

Summary
Inactivation of selected neurons in vivo can define their contribution to specific developmental outcomes, circuit functions, and behaviors. Here, we show that the optogenetic tool KillerRed selectively, rapidly, and permanently inactivates different classes of neurons in C. elegans in response to a single light stimulus, through generation of reactive oxygen species (ROS). Ablation scales from individual neurons in single animals to multiple neurons in populations, and can be applied to freely behaving animals. Using spatially restricted illumination, we demonstrate that localized KillerRed activation in either the cell body or the axon triggers neuronal degeneration and death of the targeted cell. Finally, targeting KillerRed to mitochondria results in organelle fragmentation without killing the cell, in contrast to cell death observed when KillerRed is targeted to the plasma membrane. We expect this genetic tool to have wide-ranging applications in studies of circuit function, as well as of sub-cellular responses to ROS.
Introduction
Optogenetic approaches to studying the function of specific neurons in vivo generally involve using light to acutely either activate or inactivate the cells of interest. In particular, inactivation in C. elegans of specific neurons in vivo can be used to investigate their contribution to specific network functions and also to study compensation or homeostasis in remaining cells. However, the best-described optogenetic tools for silencing neurons in C. elegans, halorhodopsin (Zhang et al., 2007) and archaerhodopsin (Chow et al., 2010; Okazaki et al., 2012), are light-driven ion pumps (chloride and protons, respectively) that require constant stimulation to keep neurons inactive, and may not be suitable for studying behavior, compensation or homeostasis over longer time scales.
An alternative way to acutely and permanently inactivate specific neurons in vivo is to kill them. This approach has been extensively utilized in C. elegans to define the function of a wide variety of cells, especially neurons, with ablations performed using a laser microbeam (Avery and Horvitz, 1989; 1987; Fang-Yen et al., 2012). However, application of the laser ablation approach is limited because of the difficulty of ablating multiple target cells without damaging surrounding cells or tissues, and the extensive amount of time required to perform surgery on a large number of animals. Thus, the development of optogenetic ablation tools that can be spatially and temporally controlled is highly desirable. An optimal optogenetic ablation tool should kill cells rapidly, avoid collateral damage to neighboring cells and tissues, and scale in application from single neurons to groups of cells in single animals or populations.
KillerRed is a dimeric red fluorescent protein that produces high levels of ROS upon illumination with green light (540-580nm), thereby inducing cell death (Bulina et al., 2006). Structurally, KillerRed resembles other green fluorescence protein (GFP)-like proteins, which comprise an 11 strand antiparallel β-barrel that surrounds a centrally located chromophore (Carpentier et al., 2009; Pletnev et al., 2009). However, KillerRed is 1000 times more toxic than other fluorescent proteins. This higher toxicity is due to the presence of a long water-filled channel in KillerRed that allows diffusion of molecular oxygen near the chromophore, and is thought to provide a path for electron transfer during the production of superoxide radicals (Roy et al., 2010; Serebrovskaya et al., 2009). Similar to generation of GFP fluorescence, the phototoxic activity of KillerRed is effective in vivo and can be induced in mammalian and zebrafish cells without co-expression of other factors (Bulina et al., 2006; Teh et al., 2010).
Here, we demonstrate that KillerRed can efficiently kill a variety of neurons in C. elegans, including sensory neurons, interneurons and motor neurons. We show that the effect of KillerRed is cell-autonomous and does not spread to nearby neurons. Our results indicate that KillerRed disruption of neuronal function is rapid, and can be used to alter circuits and behavior of freely moving animals. In addition the tool is highly modifiable, with ablation efficiency dependent upon illumination duration and intensity. We also demonstrate that certain neurons are resistant to KillerRed damage, raising the intriguing possibility that some cells possess increased intrinsic protection mechanisms against ROS. We further show that subcellular localization of KillerRed dramatically alters its function, with mitochondria localization resulting in specific disruption of organelle morphology. Finally, using selective illumination, we show that exposure of individual neuronal compartments, such as the cell body or the axon, is sufficient to induce neuronal damage and death.
Results
Expressing KillerRed in C. elegans GABA neurons results in light-dependent cell death
We established the consequence of KillerRed activation in different sets of C. elegans neurons, including motor neurons, mechanosensory neurons, chemosensory neurons and interneurons (details of KillerRed constructs and illumination parameters can be found in Table S1). We first generated transgenic animals expressing cytosolic KillerRed under the control of the GABAergic neuron-specific promoter (Punc-47); this promoter drives expression in 26 motor neurons required for coordinated movement (McIntire et al., 1997). Cytosolic KillerRed activation in GABAergic neurons caused a ‘Shrinker’ phenotype (longitudinal shortening of the body upon head touch) in the animals 24 hours after illumination, consistent with loss of GABAergic neuronal function (Figure 1A) (McIntire et al., 1993). Furthermore, neuronal function remained disrupted 72 hours after KillerRed activation (Figure 1A), suggesting that the affected neurons were dead rather than merely damaged. To examine the morphology of KillerRed-expressing neurons before and after KillerRed activation, we co-expressed GFP in the targeted cells (Figure 1B). Twenty-four hours after KillerRed activation, we found that the morphology of the GABAergic neurons was severely disrupted, with axonal processes that were fragmented and broken, often with large blebs, or missing altogether (Figure 1B′). Cell bodies of affected neurons were round, swollen, and appeared smaller than in non-illuminated animals, consistent with neuronal death. To confirm that the morphological changes observed based on GFP fluorescence reflected cell death, we analyzed the GABAergic neurons by DIC microscopy and found that the cell bodies of affected neurons displayed swelling, presence of vacuole-like structures, and nuclear changes (Figure 1C), all of which are phenotypes consistent with cell death.
Figure 1. KillerRed activation disrupts the function and morphology of targeted neurons and results in cell death.
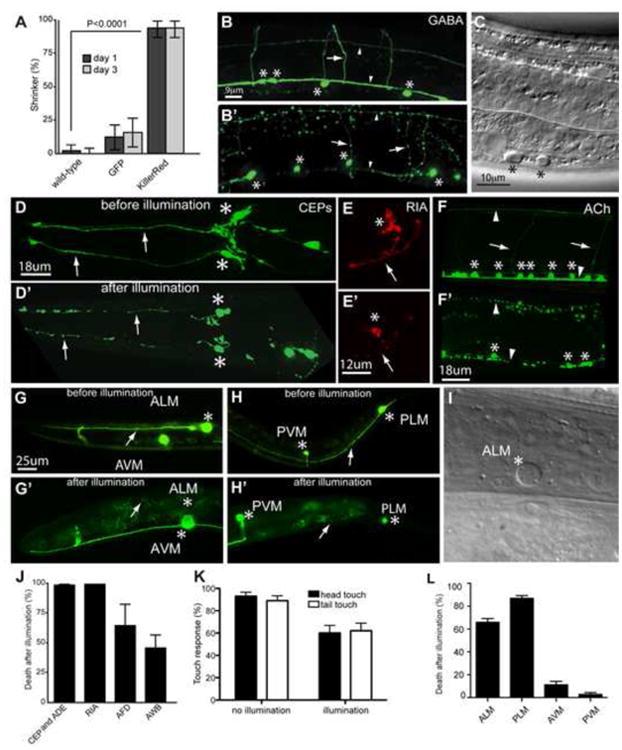
(A) KillerRed activation induces behavioral defects. Animals were illuminated with white light, then scored 24 and 72 hrs later for the shrinker phenotype caused by GABAergic neuron dysfunction. N = 50 for KillerRed and GFP, and 100 for wild type. (B-B′) Confocal images of the GABAergic nervous system of GABA∷KillerRed (Punc-47 ∷KillerRed) animals before (B) and 24 hours after (B′) illumination. (C) Nomarski image of affected GABAergic neuron cell bodies 24 hours after KillerRed activation. (D-D′) CEPs before (D) and 24 hours after (D′) illumination. After illumination the dendrites of CEP neurons (arrows) are fragmented and the cell bodies (asterisks) are smaller and rounded. (E-E′) RIA interneuron before (E) and 24 hours after (E′) illumination, with the cell body and axon degenerated in E′. (F-F′) Cholinergic nervous system before (F) and 24 hours after (F′) illumination. Activation results in fragmentation of neuronal processes and fewer, as well as smaller, cell bodies. (G-H′) Fluorescent images of axonal and neuronal damage in ALM and PLM before (G, H) and 24 hours after illumination (G′, H′). After illumination, ALM is completely cleared (G′), and the PLM axon has disappeared (H′). (I) Nomarski image of ablated ALM cell body 24 hours after illumination. (J) Quantification of cell death 24 hours after illumination. N = 155 cells (CEP and ADE), 48 cells (RIA), 74 cells (AWB), 17 cells (AFD). (K) Light touch response 24 hours after illumination in wild-type and KillerRed expressing animals. After KillerRed activation there is a reduction in the response to light touch in both the head and the tail of the animal. N = 50 animals. (L) Quantification of cell death in touch neurons 24 hours after illumination. N = 107 cells (AVM and PVM), 214 cells (ALM and PLM). In all figures, asterisks mark neuron cell bodies, arrows mark processes, and arrowheads mark dorsal and ventral nerve cords. Error bars indicate 95% confidence intervals.
The observed structural and behavioral defects were specific and dependent on KillerRed expression and activation. Illumination of either wild-type animals, or animals expressing only GFP in GABAergic neurons, did not elicit a shrinker phenotype (Figure 1A). Moreover, expression and illumination of the spectrally similar fluorophore mCherry did not cause a detectable behavioral defect (data not shown). Furthermore, prior to illumination, neurons expressing KillerRed displayed no morphological or functional deficits, indicating that neuronal death was dependent upon KillerRed activation (Figures 1A and 1B). Thus, KillerRed activation in GABAergic neurons permanently disrupts function and results in cell death.
KillerRed can be used to efficiently ablate multiple classes of neurons
Our experiments demonstrated that KillerRed can disrupt neuronal function. Next, we asked whether we could improve the efficiency of disruption. KillerRed is dimeric (Bulina et al., 2006; Carpentier et al., 2009; Pletnev et al., 2009), and we reasoned that increased efficiency might result from expression as a tandem dimer, as for other multimeric fluorescent proteins (Campbell et al., 2002; Shaner et al., 2004). We generated a tandem-dimer version of KillerRed (tdKillerRed) and found that it was significantly more effective than monomeric KillerRed at disrupting GABA neuron function: after illumination with green light (5.1mW/mm2 for 5 min), 100% of tdKillerRed animals (N = 11) exhibited a shrinker phenotype compared to only 14% for monomeric KillerRed (N = 14) (P < 0.0001). We also localized the tandem-dimer form of KillerRed to the plasma membrane using a myristylation tag (myr-tdKillerRed). Membrane targeted dimeric KillerRed had efficiency similar to soluble dimeric KillerRed (92% shrinker, N = 13, P = 1.0). We used myr-tdKillerRed for some of the subsequent experiments (Table S1).
To determine whether other neurons could be targeted by KillerRed, we generated different transgenic strains expressing KillerRed in the dopaminergic neurons CEP, ADE and PDE (Pdat-1∷myr-tdKillerRed), the interneuron RIA (Pglr-3∷myr-tdKillerRed), the cholinergic neurons (approximately 120 neurons; Punc-17∷myr-tdKillerRed), the amphid sensory neurons AWB (Pstr-1∷myr-tdKillerRed) and AFD (Pgcy-8∷myr-tdKillerRed), and the six mechanosensory neurons ALM (L/R), PLM(L/R), AVM and PVM (Pmec-4∷KillerRed). Like the GABAergic motor neurons, the dopaminergic neurons CEP and ADE and the interneuron RIA were efficiently killed after KillerRed activation (Figures 1D, 1E, and 1J). The cholinergic neurons were also efficiently killed, although the maximum efficiency could not be determined as the animals died (Figure 1F). This lethal phenotype is consistent with a requirement for cholinergic neuron function in survival of animals at early developmental stages (Alfonso et al., 1993). By contrast to these cell types, the AWB and AFD amphid neurons were killed with lower efficiency (Figure 1J). The dopaminergic PDE neurons were also killed with lower efficiency (26 out of 60 cells), even though the other dopaminergic neurons were efficiently killed in the same animals. We also tested the efficiency of KillerRed in the mechanosensory neurons, which mediate a specific behavior — response to light touch. We found that expression and activation of KillerRed in the mechanosensory neurons caused deficits in mechanosensation (Figure 1K). Consistent with this loss of touch sensitivity, KillerRed activation also caused efficient neuronal cell death in ALM and PLM neurons (Figures 1G-I and 1L). However, the AVM and PVM neurons were refractory to axonal degeneration or cell death. Thus, KillerRed can ablate many neuronal types in C. elegans, and is effective at targeting single neurons as well as entire neuronal classes, with selected neurons specifically resistant to disruption.
KillerRed induces immediate inactivation of illuminated neurons
Although the morphological effects of KillerRed activation in neuronal cell bodies and processes are not apparent immediately after activation, it is possible that KillerRed disrupts neuronal function more rapidly than its effect on neuronal morphology. To test this, we used two strategies. First, we took advantage of the profound consequence of KillerRed activation in cholinergic neurons, which triggers paralysis in a coiled posture, similar to animals that lack cholinergic neurotransmission (Rand, 1989). To determine the time necessary for neuronal function to be disrupted by KillerRed activation, we quantified this uncoordinated behavior at various times after illumination. This revealed that functional disruption was both immediate and permanent (Figure 2A). Immediately after completion of the 5 minute illumination period, animals exhibited a robust uncoordinated phenotype consistent with loss of cholinergic neuron function. This functional deficit persisted long after activation, suggesting that disruption was not due to temporary inactivation but rather to permanent disruption of neuronal function followed by cell death.
Figure 2. KillerRed activation results in immediate disruption of neuronal function.
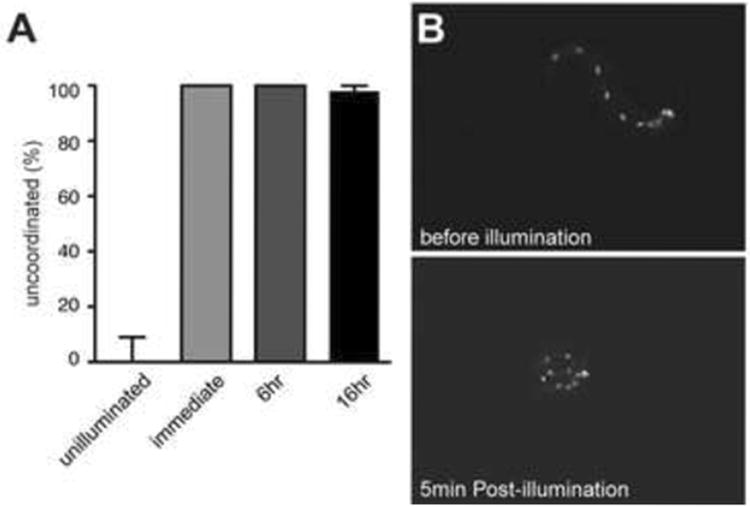
(A) Illumination of animals expressing KillerRed in cholinergic neurons results in immediate and permanent disruption of movement. N = 50 for unilluminated; N = 20 for immediate (just after the end of illumination) and 6 hour timepoints; N = 41 for 16 hour timepoint. Error bars indicate 95% confidence intervals. (B) A freely swimming L1 larva before KillerRed activation in GABAergic motor neurons (top panel). The same larva immediately following 5 min exposure to green light illumination (∼40 mW/mm2) displays uncoordinated locomotion (bottom panel).
Second, we used an optogenetic illumination system that allowed us to expose single freely swimming animals to intense green laser light while monitoring behavior before, during, and after illumination (Leifer et al., 2011). When this system was used to analyze the effect of KillerRed activation in GABAergic motor neurons of freely swimming L1 larvae, we found that activation of KillerRed caused severe deficit in locomotion, observed immediately after the end of the illumination period (Figure 2B; Movies S1 and S2). Together, these experiments demonstrate that KillerRed activation can rapidly and permanently disrupt neuronal function. In addition, our inactivation experiments in the GABAergic neurons of L1 animals suggest that these neurons make a greater contribution to normal locomotion of L1 animals than previously appreciated.
KillerRed-dependent neuronal death is caspase-independent and can be improved by mutating ROS detoxifying enzymes
KillerRed is thought to act as a type II photosensitizer, producing superoxide anion radicals rather than singlet oxygen (Pletnev et al., 2009; Shu et al., 2011). Superoxide is scavenged by superoxide dismutases, which catalyze the conversion of superoxide radicals to hydrogen peroxide and thus limit cellular damage. There are a total of five superoxide dismutases (SODs) in C. elegans, two of which (sod-1 and sod-5) are cytoplasmic Cu/Zn SODs (the remaining three SODs are mitochondrial Fe/Mn SODs (sod-2 and sod-3), or an extracellular Cu/Zn SOD (sod-4) (Landis and Tower, 2005). We found that a sod-1 mutant background, which eliminates one of the cytoplasmic superoxide dismutases, increased the efficiency of ablation in AWB, a neuron normally refractory to KillerRed damage (Figure 3A). These data support a model in which KillerRed exerts its effect via superoxide and demonstrate that the effect of KillerRed in normally resistant cells can be enhanced by specific genetic mutations.
Figure 3. KillerRed phototoxicity is enhanced in superoxide dismutase mutants, and does not require the caspase CED-3.
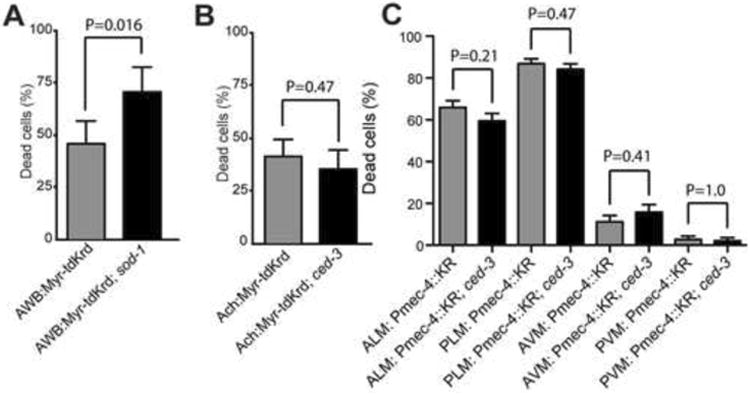
(A) Myr-tdKillerRed activation ablates AWB amphid neurons more effectively in sod-1(tm776) mutant animals. N = 74 cells (wild type); N = 38 cells (sod-1). (B) Neuronal cell death induced by Myr-tdKillerRed activation in cholinergic neurons does not require ced-3(n717). The illumination protocol was adjusted to avoid killing the animals (see Table S1). N = 132 cells (wild type); N = 79 (ced-3). (C) Neuronal cell death induced by cytosolic KillerRed activation in mechanosensory neurons does not require ced-3(n717). For ced-3, N = 95 cells (AVM, PVM), 190 cells (ALM, PLM); for wild type, N = 107 cells (AVM and PVM), 214 cells (ALM and PLM) (same data as Figure 2I). Phenotypes were determined 24 hours after illumination. In all panels, error bars indicate 95% confidence intervals.
In C. elegans, apoptotic cell death requires the caspase CED-3, whereas necrotic cell death does not (Ellis, 1986; Hall et al., 1997). We tested if KillerRed-mediated neuronal cell death occurred through apoptosis by examining the requirement for ced-3. Cell death, as well as the other morphological and behavioral consequences of KillerRed activation, was as robust in ced-3 mutants as in wild type for both acetylcholine motor neurons and mechanosensory neurons (Figures 3B and 3C). Further, the pathology of neuronal cell death induced by KillerRed includes cell swelling, vacuolation, and nuclear changes (Figures 1C and 1I) that are similar to previous observations of necrosis-like neuronal cell death (Hall et al., 1997), in contrast to the compact morphology of apoptotic cells. These observations suggest that KillerRed does not act via an apoptotic pathway and might instead kill neurons through a necrotic, caspase-independent mechanism.
KillerRed effects are cell autonomous and do not spread to adjacent neurons
To test the cell-autonomy of KillerRed cytotoxicity, we examined the morphology of neurons located adjacent to KillerRed targeted cells in three different experiments. First, we examined the AWA and AWB sensory neurons in the head of the animal. The cell bodies of the AWA and AWB neurons are located next to each other within the lateral ganglia, their axons project into the nerve ring, and their dendrites fasciculate with each other (White et al., 1986). We found that activation of KillerRed in the AWB neuron resulted in its structural disruption after illumination, but had no effect on the structure of AWA (Figures 4A and 4B). Second, we examined cholinergic and GABAergic motor neurons. These neurons are synaptic partners; their cell bodies are located near the ventral nerve cord, and their processes fasciculate together in the dorsal and ventral nerve cords (White et al., 1986). We found that KillerRed activation in the GABAergic neurons did not affect the morphology of the cholinergic neurons (Figures 4C and 4D). Furthermore, KillerRed activation in GABAergic neurons caused a GABAergic-specific functional defect (Figure 1A), rather than the coiled paralysis defect caused by activation in cholinergic neurons (Figures 1F and 2). Similar specificity was observed when GABA neurons were imaged after KillerRed activation in the cholinergic neurons — that is, activation in cholinergic neurons did not affect the GABA neurons. Finally, we tested if KillerRed activation in the PLM mechanosensory neuron had any effect on the morphology of the nearby PLN neuron. Both PLM and PLN neurons have their cell bodies in the lumbar ganglia, and their processes run adjacent to each other for the entire posterior half of the animal's body (White et al., 1986). Activation of KillerRed in PLM had no effect on the axon or cell body of PLN (Figures 4E-4G). Together these results indicate that KillerRed-mediated functional disruption and cell damage is cell-autonomous and does not spread beyond the targeted cell (or cells).
Figure 4. KillerRed phototoxicity does not spread to neighboring neurons.
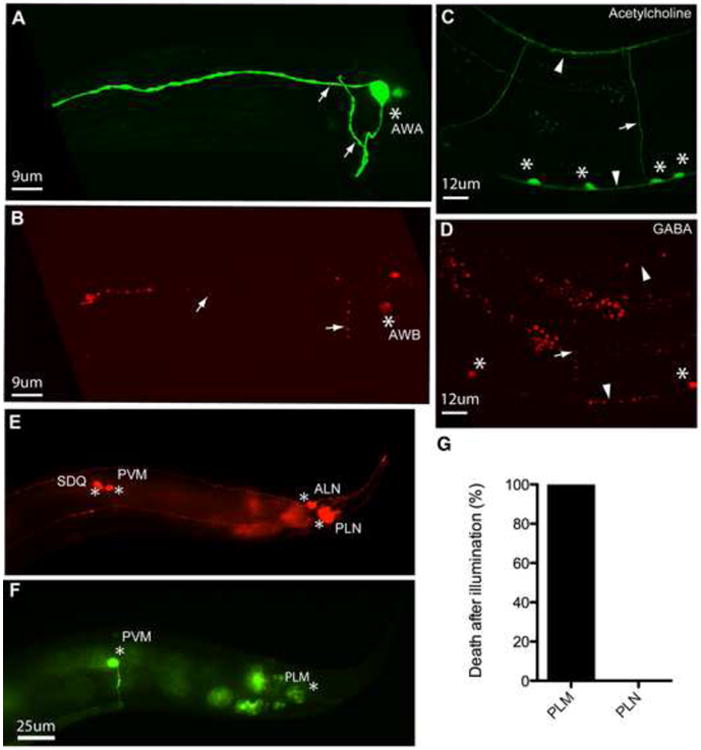
(A) AWA neuron intact after illumination (green = Podr-10 ∷GFP). (B) AWB degenerated neuron in the same animal as (A) (red = Pstr-1 ∷myr-tdKillerRed). (C) Cholinergic neurons after illumination (green = Punc-17∷GFP). (D) Affected GABAergic neurons after illumination in the same animal as (C) (red = Punc-47 ∷myr-tdKillerRed). (E) PLN neuron after illumination (red = Plad-2 ∷mCherry). (F) PLM neurons expressing KillerRed after illumination in the same animal as (E) (green = Pmec-4∷GFP). In all panels, asterisks mark cell bodies, arrows mark processes, arrowheads mark dorsal (top) and ventral (bottom) nerve cords. (G) Quantification of cell death in PLM and PLN neurons in animals expressing KillerRed in PLM only. Animals were scored for PLN death only if PLM had died; thus, PLM death is 100%. N = 25 animals in which both PLM and PLN were scored. In all panels, phenotypes were determined 24 hours after illumination.
Selective illumination results in neuronal cell death
We next investigated the effect of selective illumination on different regions of the animal's body, or on defined regions of individual neurons. For these experiments we chose the mechanosensory neurons ALM and PLM and used a spatially restricted illumination scheme similar to that used previously for optogenetic stimulation and inhibition of neurons (Stirman et al., 2011). Specifically, we first imaged animals for GFP to locate the neurons expressing KillerRed without inducing cell damage; then, using an LCD projector controlled by a LabVIEW program, we illuminated selected regions (such as the anterior half or posterior half of the animal's body, or the cell body or the axon of a single neuron) (Figure 5). Using high magnification lenses with a small focal depth (e.g. 100×), we were able to confine illumination with precision and reproducibility. Targeting the anterior or posterior half of the animal induced cell death of the specific neurons included in respective illuminated regions (Figures 5A-5C). As with the non-targeted illumination, AVM and PVM were resistant to KillerRed-induced damage. When activation was confined to a limited portion of a single neuron, we found that targeting only the cell body of the ALM mechanosensory neuron resulted in cell damage and death (Figures 5D and 5E). Similarly, when the illumination was directed selectively to the axon, we observed robust cell death (>40% of cases), with the remaining animals presenting axonal degeneration in regions beyond the irradiated sections. Thus, even though expression of KillerRed was not spatially limited, regional illumination is enough to produce KillerRed-mediated cell damage and death.
Figure 5. Selected region illumination can be used in combination with KillerRed to induce cell specific killing.
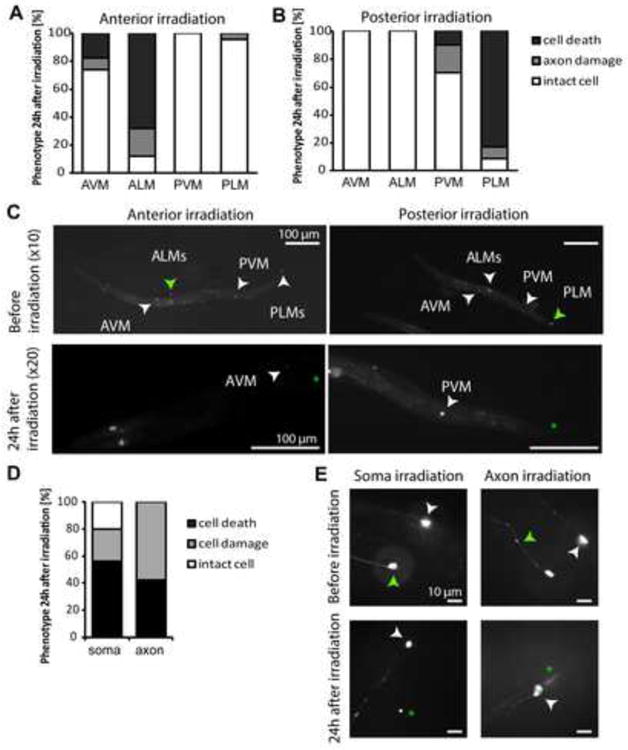
(A, C) Illumination of the anterior half body of the animal causes selective ablation of ALM neurons, while PLM neurons are intact. N = 25 animals. (B, D) Illumination of the posterior half of the animal's body ablated PLM neurons leaving intact ALMs. N = 24 animals. (D, E) Selective illumination, either of the soma or of the axon of ALM neurons, can cause damage and neuronal ablation. N = 25 animals for soma and 19 for axon illumination. In all the images, green arrowhead points to the targeted neuron or axon before illumination, and asterisks to the same cell 24 hours post illumination; white arrowheads point to different neuronal cell bodies in the animal.
Mitochondrial targeting results in organelle-specific effects
The experiments described above show that both membrane targeted and cytoplasmic KillerRed can disrupt neuronal function and kill neurons. To test whether KillerRed function is dependent on subcellular localization, we targeted tandem dimeric KillerRed to mitochondria using the tom-20 targeting sequence, which results in localization of KillerRed to the outer surface of the mitochondrion (Ichishita et al., 2008; Kanaji et al., 2000). We performed these experiments in body wall muscles, because mitochondria in these cells have a well-defined morphology, enabling us to assess the effect of KillerRed on cell structure and function and on the mitochondria themselves. We found that KillerRed was efficiently targeted to mitochondria, and that mitochondria labeled with KillerRed (without activation) had normal morphology consisting of a reticulated network (Figure 6A). Further, animals expressing mitochondria-targeted KillerRed (mtKillerRed) in all their body wall muscles displayed normal movement in the absence of light activation, demonstrating that muscle function was not disrupted by mtKillerRed expression (data not shown). By contrast, when mtKillerRed-expressing animals were exposed to green light, we observed an immediate and dramatic change in mitochondria morphology in muscle. mtKillerRed activation disrupted the reticulated network of mitochondria, and resulted in small spherical mitochondria of various sizes (Figure 6B). Surprisingly, we did not observe behavioral deficits in these animals, suggesting that activation of mtKillerRed did not kill muscle cells (Figure 6G). By contrast, expression of plasma membrane-targeted KillerRed (myrKillerRed) in the same muscle cells resulted in cell death and immediate behavioral deficits after activation (Figures 6E and 6F). Since mitochondrial KillerRed did not kill muscles, we performed studies to test whether mitochondrial morphology could recover after KillerRed-mediated disruption. We found that by 48 hours after disruption, mitochondria had recovered normal morphology (Figures 6C and 6D). Together, these results demonstrate that subcellular targeting of KillerRed enables the generation of local and specific cell-biological phenotypes.
Figure 6. KillerRed activation at mitochondria results in organelle damage but cell survival.
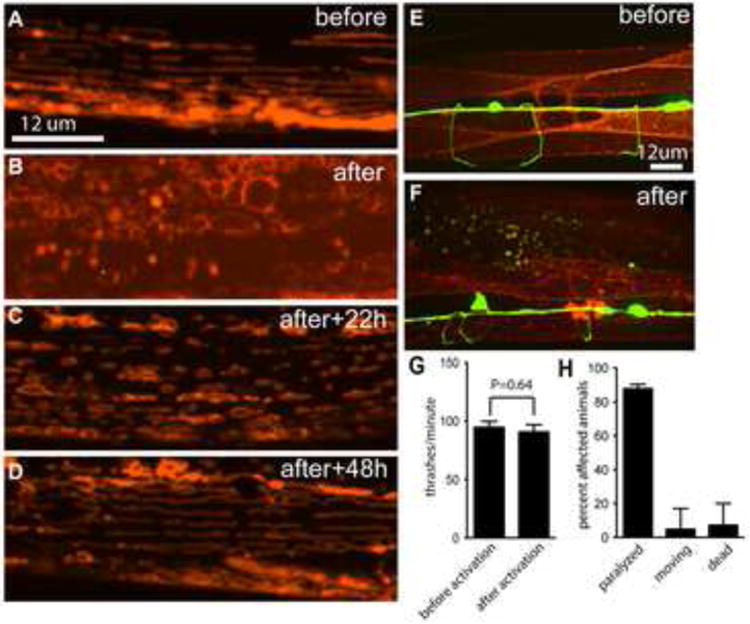
(A-D) Mitochondria in body wall muscles express mtKillerRed and were imaged before (A) or at various times after (B-D) illumination. (E, F) Body wall muscles express plasma membrane KillerRed and were imaged before (E) or 16 hours after (F) illumination. (GABA neurons express GFP in (e) and (f)) (G) Average thrash rate in liquid of animals expressing mitochondria KillerRed in muscles, before and after illumination, corresponding to panels (A) and (B). N = 9 for before illumination and 10 for after illumination; error bars indicate SEM. (H) Phenotype distribution of animals expressing plasma membrane KillerRed in muscles after illumination, corresponding to panel (F). N = 41.
Use of KillerRed with other optical tools
A number of optical tools are currently in use in C. elegans, including the optogenetic tool channelrhodopsin (Nagel et al., 2005) and the reactive oxygen generator miniSOG (Qi et al., 2012). Because KillerRed is excited by green light while these other tools are excited by blue light, we considered that it might be possible to address them separately in a single animal by using different illumination wavelengths. To determine the feasibility of this idea, we compared the requirements for activation of KillerRed, channelrhodopsin, and miniSOG in the cholinergic motor neurons. The activity of all three tools has been described in these cells (Figures 1F and 2) (Liu et al., 2009a; Qi et al., 2012; Zhang et al., 2007). First, we determined the feasibility of using KillerRed in combination with miniSOG. We irradiated animals expressing miniSOG in cholinergic motor neurons (CZ14527), using 0.57mW/mm2 of blue light for 30 minutes as described (Table S1) (Qi et al., 2012). This treatment affected 100% of the animals: 42% were paralyzed, and the remaining animals could move but were sluggish. Similarly, we irradiated animals expressing KillerRed in cholinergic neurons with the same intensity (0.57 mW/mm2) and time of green light and also observed 100% affected animals, with 76% paralysis and 24% sluggish movement (Figure 7A). Next, we swapped the illumination conditions — that is, we illuminated miniSOG animals with 0.57 mW/mm2 of green light and KillerRed animals with 0.57 mW/mm2 of blue. This treatment had little to no effect, although approximately 6% of KillerRed animals did react to blue light illumination (Figure 7A). Finally, we asked whether higher-intensity activation could be used for miniSOG. We had previously observed that C. elegans can tolerate green light at 46mW/mm2 for 15 minutes with no adverse effects (other than KillerRed activation) (Figure 1). However, exposing wild type worms (N = 20) to blue light at 8mW/mm2 for 10 minutes resulted in 100% lethality, consistent with previous results (Edwards et al., 2008). Together, these data suggest that KillerRed and miniSOG can be separately addressed in vivo.
Figure 7. KillerRed can be used in multimodal optical experiments.
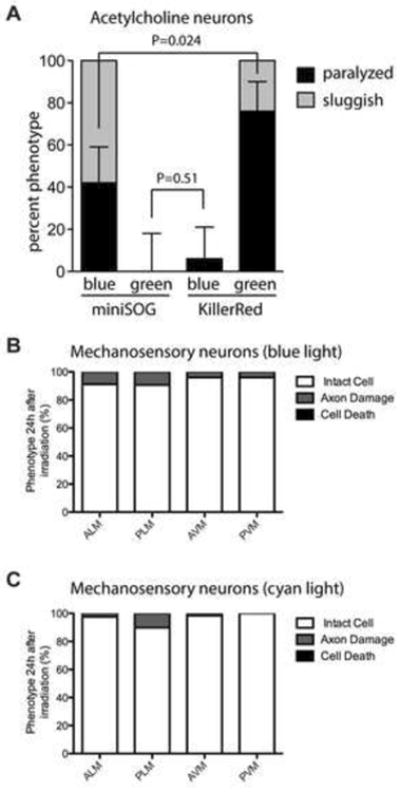
(A) Comparasion of effects of KillerRed and miniSOG in acetylcholine neurons, when illuminated with either blue or green light. N = 33 for miniSOG/blue; N = 21 for miniSOG/green; N = 32 for KillerRed/blue; N = 21 for KillerRed/green. Error bars show 95% confidence interval for the paralyzed phenotype. Phenotypes were determined 10 minutes after illumination. (B-C) Effect of KillerRed in mechanosensory neurons, when illuminated with either blue (b) or cyan (c) light. N = 102 for blue light; N = 64 for cyan light. Phenotypes were determined 24 hours after illumination.
Next, we determined the feasibility of using KillerRed in conjunction with channelrhodopsin. Again, we focused on the cholinergic motor neurons, which are a well-descibed site for channelrhodopsin activation (Liu et al., 2009b; Zhang et al., 2007). We tested whether activation of channelrhodopsin could be performed independently of KillerRed activation. We activated channelrhodopsin in cholinergic motor neurons using 0.47 mW/mm2 of blue light, which caused 100% of animals to stop moving (N = 23). By contrast, 2.2 mW/mm2 of green light had no effect on these animals (N = 22). As we previously determined that an even lower intensity of green light could be used to activate KillerRed, while a higher intensity of blue light could be used without activating KillerRed (Figure 7A), these experiments indicate that KillerRed can be used in multimodal experiments with channelrhodopsin. Finally, we confirmed these experiments in mechanosensory neurons. As in the cholinergic neurons, we found that prolonged illumination of KillerRed strains at the appropriate wavelength and intensity to activate channelrhodopsin had very little effect, although axon damage was observed in a small percentage of animals (Figure 7B and 7C). These data confirm that KillerRed can be used in conjunction with channelrhodopsin in C. elegans, as well as in conjunction with other fluorescent tools that are excited by blue or cyan light.
Discussion
KillerRed is a new tool in the C. elegans optogenetic toolkit that can kill and inactivate cells in populations of animals with extremely high temporal and spatial control. The effect of KillerRed is immediate and cell-autonomous with no spreading of killing, or even damage, to surrounding cells or tissues. This critical property makes KillerRed a powerful biological tool that can be applied in a wide variety of cellular perturbation studies in vivo, at scales ranging from single to multiple cells in both single animals and populations. For example, the rapidity and efficacy of this genetically encoded cell-ablation reagent now allows investigators to examine the acute effects of the disruption of specific neurons during behavioral performance. KillerRed can also be used for longer behavioral studies, since ablated neurons never recover function. For example, our data indicate that the GABA neurons have an essential function in the locomotion of L1 larvae. Similarly, our data indicate that ablation of acetylcholine neurons in L4 stage animals results in relatively rapid lethality (within 24 hours of ablation), suggesting that C. elegans has an acute requirement for cholinergic neurotransmission for viability. One potential concern for such behavioral studies is that because KillerRed damages and kills neurons, rather than merely silencing them, it is possible that secondary effects on the remaining cells in the network might complicate analysis. However, our data demonstrate that, at least for the neurons assessed here, the effects of ablation are restricted to the cells that express KillerRed.
Further, because KillerRed is excited by green light, it can be used in combination with other optogenetic tools that are excited by blue light, such as GFP, GCaMP, Channelrhodopsin, and miniSOG. The green light activation of KillerRed may have some additional practical advantages in C. elegans, as worms tolerate high levels of green light, but actively avoid — and can be killed by — equivalent amounts of blue light (Edwards et al., 2008). Two additional applications stemming from our selective illumination experiments are (i) the possibility of ablating a neuron “from a distance” by targeting a region of its axon (this is particularly important when using KillerRed expressed in cells whose cell bodies are in very close proximity and therefore more difficult to target individually), and (ii) the possibility of investigating ROS effects in axonal sections.
Our results also indicate that subcellular localization of KillerRed can be exploited to study specific cell-biological functions. Targeting and activating KillerRed at the plasma membrane is sufficient to inactivate and kill cells. This rapid and lethal effect could be due to the susceptibility of membrane lipids to oxidation. For example, unsaturated lipids are highly susceptible to oxidative stress, resulting in the generation of toxic compounds such as hydroxynonenal and acrolein (Catalá, 2010; Fatokun et al., 2008). In addition to causing loss of membrane fluidity and destruction of the plasma membrane barrier, these toxic compounds react with membrane proteins resulting in inactivation of critical proteins such as the neuronal glucose transporter, the glutamate transporter, and Na+/K+ ATPases (Barnham et al., 2004; Keller et al., 2002; Mark et al., 1995; Smith et al., 2009). By contrast, targeting and activating KillerRed at mitochondria results in specific disruption (and eventual recovery) of organelle morphology, without killing cells. A potential explanation for the local effect of KillerRed on mitochondria is that ROS are quenched rapidly and thus the effective diffusion rate is very low. Alternatively, it is possible that the TOM20 tag affects KillerRed function, allowing for mitochondrial damage but preventing cell killing. Interestingly, another optogenetic reactive oxygen generator, miniSOG, requires mitochondria targeting to kill neurons (Qi et al., 2012). miniSOG generates mainly singlet oxygen, while KillerRed generates mainly superoxide (Pletnev et al. 2009; Shu et al. 2011; this work). These data suggest that different reactive oxygen species may affect mitochondria and cell function in different ways. KillerRed thus enables the dissection of local effects of reactive oxygen species.
ROS are toxic cellular molecules that need to be kept in check for a neuron to function over a long period of time, and ROS accumulation correlates with a variety of neurodegenerative diseases (DiMauro and Schon, 2008; Uttara et al., 2009). The nature of the cytotoxicity of KillerRed, a generator of ROS, therefore offers a significant opportunity to genetically investigate the mechanisms that regulate cellular responses to ROS. Our findings show that selective neurons, such as AVM, PVM and AWB, are resistant to damage caused by KillerRed activation and ROS. KillerRed expression in PVM is weaker than that in other neurons, so the reduced killing effect could be due to the presence of fewer molecules in the cell. However, AVM and AWB express KillerRed at levels comparable to those of other neurons, suggesting that they might have a higher intrinsic capacity to buffer ROS. One such mechanism could be a higher expression of molecules, such as SOD-1, that are able to catabolize ROS into non-toxic elements. Our data demonstrating that a mutation in SOD-1 is able to increase the cytotoxic effect of KillerRed activation in AWB provide support for this idea, although additional mechanisms might also be in place. We expect the application of KillerRed shown here will allow a greater flexibility and control for genetic studies of both neuronal function and ROS-induced neuronal damage in vivo.
Experimental Procedures
Strains and Molecular Biology
Nematodes were cultured using standard methods. All experiments were performed at 22°C except where otherwise noted. A C. elegans version of the KillerRed coding sequence was synthesized (GenScript) using worm-optomized codons and synthetic introns (Fire et al., 1990; Stenico et al., 1994). Flanking regions for Multisite Gateway (Invitrogen) were included and the KillerRed construct was placed in pDONR221. Tandem dimer KillerRed (tdKillerRed) was generated from this monomeric construct using the same linker sequences as tdTomato (Serebrovskaya et al., 2009; Shaner et al., 2004). tdKillerRed was targeted to the plasma membrane through the addition of a myristoylation tag to generate myr-tdKillerRed (Gitai et al., 2003). Promoters were cloned into the [4-1] slot of Multisite Gateway. Final expression constructs were generated with LR clonase into the destination vector pCFJ150 (Frøkjær-Jensen et al., 2010). For experiments using the mechanosensory neurons the following transgenes and mutations were used: zdIs5(Pmec-4 ∷GFP), vdEx405[Pmec-4∷KillerRed (20ng/μl) + odr-1∷DsRED (30ng/μl)], LGIV ced-3(n717), vdEx167[Plad-2∷mCherry (25ng/μl) + Podr-1∷dsRED (30 ng/μl)] (Neumann et al., 2011). Standard molecular biology methods were used. The Pmec-4∷KR construct was generated by cloning a KillerRed cDNA (Evrogen) into a pSM vector (a derivative of pPD49.26 (Fire et al., 1990), a kind gift from Steve McCarroll and Cori Bargmann) containing Pmec-4. KillerRed was amplified with primers containing BamHI restrictions sites and inserted into pSM Pmec-4 digested with the same enzyme.
DNA constructs were injected into the germline using standard methods (Mello et al., 1991). Expression of KillerRed in the correct cells was verified by fluorescence microscopic examination.
KillerRed activation and imaging
Light from a 200 W mercury bulb (PhotoFluor II, 89 North) was filtered with a 562/20 nm filter. The resulting green light was trained into a 5 mm liquid-light guide. The output from the liquid-light guide was directed through a collimating lens (LA1951-A, ThorLabs) and a focusing lens (LA1027-A). Light intensity was measured with a powermeter (Thor Labs, PM100A). Illuminations were done in upside-down PCR tube caps in 30μl of M9 medium on ice to minimize sample heating. L4 stage transgenic worms were used in all experiments. Animals were allowed to crawl on unseeded plates prior to the experiment to remove excess bacteria. Different neuronal cell types required different illumination protocols to result in optimal killing. Transgenic lines carrying myr-tdKillerRed, illumination times and light intensities are shown in Table S1. An UltraVIEW VoX (PerkinElmer) spinning disc mounted on a Nikon Ti-E Eclipse inverted microscope and a 60× CFI Plan Apo, NA 1.0 oil objective were used to capture Z stacks of non-illuminated and illuminated neurons. Cellular damage in illuminated neurons was documented 18-24 hrs after illumination.
Illumination of mechanosensory neurons was performed on animals immobilized on NGM agar plates using tetramisole hydrochloride (0.03%), using a Leica MZ10F fluorescence dissecting microscope using an EtDsRed filter (green light 530-560 nm) with an 80× magnification (intensity 3.8mW/mm2). 24 hrs after irradiation animals were mounted on 4% agar pads and epifluorescence was used to visualize animals with a Zeiss Axioimager Z1 and a Zeiss Axioimager A1 microscope. A Photometrics camera, Cool snap HQ2, was used for imaging. Metamorph software was used to analyze the collected images.
The selective illumination using the LCD projector system is as described in Stirman et al., 2011 Nat Meth. GFP fluorescence was used to image the cells and define the location of the soma and axon. A green filter (543-593nm) was used to activate KR in regions of interest selected after imaging. Typical power densities are 2-3 mW/mm2. In all experiments performed using this system illumination time was 1 hr. The length of the axonal processes that were selectively illuminated was 80 μm in all cases. Extensive degeneration was defined as damage that extended beyond 10% of the illuminated length along the axon.
Acute KillerRed activation using freely swimming worms was performed on a modified version of the COLBERT system (Leifer et al., 2011). Briefly, a freely moving L1 larva was imaged with a 20× microscope objective on an inverted Nikon microscope (TE2000). The fluorescence video was taken by a CoolSNAP CCD camera (Photometrics, Tucson, Arizona). A digital micromirror device was used to reflect green laser light to illuminate targeted regions of the worm, inducing KillerRed activation in GABAergic motor neurons.
Activation of miniSOG was performed using the same Photofluor II system as for KillerRed, but with a filter at the appropriate wavelength (Table S1).
Statistics
Statistical comparisons were made using the two-tailed Fisher's exact test (http://www.graphpad.com/quickcalcs/contingency1/). 95% confidence intervals were calculated using http://www.graphpad.com/quickcalcs/confInterval1/.
Supplementary Material
Highlights.
KillerRed activation causes immediate and permanent silencing of neuronal function
KillerRed can be used to acutely manipulate neuronal circuits
KillerRed activation results in cell death of targeted neurons
Localizing KillerRed to mitochondria produces organelle-specific effects
Acknowledgments
We thank Brent Neumann, Rosina Giordano-Santini, and Rowan Tweedale for reading the manuscript, and Luke Hammond for technical assistance.
Work in the Hammarlund lab is supported by the Ellison Medical Foundation and NIH R01NS066082 to M.H. Work in the Hilliard lab is supported by the NHMRC (Project Grants # 569500 and 631634), and ARC (Future Fellowship to M.A.H.); an Australian Postgraduate Award supported S.C. Work in the Lu lab is supported by the NSF, NIH (R01GM088333, R21EB012803, R01AG035317), Sloan Foundation, and Human Frontiers Science Program to H.L. Work in the Samuel lab is supported by an NIH Pioneer Award, the NSF, and the Harvard-MIT Joint Research Grants Program. The American Cancer Society PF-07-037-01-CSM supported D.C.W.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alfonso A, Grundahl K, Duerr JS, Han HP, Rand JB. The Caenorhabditis elegans unc-17 gene: a putative vesicular acetylcholine transporter. Science. 1993;261:617–619. doi: 10.1126/science.8342028. [DOI] [PubMed] [Google Scholar]
- Avery L, Horvitz HR. Pharyngeal pumping continues after laser killing of the pharyngeal nervous system of C. elegans. Neuron. 1989;3:473–485. doi: 10.1016/0896-6273(89)90206-7. [DOI] [PubMed] [Google Scholar]
- Avery L, Horvitz HR. A cell that dies during wild-type C. elegans development can function as a neuron in a ced-3 mutant. Cell. 1987;51:1071–1078. doi: 10.1016/0092-8674(87)90593-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnham KJ, Masters CL, Bush AI. Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov. 2004;3:205–214. doi: 10.1038/nrd1330. [DOI] [PubMed] [Google Scholar]
- Bulina ME, Chudakov DM, Britanova OV, Yanushevich YG, Staroverov DB, Chepurnykh TV, Merzlyak EM, Shkrob MA, Lukyanov S, Lukyanov KA. A genetically encoded photosensitizer. Nat Biotechnol. 2006;24:95–99. doi: 10.1038/nbt1175. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier P, Violot S, Blanchoin L, Bourgeois D. Structural basis for the phototoxicity of the fluorescent protein KillerRed. FEBS Lett. 2009;583:2839–2842. doi: 10.1016/j.febslet.2009.07.041. [DOI] [PubMed] [Google Scholar]
- Catalá A. A synopsis of the process of lipid peroxidation since the discovery of the essential fatty acids. Biochemical and Biophysical Research Communications. 2010;399:318–323. doi: 10.1016/j.bbrc.2010.07.087. [DOI] [PubMed] [Google Scholar]
- Chow BY, Han X, Dobry AS, Qian X, Chuong AS, Li M, Henninger MA, Belfort GM, Lin Y, Monahan PE, et al. High-performance genetically targetable optical neural silencing by light-driven proton pumps. Nature. 2010;463:98–102. doi: 10.1038/nature08652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMauro S, Schon EA. Mitochondrial Disorders in the Nervous System. Annu Rev Neurosci. 2008;31:91–123. doi: 10.1146/annurev.neuro.30.051606.094302. [DOI] [PubMed] [Google Scholar]
- Edwards SL, Charlie NK, Milfort MC, Brown BS, Gravlin CN, Knecht JE, Miller KG. A Novel Molecular Solution for Ultraviolet Light Detection in Caenorhabditis elegans. PLoS Biol. 2008;6:e198. doi: 10.1371/journal.pbio.0060198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis H. Genetic control of programmed cell death in the nematode C. elegans. Cell. 1986;44:817–829. doi: 10.1016/0092-8674(86)90004-8. [DOI] [PubMed] [Google Scholar]
- Fang-Yen C, Gabel CV, Samuel ADT, Bargmann CI, Avery L. Laser microsurgery in Caenorhabditis elegans. Methods Cell Biol. 2012;107:177–206. doi: 10.1016/B978-0-12-394620-1.00006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatokun AA, Stone TW, Smith RA. Oxidative stress in neurodegeneration and available means of protection. Front Biosci. 2008;13:3288–3311. doi: 10.2741/2926. [DOI] [PubMed] [Google Scholar]
- Fire A, Harrison SW, Dixon D. A modular set of lacZ fusion vectors for studying gene expression in Caenorhabditis elegans. Gene. 1990;93:189–198. doi: 10.1016/0378-1119(90)90224-f. [DOI] [PubMed] [Google Scholar]
- Frøkjær-Jensen C, Davis MW, Hollopeter G, Taylor J, Harris TW, Nix P, Lofgren R, Prestgard-Duke M, Bastiani M, Moerman DG, et al. Targeted gene deletions in C. elegans using transposon excision. Nat Meth. 2010;7:451–453. doi: 10.1038/nmeth.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitai Z, Yu TW, Lundquist EA, Tessier-Lavigne M, Bargmann CI. The netrin receptor UNC-40/DCC stimulates axon attraction and outgrowth through enabled and, in parallel, Rac and UNC-115/AbLIM. Neuron. 2003;37:53–65. doi: 10.1016/s0896-6273(02)01149-2. [DOI] [PubMed] [Google Scholar]
- Hall DH, Gu G, García-Añoveros J, Gong L, Chalfie M, Driscoll M. Neuropathology of degenerative cell death in Caenorhabditis elegans. J Neurosci. 1997;17:1033–1045. doi: 10.1523/JNEUROSCI.17-03-01033.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichishita R, Tanaka K, Sugiura Y, Sayano T, Mihara K, Oka T. An RNAi screen for mitochondrial proteins required to maintain the morphology of the organelle in Caenorhabditis elegans. J Biochem. 2008;143:449–454. doi: 10.1093/jb/mvm245. [DOI] [PubMed] [Google Scholar]
- Kanaji S, Iwahashi J, Kida Y, Sakaguchi M, Mihara K. Characterization of the signal that directs Tom20 to the mitochondrial outer membrane. Journal of Cell Biology. 2000;151:277–288. doi: 10.1083/jcb.151.2.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP. Impairment of Glucose and Glutamate Transport and Induction of Mitochondrial Oxidative Stress and Dysfunction in Synaptosomes by Amyloid β-Peptide: Role of the Lipid Peroxidation Product 4-Hydroxynonenal. J Neurochem. 2002;69:273–284. doi: 10.1046/j.1471-4159.1997.69010273.x. [DOI] [PubMed] [Google Scholar]
- Landis GN, Tower J. Superoxide dismutase evolution and life span regulation. Mech Ageing Dev. 2005;126:365–379. doi: 10.1016/j.mad.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Leifer AM, Fang-Yen C, Gershow M, Alkema MJ, Samuel ADT. Optogenetic manipulation of neural activity in freely moving Caenorhabditis elegans. Nat Meth. 2011;8:147. doi: 10.1038/nmeth.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Hollopeter G, Jorgensen EM. Graded synaptic transmission at the Caenorhabditis elegans neuromuscular junction. Proc Natl Acad Sci USA. 2009a;106:10823–10828. doi: 10.1073/pnas.0903570106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Hollopeter G, Jorgensen EM. Graded synaptic transmission at the Caenorhabditis elegans neuromuscular junction. Proc Natl Acad Sci USA. 2009b;106:10823–10828. doi: 10.1073/pnas.0903570106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid beta-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntire SL, Jorgensen E, Kaplan J, Horvitz HR. The GABAergic nervous system of Caenorhabditis elegans. Nature. 1993;364:337–341. doi: 10.1038/364337a0. [DOI] [PubMed] [Google Scholar]
- McIntire SL, Reimer RJ, Schuske K, Edwards RH, Jorgensen EM. Identification and characterization of the vesicular GABA transporter. Nature. 1997;389:870–876. doi: 10.1038/39908. [DOI] [PubMed] [Google Scholar]
- Nagel G, Brauner M, Liewald JF, Adeishvili N, Bamberg E, Gottschalk A. Light Activation of Channelrhodopsin-2 in Excitable Cells of Caenorhabditis elegans Triggers Rapid Behavioral Responses. Current Biology. 2005;15:2279–2284. doi: 10.1016/j.cub.2005.11.032. [DOI] [PubMed] [Google Scholar]
- Neumann B, Nguyen KCQ, Hall DH, Ben-Yakar A, Hilliard MA. Axonal regeneration proceeds through specific axonal fusion in transected C. elegans neurons. Dev Dyn. 2011;240:1365–1372. doi: 10.1002/dvdy.22606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki A, Sudo Y, Takagi S. Optical Silencing of C. elegans Cells with Arch Proton Pump. PLoS ONE. 2012;7:e35370. doi: 10.1371/journal.pone.0035370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pletnev S, Gurskaya NG, Pletneva NV, Lukyanov KA, Chudakov DM, Martynov VI, Popov VO, Kovalchuk MV, Wlodawer A, Dauter Z, et al. Structural basis for phototoxicity of the genetically encoded photosensitizer KillerRed. J Biol Chem. 2009;284:32028–32039. doi: 10.1074/jbc.M109.054973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi YB, Garren EJ, Shu X, Tsien RY, Jin Y. Photo-inducible cell ablation in Caenorhabditis elegans using the genetically encoded singlet oxygen generating protein miniSOG. Proc Natl Acad Sci USA. 2012;109:7499–7504. doi: 10.1073/pnas.1204096109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand JB. Genetic analysis of the cha-1-unc-17 gene complex in Caenorhabditis. Genetics. 1989;122:73–80. doi: 10.1093/genetics/122.1.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A, Carpentier P, Bourgeois D, Field M. Diffusion pathways of oxygen species in the phototoxic fluorescent protein KillerRed. Photochem Photobiol Sci. 2010;9:1342–1350. doi: 10.1039/c0pp00141d. [DOI] [PubMed] [Google Scholar]
- Serebrovskaya EO, Edelweiss EF, Stremovskiy OA, Lukyanov KA, Chudakov DM, Deyev SM. Targeting cancer cells by using an antireceptor antibody-photosensitizer fusion protein. Proc Natl Acad Sci USA. 2009;106:9221–9225. doi: 10.1073/pnas.0904140106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BNG, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Shu X, Lev-Ram V, Deerinck TJ, Qi Y, Ramko EB, Davidson MW, Jin Y, Ellisman MH, Tsien RY. A genetically encoded tag for correlated light and electron microscopy of intact cells, tissues, and organisms. PLoS Biol. 2011;9:e1001041. doi: 10.1371/journal.pbio.1001041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AJ, Smith RA, Stone TW. 5-Hydroxyanthranilic Acid, a Tryptophan Metabolite, Generates Oxidative Stress and Neuronal Death via p38 Activation in Cultured Cerebellar Granule Neurones. Neurotox Res. 2009;15:303–310. doi: 10.1007/s12640-009-9034-0. [DOI] [PubMed] [Google Scholar]
- Stenico M, Lloyd AT, Sharp PM. Codon usage in Caenorhabditis elegans: delineation of translational selection and mutational biases. Nucleic Acids Res. 1994;22:2437–2446. doi: 10.1093/nar/22.13.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirman JN, Crane MM, Husson SJ, Wabnig S, Schultheis C, Gottschalk A, Lu H. Real-time multimodal optical control of neurons and muscles in freely behaving Caenorhabditis elegans. Nat Meth. 2011;8:153. doi: 10.1038/nmeth.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh C, Chudakov DM, Poon KL, Mamedov IZ, Sek JY, Shidlovsky K, Lukyanov S, Korzh V. Optogenetic in vivo cell manipulation in KillerRed-expressing zebrafish transgenics. BMC Dev Biol. 2010;10:110. doi: 10.1186/1471-213X-10-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uttara B, Singh AV, Zamboni P, Mahajan RT. Oxidative Stress and Neurodegenerative Diseases: A Review of Upstream and Downstream Antioxidant Therapeutic Options. Current Neuropharmacology. 2009;7:65–74. doi: 10.2174/157015909787602823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JG, Southgate E, Thomson JN, Brenner S, White JG, Southgate E, Thomson JN, Brenner S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos Trans R Soc Lond, B, Biol Sci. 1986;314:1–340. doi: 10.1098/rstb.1986.0056. [DOI] [PubMed] [Google Scholar]
- Zhang F, Wang LP, Brauner M, Liewald JF, Kay K, Watzke N, Wood PG, Bamberg E, Nagel G, Gottschalk A, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.