-4522(99)00568-0. [DOI] [PubMed] [Google Scholar]
- Torp R, et al. Identification of neuronal plasma membrane microdomains that colocalize beta-amyloid and presenilin: implications for beta-amyloid precursor protein processing. Neuroscience. 2003;120:291–300. doi: 10.1016/s0306-4522(03)00320-8. [DOI] [PubMed] [Google Scholar]
- Tudor C, et al. Biliverdin reductase is a transporter of haem into the nucleus and is essential for regulation of HO-1 gene expression by haematin. Biochem J. 2008;413:405–16. doi: 10.1042/BJ20080018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaya J, et al. Effects of heme oxygenase-1 expression on sterol homeostasis in rat astroglia. Free Radic Biol Med. 2007;42:864–71. doi: 10.1016/j.freeradbiomed.2006.12.022. [DOI] [PubMed] [Google Scholar]
- Vitek L. The role of bilirubin in diabetes, metabolic syndrome, and cardiovascular diseases. Front Pharmacol. 2012;3:55. doi: 10.3389/fphar.2012.00055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, et al. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Wolozin B, et al. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–43. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- Wolozin B, et al. Simvastatin is associated with a reduced incidence of dementia and Parkinson’s disease. BMC Med. 2007;5:20. doi: 10.1186/1741-7015-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev. 2005;57:585–630. doi: 10.1124/pr.57.4.3. [DOI] [PubMed] [Google Scholar]
- Yamashita K, et al. Biliverdin, a natural product of heme catabolism, induces tolerance to cardiac allografts. FASEB J. 2004;18:765–7. doi: 10.1096/fj.03-0839fje. [DOI] [PubMed] [Google Scholar]
- Zafrilla P, et al. Oxidative stress in Alzheimer patients in different stages of the disease. Curr Med Chem. 2006;13:1075–83. doi: 10.2174/092986706776360978. [DOI] [PubMed] [Google Scholar]
- Zamrini E, et al. Association between statin use and Alzheimer’s disease. Neuroepidemiology. 2004;23:94–8. doi: 10.1159/000073981. [DOI] [PubMed] [Google Scholar]
- Zenke-Kawasaki Y, et al. Heme induces ubiquitination and degradation of the transcription factor Bach1. Mol Cell Biol. 2007;27:6962–71. doi: 10.1128/MCB.02415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, et al. Carbon monoxide produced by isolated arterioles attenuates pressure-induced vasoconstriction. Am J Physiol Heart Circ Physiol. 2001;281:H350–8. doi: 10.1152/ajpheart.2001.281.1.H350. [DOI] [PubMed] [Google Scholar]
- Zhang F, et al. CO modulates pulmonary vascular response to acute hypoxia: relation to endothelin. Am J Physiol Heart Circ Physiol. 2004;286:H137–44. doi: 10.1152/ajpheart.00678.2002. [DOI] [PubMed] [Google Scholar]