

Abstract
Parkinson’s disease associated mutations in leucine rich repeat kinase 2 (LRRK2) impair mitochondrial function and increase the vulnerability of induced pluripotent stem cell (iPSC)-derived neural cells from patients to oxidative stress. Since mitochondrial DNA (mtDNA) damage can compromise mitochondrial function, we examined whether LRRK2 mutations can induce damage to the mitochondrial genome. We found greater levels of mtDNA damage in iPSC-derived neural cells from patients carrying homozygous or heterozygous LRRK2 G2019S mutations, or at-risk individuals carrying the heterozygous LRRK2 R1441C mutation, than in cells from unrelated healthy subjects who do not carry LRRK2 mutations. After zinc finger nuclease-mediated repair of the LRRK2 G2019S mutation in iPSCs, mtDNA damage was no longer detected in differentiated neuroprogenitor and neural cells. Our results unambiguously link LRRK2 mutations to mtDNA damage and validate a new cellular phenotype that can be used for examining pathogenic mechanisms and screening therapeutic strategies.
Keywords: Parkinson’s disease, LRRK2, mitochondrial DNA damage, stem cells
Introduction
Mutations in leucine-rich repeat kinase 2 (LRRK2) are associated with sporadic and familial forms of Parkinson’s disease (PD) (Paisan-Ruiz et al., 2004; Satake et al., 2009; Simon-Sanchez et al., 2009; Zimprich et al., 2004). Mitochondrial impairment is considered to be a critical factor in the pathogenesis of both sporadic and genetic forms of PD (Henchcliffe and Beal, 2008). Recently, using PD patient-specific induced-pluripotent stem cells (iPSCs) carrying LRRK2 G2019S and R1441C mutations, both LRRK2 mutations were linked to compromised oxidative phosphorylation and mitochondrial dynamics, rendering neural cells more vulnerable to mitochondria-associated stress (Cooper et al., 2012). However, the mechanisms by which LRRK2 mutations lead to a loss of mitochondrial function are poorly understood.
In PD, reactive oxygen species (ROS) damage lipids and proteins (Sherer and Greenamyre, 2005), but less is known about damage to mtDNA (Sanders and Timothy Greenamyre, 2013). DNA damage, defined as any modification of DNA that can alter its coding properties or interfere with normal function in transcription or replication (Lindahl, 1993; Rao, 1993), is distinct from mutations, which are a change in the base sequence of the DNA. However, damage to mtDNA may lead to mtDNA mutations. The mitochondrial genome is particularly susceptible to oxidative damage, likely due to the proximity of mtDNA to ROS production at the inner mitochondrial membrane and the lack of protection afforded by histones (Yakes and Van Houten, 1997). Mitochondrial DNA damage can compromise metabolic functions, predispose to ROS generation and trigger cell death. Accumulation of mtDNA damage is a particular problem for the brain because neurons are post-mitotic and long-lived. In order to study these issues in a neuronal context, we applied cellular reprogramming technology to examine whether LRRK2 mutations lead to mtDNA damage.
Materials and Methods
Induced pluripotent stem cell (iPSC) generation, propagation and differentiation and repair of the LRRK2 G2019S mutation
Institutional Review Boards approved the study. The subjects gave written, informed consent for participation in this study. Fibroblasts from a 4 mm skin punch biopsy were cultured using standard techniques (Byers et al., 2011; Cooper et al., 2012). The fibroblasts were reprogrammed using OCT4, SOX2, KLF4 and CMYC and standard methods (Chan et al., 2009). The additional iPSC lines are available from the Coriell Stem Cell Biobank (LRRK2 R1441C ND34394, ND34393, ND35884; LRRK2 G2019S ND35367) (Cooper et al., 2012). The iPSCs were propagated on irradiated/mitomycin-C inactivated mouse embryonic fibroblasts (Global Stem, Rockville, MD) and manually passaged (Cooper et al., 2012; Nguyen et al., 2011). The details of individual iPSC clones are listed in Supplemental Table 1. ZFN-mediated genomic repair of LRRK2 G2019S in iPSCs was performed.
Assessment of iPSC pluripotency
Teratoma analyses of selected iPSC clones were performed in nonobese diabetic/severe combined immunodeficient mice (Applied StemCell). Karyotypes were analyzed by GTW banding at >400 band resolution (Cytogenetics Laboratory, Stanford University School of Medicine). Immunocytochemistry was performed as described (Mak et al., 2012) using primary antibodies raised against OCT4, SOX2, Tra1-60 and SSEA4 (All from Millipore). Images of the immunofluorescence were taken using the Odyssey Infrared Imaging System (LI-COR Biosciences, USA). For RT-PCR, total RNA was extracted using the RNeasy Micro kit (Qiagen, Valencia, CA) and 150ng RNA was used for reverse-transcription into cDNA using the iScript cDNA Synthesis Kit (BioRad, Hercules, CA). Total reaction volume was 20 μl; the resulting cDNA sample was diluted such that the final cDNA concentration was 18.75ug/uL. 4 μl of the diluted cDNA sample was used as template for qPCR amplification. qPCR was performed using the Biorad CFX96 Real-Time system using Applied Biosystems TaqMan Gene Expression Assays for the following probes: EOMES (Hs00172872_m1), DNMT3B (Hs00171876_m1), FOXD3 (Hs00255287_s1) and FOXA2 (Hs00232764_m1).
iPSC differentiation into neuroprogenitor cells and neural cells
Two differentiation protocols were used to generate cells for analysis of mtDNA damage. The analysis of mtDNA damage across neural cells from multiple patients and healthy subjects (Fig. 1) used a differentiation protocol that had previously been used to determine mitochondrial deficits in neural cells (Cooper et al., 2012). The differentiation of immature neuroprogenitor cells and more mature neural cells from repaired iPSCs for analyses of mtDNA damage was performed as described (Mak et al., 2012). MtDNA damage was increased in iPSC-derived cells carrying LRRK2 mutations differentiated with either protocol.
Fig. 1.
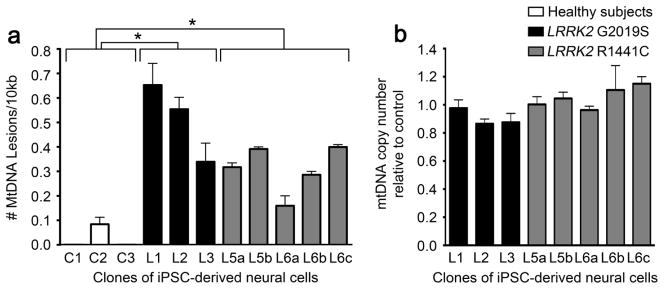
LRRK2 neural cells exhibit greater levels of mtDNA damage than neural cells from healthy subjects. (A) Neural cells were differentiated from iPSCs derived from LRRK2 mutation carriers with the G2019S (black bars) and R1441C (gray bars) mutations and from healthy subjects (white bars). Mitochondrial DNA lesions were increased in neural cells from individual iPSC clones carrying LRRK2 mutations (clones L1–3 and L5–6, Supplemental Table 1) relative to neural cells from healthy subjects’ iPSCs (clones C1–3, Supplemental Table 1). (B) In parallel, neural cells from individuals carrying LRRK2 mutations (black and grey bars) contained a similar number of mtDNA copies as neural cells from healthy subjects. QPCR-based assay was performed in triplicate with three experimental replicates. Data are presented as mean ± SEM.
Immunocytochemistry
Cells were fixed with 4% PFA (EMS, RT15713) for 10 min, washed with PBS (Sigma, T8787), permeabilized with 0.3% Triton x-100 (Sigma, X-100) in PBS for 5 min, and then blocked with 5 % serum (Vector, S-10000) after washing. Cells were incubated with primary antibody overnight at 4°C then incubated with Alexa Fluor 488 (1:200, Invitrogen Molecular Probes, A11029) or Alexa Fluor 555 (1:200, Invitrogen Molecular Probes, A21429). Coverslips were mounted with Vectashield Mounting Medium for Fluorescence with DAPI (Vector Laboratories, H-1200) and imaged on a Nikon Eclipse Ti inverted fluorescence microscope. Antibodies that were used are summarized in (Supplemental Table 2).
DNA isolation and quantification for analysis of mtDNA damage
Coded, pelleted samples of cells were received for blinded analysis. DNA was isolated using a high molecular weight genomic DNA purification kit (QIAGEN Genomic tip). DNA was quantified using the Picogreen dsDNA quantification assay (Molecular Probes). The fluorescence from the Picogreen was measured with a 485 nm emission filter and a 530 nm excitation filter using a microplate reader (SpectraMax Gemini EM). Lambda DNA was used to construct a standard curve in order to determine the concentration of unknown samples. Quality of the DNA prior to QPCR analysis was verified by running the DNA on a 0.6% ethidium bromide-stained agarose gel. Only DNA containing single bands of intact high molecular weight, and which showed negligible evidence of degradation, were used in the DNA damage assays. DNA samples were aliquoted and stored at −20°C. Samples were thawed only once prior to downstream assays.
Quantifying mtDNA damage using the quantitative polymerase chain reaction (QPCR) based assay
Each cell line was independently differentiated three separate times. These three independently generated cell lines were analyzed blinded. The QPCR assay was performed in technical triplicate, which were then averaged, and the statistics performed on the means generated from the three independent experimental values. Cells were collected in lysis buffer, centrifuged at 10,000 × g for 20 min at 4°C. The DNA was precipitated with isopropanol for 48 h at −80°C. The QPCR assay was performed as described (Ayala-Torres et al., 2000; Santos et al., 2006). To confirm that QPCR assays were performed in the linear range, optimal number of cycles for each template was conducted to show that 50% reduction in the amount of template resulted in about 50% reduction in amplification. QPCR products that demonstrated 40–60% in the amplification of the target sequence when using 50% of the original template were considered acceptable.
The PCR amplification for the 16.2 kb fragment of human mtDNA had an annealing/extension time of 14 min for 28 cycles for NPCs and 33 cycles for iPSC-derived neural cells (Ayala-Torres et al., 2000). 10 ng was used to amplify the human large mtDNA fragment and the primer nucleotide sequences were 14841 and 15149 (Ayala-Torres et al., 2000). The final concentration of magnesium in the PCR for human mtDNA fragments was 1.1 mM. To ensure the quality and specificity, all PCR products were resolved on a 0.6% agarose gel and UV light used to visualize ethidium bromide-stained gels. Relative fluorescence of PCR products was quantified using Picogreen. Assuming a random distribution, the Poisson equation was used to calculate the number of DNA lesions. Based on this equation, the amplification is directly proportional to the fraction of undamaged DNA templates. As such, the average lesion frequency per strand is calculated as –lnAD/AO, where AD is the amplification of the damaged or experimental template, while AO is the amplification of the undamaged or control template. Therefore, the results are shown as the number of lesions per strand normalized to 10kb. DNA damage can then be expressed mathematically as the number of DNA lesions per kilobase. All samples were analyzed in a blinded fashion.
QPCR of a small mtDNA fragment
To ensure that the amplification of the large mtDNA fragment was not due to possible changes in mtDNA steady state levels, we amplified a human small mtDNA fragment (248 bp), since the amplification of this small fragment should be independent of damage. The profile for this PCR amplification was as follows: hot start for 10 min at 75°C when the DNA polymerase was added, an initial denaturation step for 1 min at 94°C, followed by 23 cycles for NPCs and iPSC-derived neural cells of denaturation for 1 min at 94°C and then annealing at 60°C for 45 seconds and extension at 72°C for 45 seconds. To complete the profile, a final extension for 10 min at 72°C was performed. The primer nucleotide sequences 14841 and 14620 were used to amplify the human 248-bp mtDNA fragment (Ayala-Torres et al., 2000). 10 ng of DNA template was used to amplify the human small mtDNA fragment. The PCR product was resolved on a 1.5 % agarose gel and UV light used to visualize ethidium bromide-stained gels. Relative fluorescence of PCR products were quantified using Picogreen.
Statistical analysis
Data were analyzed by Student’s t test or ANOVA using computer software (Prism, GraphPad, La Jolla, CA), and p < 0.05 was considered statistically significant.
Results and Discussion
Given the mitochondrial deficits of iPSC-derived neural cells from subjects carrying LRRK2 mutations and the fact that mtDNA damage compromises mitochondrial and neuronal function, we carried out experiments to determine if LRRK2 PD iPSC-derived neural cells accumulate mtDNA damage (Cooper et al., 2012). iPSCs were derived from three patients carrying the homozygous or heterozygous LRRK2 G2019S mutation, two asymptomatic subjects carrying the heterozygous LRRK2 R1441C mutation, and three age-matched healthy subjects without LRRK2 mutations (Supplemental Table 1, Supplemental Fig 1) (Cooper et al., 2012). Multiple iPSC clones were examined from each individual carrying the LRRK2 R1441C mutation. We have previously shown that our protocol for iPSC-derived neural cells yields similar numbers of dopaminergic vs non-dopaminergic functional neurons (Cooper et al., 2012). Upon neuronal differentiation of the iPSC lines, cultures were harvested, pelleted and coded for blinded analysis. After receipt of the coded samples, DNA was purified and a quantitative polymerase chain reaction (QPCR)-based assay specific for the mitochondrial genome determined mtDNA damage. This method is based on the principle that various forms of DNA damage have the propensity to slow down or block DNA polymerase progression (Santos et al., 2006). Thus, if equal amounts of mtDNA from experimental and control specimens are amplified under identical conditions, the mtDNA sample with the least mtDNA damage will produce the greatest amount of PCR product (Santos et al., 2006). Using this approach, a significant increase in levels of mtDNA damage was found in neural cells derived from individuals carrying either the homozygous or heterozygous LRRK2 G2019S (black bars, p<0.0001 ANOVA) or heterozygous R1441C (grey bars, p<0.0001 ANOVA) mutations compared to neural cells from healthy subjects (white bars, Fig 1a). Mitochondrial DNA copy number was similar across all clones (Fig 1b).
While increased levels of mtDNA damage in LRRK2 neural cells were observed across the different pathogenic mutations, across multiple clones from single individuals and in siblings carrying the same mutation (Fig 1), we used two additional approaches to further strenghten our interpretation that LRRK2 mutations induce mtDNA damage. First, we measured mtDNA damage in iPSC-derived neuroprogenitor cells (NPCs) from two brothers. NPCs from a PD patient carrying the heterozygous LRRK2 G2019S mutation (iPSC clone L4a) showed greater levels of mtDNA damage compared to his healthy brother who did not carry the LRRK2 mutation (iPSC clone C3; L4a; Fig 2, p = 0.0002). Second, we addressed the issue of genetic and biological variability between patient and healthy subject control iPSCs. Ideally, isogenic cell lines that differ from the original culture lines only by a disease-causing mutation should be used for study. Without such isogenic lines, there may be difficulties in data interpretation, because personal genomic variation may cause functionally relevant differences between individuals. We therefore used zinc finger nucleases (ZFNs) to repair the LRRK2 G2019S mutation (iPSC clone L4bWT/WT, Supplemental Table 1). In parallel, we used an iPSC clone from the same parental iPSC line (iPSC clone L4a) that was not modified during the ZFN process and had retained the LRRK2 G2019S mutation (unmodified mutant iPSC clone L4eUnmod, Supplemental Table 1). Before assessing mtDNA damage, we differentiated repaired and control unmodified LRRK2 G2019S iPSC clones into neuroprogenitor cells (NPCs) and neural cells (Mak et al., 2012). Immunocytochemistry revealed that while differentiated cultures expressed general neuronal markers (synapsin and alpha-synuclein), there was robust expression of markers for dopaminergic neurons (TH and VMAT2) in both mutant and gene-corrected cultures (Fig 3). Additionally, some neurons expressed GABA, but astroglial cells were not seen. The NPCs and neural cells differentiated from the repaired LRRK2 G2019S iPSCs showed less mtDNA damage than otherwise isogenic cells from the control LRRK2 G2019S unmodified iPSCs (p<0.0001 and p<0.002, respectively; Fig 4a,b). The number of mtDNA genomes in neural cells and NPCs differentiated from repaired and control LRRK2 G2019S iPSCs was similar (1.0 ± 0.009, 1.0 ± 0.002 respectively). Mitochondrial DNA damage levels were also reversed to healthy control levels (Supplemental Fig 2), using a second independently generated ZFN corrected line (iPSC clone L4fWT/WT Supplemental Table 1).
Fig. 2.

Neuroprogenitor cells carrying the LRRK2 G2019S mutation show more mtDNA damage than neuroprogenitor cells from iPSCs of a healthy sibling without the LRRK2 G2019S mutation. (A) Neuroprogenitor cells were differentiated from iPSCs carrying the LRRK2 G2019S mutation (black bar) and from a healthy sibling (white bar). mtDNA lesions were increased in neuroprogenitor cells carrying the LRRK2 G2019S mutation (clone L4a, Supplemental Table 1) relative to mtDNA from healthy sibling (clone C3, Supplemental Table 1). QPCR-based assay was performed in triplicate with three experimental replicates. Data are presented as mean ± SEM, *p = 0.0002.
Fig. 3.

(A–D) Representative images of immunocytochemistry show that differentiated neural cells derived from both mutant LRRK2G2019S/WT and gene-corrected LRRK2WT/WT iPSCs show equivalent expression of general neuronal markers (synapsin and alpha-synuclein) as well as robust expression of dopaminergic markers (TH & VMAT2). While GABA-containing neurons were also seen, astroglia (GFAP) were not (data not shown). Also note that our previous work provided compelling electrophysiological evidence that our protocol produces a true neuronal phenotype (Cooper et al., 2012).
Fig. 4.
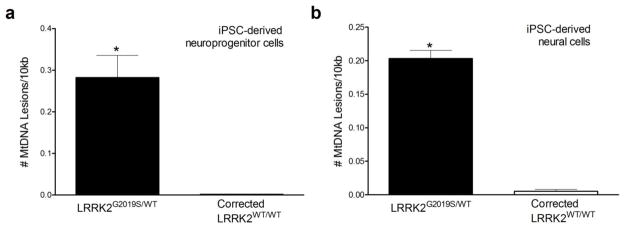
Genomic repair of the LRRK2 G2019S mutation reduced mtDNA damage in neuroprogenitor cells and neural cells. NPCs (A) and neural cells (B) differentiated from iPSCs that retained LRRK2 G2019S mutation after ZFN transfection (clone L4eUnmod, black bar, Supplemental Table 1) exhibited greater levels of mtDNA damage than cells differentiated from ZFN-corrected iPSCs (clone L4bWT/WT, white bar, Supplemental Table 1, *p < 0.002). QPCR-based assay was performed in triplicate with three experimental replicates. Data are presented as mean ± SEM.
Next, we examined whether cellular reprogramming or a neural phenotype was required for the mtDNA damage phenotype observed in differentiated NPCs and neural cells from LRRK2 G2019S carriers. No differences in mtDNA damage levels between healthy subjects and LRRK2 G2019S mutation carriers were detected in either fibroblasts (FBCs clones 1815 and 1828, −0.01 ± 0.005 lesions/10kb vs. −0.01 ± 0.003 lesions/10kb; p = 0.2) or undifferentiated iPSCs (iPSC clones L4bWT/WT and L4eUnmod Supplemental Table 1, −0.01 ± 0.02 lesions/10kb vs. −0.01 ± 0.02 lesions/10kb; p = 0.99). Mitochondrial DNA copy number was similar across all clones (1.02 ± 0.007, 1.0 ± 0.002 FBCs, iPSCs respectively). Therefore, mtDNA damage is only observed in iPSC derived-NPCs and neural cells from individuals carrying the LRRK2 G2019S mutation, and not in FBCs or undifferentiated iPSCs. Thus, the observed mtDNA damage is a neural specific phenotype.
Here we demonstrate that LRRK2 mutations are associated with mtDNA damage, even in neural cells derived from presymptomatic mutation carriers. Thus, mtDNA damage could be an early event in the pathogenesis of PD. The use of ZFNs to repair the genetic mutation in otherwise isogenic cells abrogated the mitochondrial phenotype, thereby providing strong evidence that LRRK2 mutations cause mtDNA damage in neural cells. The mechanisms that result in mitochondrial dysfunction in PD include intrinsically high levels of reactive oxygen species (ROS) (Guzman et al., 2010) and environmental factors that can cause significant oxidative damage and neurodegeneration (Betarbet et al., 2000; Langston et al., 1983). Whether the robust and reproducible mtDNA damage phenotype causes LRRK2 associated mitochondrial dysfunction or is a consequence thereof requires further investigation; however, LRRK2-induced mtDNA damage is a new cellular phenotype and molecular marker of disease-causing LRRK2 mutations. Having an easily quantifiable phenotype will now allow us to dissect cause-effect relationships, underlying mechanisms of LRRK2 pathogenesis and potential responses to therapeutic interventions.
To date, a few reports suggest a modest increase in ROS as a result of pathogenic LRRK2 mutations (Angeles et al., 2011; Heo et al., 2010). In addition, pathogenic LRRK2 mutations can increase susceptibility to oxidative stress (Cooper et al., 2012; Ng et al., 2009; Nguyen et al., 2011). While LRRK2 mutations were not associated with problems regulating O2− levels by iPSC-derived neural cells (Cooper et al., 2012), measurement of ROS levels at different stages of iPSC differentiation would elucidate whether levels of ROS are correlated with levels of mtDNA damage observed in the current study.
Because LRRK2 mutations are a common cause of both sporadic and autosomal dominant PD, examination of the molecular targets of LRRK2 and their potential roles in mtDNA damage is likely to provide critical mechanistic and therapeutic insights into both LRRK2-related and idiopathic PD. The QPCR-based assay we used simultaneously detects a wide variety of types of mtDNA damage, including strand breaks, apurinic/apyrimidinic sites, modified purines and pyridines and DNA repair intermediates. Future studies to identify the specific types of mtDNA damage in mutant LRRK2 neural cells will help identify the critical DNA repair pathways involved and may suggest additional therapeutic targets.
In conclusion, our data demonstrate that mtDNA damage is induced in neural cells by PD-associated mutations in LRRK2, and this phenotype can be functionally reversed or prevented by ZFN-mediated genome editing in iPSCs. These results indicate that mtDNA damage is likely a critical early event in the neuronal dysfunction that leads ultimately to LRRK2-related PD. Moreover, the cellular phenotype described here suggests new avenues for mechanistic exploration and provides a novel platform for screening therapeutics.
Supplementary Material
Highlights.
LRRK2 PD-associated mutations induce mtDNA damage in iPSC-derived neural cells
MtDNA damage is functionally reversed by ZFN-mediated genome editing
MtDNA damage is an early biomarker of LRRK2-related PD neuronal dysfunction
Acknowledgments
We would like to thank members of the Greenamyre, Isacson and Schüle laboratories. This work was supported by grants from the National Institutes of Health T32MH18273 (L.H.S.), 1F32ES019009-01 (L.H.S.), 1R01ES020718 (J.T.G.), the American Parkinson Disease Association (J.T.G), the California Institute for Regenerative Medicine (CIRM TRI-01246 and TTII-019665, J.W.L.), Parkinson Alliance (B.S.), Blume Foundation (B.S.), 1RC2NS070276 (O.I.), 1U24NS078338 (O.I.), the Harvard Stem Cell Institute Miller Consortium for the Development of Nervous System Therapies (O.I.), the Consolidated Anti-Aging Foundation (O.I.), the Poul Hansen Family (O.I.), the Cooper Family (O.I.) and the JPB Foundation (J.T.G.).
Footnotes
Conflict of interest
J.W.L., B.S. and S.H.Z. filed a patent application about the genomic repair of LRRK2 in fibroblasts and iPSCs. The remaining authors declare no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Angeles DC, et al. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum Mutat. 2011;32:1390–7. doi: 10.1002/humu.21582. [DOI] [PubMed] [Google Scholar]
- Ayala-Torres S, et al. Analysis of gene-specific DNA damage and repair using quantitative polymerase chain reaction. Methods. 2000;22:135–47. doi: 10.1006/meth.2000.1054. [DOI] [PubMed] [Google Scholar]
- Betarbet R, et al. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat Neurosci. 2000;3:1301–6. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- Byers B, et al. SNCA triplication Parkinson’s patient’s iPSC-derived DA neurons accumulate alpha-synuclein and are susceptible to oxidative stress. PLoS One. 2011;6:e26159. doi: 10.1371/journal.pone.0026159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan EM, et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat Biotechnol. 2009;27:1033–7. doi: 10.1038/nbt.1580. [DOI] [PubMed] [Google Scholar]
- Cooper O, et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci Transl Med. 2012;4:141ra90. doi: 10.1126/scitranslmed.3003985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman JN, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700. doi: 10.1038/nature09536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–9. doi: 10.1038/ncpneuro0924. [DOI] [PubMed] [Google Scholar]
- Heo HY, et al. LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp Cell Res. 2010;316:649–56. doi: 10.1016/j.yexcr.2009.09.014. [DOI] [PubMed] [Google Scholar]
- Langston JW, et al. Chronic parkinsonism in humans due to a product of meperidine analog synthesis. Science. 1983;219:979–980. doi: 10.1126/science.6823561. [DOI] [PubMed] [Google Scholar]
- Lindahl T. Instability and decay of the primary structure of DNA. Nature. 1993;362:709–15. doi: 10.1038/362709a0. [DOI] [PubMed] [Google Scholar]
- Mak SK, et al. Small molecules greatly improve conversion of human-induced pluripotent stem cells to the neuronal lineage. Stem Cells Int. 2012;2012:140427. doi: 10.1155/2012/140427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng CH, et al. Parkin protects against LRRK2 G2019S mutant-induced dopaminergic neurodegeneration in Drosophila. J Neurosci. 2009;29:11257–62. doi: 10.1523/JNEUROSCI.2375-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HN, et al. LRRK2 mutant iPSC-derived DA neurons demonstrate increased susceptibility to oxidative stress. Cell Stem Cell. 2011;8:267–80. doi: 10.1016/j.stem.2011.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisan-Ruiz C, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Rao KS. Genomic damage and its repair in young and aging brain. Mol Neurobiol. 1993;7:23–48. doi: 10.1007/BF02780607. [DOI] [PubMed] [Google Scholar]
- Sanders LH, Timothy Greenamyre J. Oxidative damage to macromolecules in human Parkinson disease and the rotenone model. Free Radic Biol Med. 2013;62:111–20. doi: 10.1016/j.freeradbiomed.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos JH, et al. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol Biol. 2006;314:183–99. doi: 10.1385/1-59259-973-7:183. [DOI] [PubMed] [Google Scholar]
- Satake W, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet. 2009;41:1303–7. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Sherer TB, Greenamyre JT. Oxidative damage in Parkinson’s disease. Antioxid Redox Signal. 2005;7:627–9. doi: 10.1089/ars.2005.7.627. [DOI] [PubMed] [Google Scholar]
- Simon-Sanchez J, et al. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet. 2009;41:1308–12. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc Natl Acad Sci U S A. 1997;94:514–9. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, et al. Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron. 2004;44:601–7. doi: 10.1016/j.neuron.2004.11.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.