

Abstract
An efficient diastereoselective oxa-Pictet-Spengler reaction strategy was developed to construct benzoisochroman diastereomers. The utility of the reaction was demonstrated in the context of both the total synthesis of naturally-occurring pyranonaphthoquinones (+)-frenolicin B and epi-(+)-frenolicin B as well as a range of frenolicin precursor analogs. The method is versatile and offers exquisite stereocontrol and, as such, offers a synthetic advance for the synthesis of pyranonaphthoquinone analogs.
The pyranonaphthoquinone antibiotic (+)-frenolicin B (1) was isolated from a Streptomyces roseofulvus strain AM-3867.1 Since this initial discovery, several members of the pyranonaphthoquinone family, including frenolicin B, have been explored in the context of anticoccidial, anti-cancer and anti-malarial lead development.2 As a part of a new Appalachian-based natural product discovery initiative, 3 we recently isolated a bacterial strain (Streptomyces RM-4-15, isolated from soil samples near a thermal vent of the Ruth Mullins underground coal mine fire in eastern Kentucky) capable of producing a range of both known and new pyranonaphthoquinones, some of which displayed potent cancer cell line cytotoxicity in vitro. While such studies revealed new naturally-occurring bioactive chemical entities, low yields and variability in the production of pyranonaphthoquinone analogs via fermentation prompted the need for a complementary robust synthetic strategy to provide access to pyranonaphthoquinones of interest.
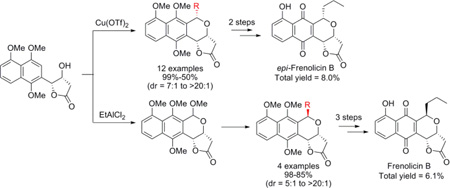
Beginning with the first asymmetric total synthesis of (+)-frenolicin B (1) by Kraus and coworkers, 4 several alternative synthetic strategies have been put forth.5 The most recent, by Fernandes et al., 6 employed a Dotz benzannulation to construct the naphthquinone scaffold from Fisher carbene 3 and chiral alkyne 4 (Figure 1). While this method achieved the desired product, the approach was limited by the number of steps and, more notably, the lack of diastereoselectivity in the late stage oxa-Pictet-Spengler reaction (the degree to which is often dependent upon the substrate architecture). 7 To address the limitations of the prior study, herein we report the optimization and application of a diastereoselective oxa- Pictet-Spengler reaction to afford benzoisochroman 6 in good to excellent yields en route to the synthesis of frenolicin B, epi-frenolicin B and precursors for a diverse array of pyranonaphthoquinone analogs.8
Figure 1.

A strategic comparsion of oxa-Pictet-Spengler-based approaches toward frenolicin B synthesis.
Following prior precedent for the synthesis of intermediate 5 by Bruckner, bromonaphthlene 7 was initially synthesized from commercially available 1,5-dihydroxy- naphthlene via a slightly modified procedure (Scheme 1). 9 Heck coupling of 7 with isobutyl but-3-enoate afforded the desired ester 9 in good yield (84%). However, subsequent direct Sharpless asymmetric dihydroxylation of 9 failed to provide 5 in >30% yield. Suspecting steric infringement to be a limiting factor, simple isobutyl- to methyl-ester substitution (9 → 10) followed by Sharpless asymmetric dihydroxylation, afforded the desired alcohol 5 in good yield and excellent ee (>99.5%).10
Scheme 1.

Synthesis of compound 5.
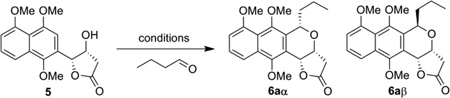
With the core reaction established, we next explored optimization of the oxa-Pictet-Spengler reaction by first assessing the potential of the Lewis acid to favor the production of the α- or β-configured product. Among the Lewis acids tested, 50 mol % Cu(OTf)2 offered the best yield and α-diastereoselectivity (Table 1, entries 2–7, see also Table S1 in Supporting Information). 11 Further investigation of solvents and reaction time in the context of the Cu(OTf)2 reaction revealed a slight improvement in yield and dr ratio with overnight stirring in dichloromethane (Table 1, entries 8–10). In contrast, FeCl3 was the only Lewis acid to favor the production β-configured product (2:1 β/α; Table 1, entry 13, and Supporting Information) and this poor diastereoselectivity could not be improved upon via further optimization (solvents, temperature and/or variant aldehyde source - data not shown).
Table 1.
Optimization of the oxa-Pictet-Spengler reaction.a
 | |||||
|---|---|---|---|---|---|
| entry | Lewis acids |
solvent | temp (ºC) |
conversion (%)b |
dr (6aα/6aβ)c |
| 1 | BF3·OEt2 | CH2Cl2 | 0-rt | 80(75) | 66:34 |
| 2 | Yb(OTf)3 | CH2Cl2 | 0-rt | 10 | 80:20 |
| 3 | Y(OTf)3 | CH2Cl2 | 0-rt | 23 | 67:33 |
| 4d | TiCl4 | CH2Cl2 | −78-rt | 70 | 70:30 |
| 5 | SnCl4 | CH2Cl2 | 0-rt | 100 | 60:40 |
| 6 | FeCl3 | CH2Cl2 | 0-rt | 100 | 52:48 |
| 7 | Cu(OTf)2 | CH2Cl2 | 0-rt | 85 | 89:11 |
| 8e | Cu(OTf)2 | CH2Cl2 | 0-rt | 95(90) | 91:9 |
| 9 | Cu(OTf)2 | DCE | 0-rt | (64) | 90:10 |
| 10 | Cu(OTf)2 | CHCl3 | 0-rt | (43) | 94:6 |
| 11f | Cu(OTf)2 | CH2Cl2 | 0-rt | (32) | 93:7 |
| 12g | Cu(OTf)2 | CH2Cl2 | 0-rt | (48) | 91:9 |
| 13 | FeCl3 | THF | 0-rt | (50) | 34:66 |
Reaction was performed with 0.2 mmol 5, 0.4 mmol aldehyde, and 50 mol % Lewis acid at 0 ºC. The temperature was allowed to subseqeuntly raise to rt over 4 h with stirring.
Conversion was determined by HPLC analysis. The data in the parentheses are the isolated yields after column chromatography.
dr ratio was determined by the proton NMR of crude products.
2 h reaction time.
Overnight.
Using 1,1-dimethoxybutane instead of butaldehyde.
Using 20 mol % Lewis acid.
Using the optimized conditions for α-configured benzoisochromane synthesis developed in Table 1, we next explored the scope of aldehyde substrates in the context of this reaction (Scheme 2). The aliphatic aldehydes tested led to the desired benzoisochromanes 6b– i with equal or better diastereoselectivity than the model n-butyraldehyde reaction (6a) with one exception - isopropionaldehyde (leading to 6g). Yields with the aliphatic set were also comparable to the model reaction (≥80%), with one exception – vinylacetaldehyde (leading to 6f), possibly due to aldehyde decomposition.12 For the aromatic aldehydes examined, the reaction afforded the desired products 6j–l with comparative yields (≥86%) but varied diastereoselectivity with dramatic improvements in α-selectivity observed upon aromatic ring substitution. Neverthless, unlike most aliphatic counterparts, all the aromatic diastereomers were readily resolved via standard silica gel chromatography.
Scheme 2.

Scope of the α-reaction with varying R group.a
a Reaction was performed with 0.2 mmol 5, 0.4 mmol aldehyde, and 50 mol % Cu(OTf)2 at 0 ºC and allowed the temp raise to room temp with overnight stirring. b Isolated yields after column chromatography; dr ratio (α/β) was determined by the proton NMR of crude products.
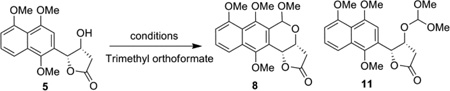
As previously indicated, FeCl3 was the only Lewis acid to favor the production β-configured product (2:1 β/α; Table 1, entry 13 and Supporting Information). Thus, focus was next shifted to developing an orthogonal route to bias the production β-configured products. Considering that the oxocarbenium formed either by the oxidation of isochroman or by the reduction of isochroman acetal could be intercepted by a range of nucleophiles, 13 we envisioned the rigid lactone ring located on the si face of the corresponding oxocarbenium generated by isochroman acetal 8 to favor re face nucleophilic attack and thereby favor formation of β-diastereomer. Several C1 cation building block (formaldehyde, dimethoxymethane, methyl chloromethyl ether, methyl formate, and trimethyl orthoformate)/Lewis acid combinations were examined for isochroman or isochroman acetal formation. Table 2 highlights optimization of the reaction containing 5 and trimethyl orthoformate ultimately affording isochroman acetal 8 in good isolated yield (70%) on reasonable scale (Table 2, entries 8–11). To test the feasibility of this strategy to access β-configured adducts, the reaction of acetal 8 with a small set of representative nucleophiles was subsequently examined (Scheme 3). As anticipated, both allyltrimethylsilane and cyanide led to β-configured products (6f and 12, respectively) in ≥85% yield, the former of which could be hydrogenated to frenolicin precursor 6aβ. Acetophenone led to a 5:1 β/α product distribution, likely due to an influence of phenylnaphthalene π-stacking upon selectivity. Notably, compound 14 was inaccessible via the previously discussed oxa-Pictet-Spengler reaction with 5.
Table 2.
Optimization of the oxa-Pictet-Spengler reaction with trimethyl orthoformate.a
 | |||||
|---|---|---|---|---|---|
| entry | Lewis acids |
solvent | temp (ºC) |
time (h) | conversion (%)b |
| 1 | FeCl3 | CH2Cl2 | 0 | 1 | 40(31) |
| 2 | BF3·OEt2 | CH2Cl2 | 0 | 1 | 20 |
| 3 | Fe(OTf)3 | CH2Cl2 | 0 | 2 | 23 |
| 4 | AlCl3 | CH2Cl2 | 0 | 1 | <10 |
| 5 | SnCl4 | CH2Cl2 | 0 | 1 | 45(33) |
| 6 | EtAlCl2 | CH2Cl2 | 0 | 1 | 60(51) |
| 7 | Et2AlCl | CH2Cl2 | 0 | 2 | <10d |
| 8 | EtAlCl2 | CH2Cl2 | −40 | 16 | 45(36) |
| 9 | EtAlCl2 | CH2Cl2 | −20 | 16 | 80(70) |
| 10 | EtAlCl2 | DCE | −20 | 16 | 74(62) |
| 11e | EtAlCl2 | CH2Cl2 | −20 | 16 | 78(65) |
Reaction was performed with 0.2 mmol 5, 0.24 mmol trimethyl orthoformate, and 100 mol % Lewis acid at 0 ºC with 1 h stirring.
Conversion was determined by HPLC analysis. The data in the parentheses are the isolated yields after column chromatography.
dr ratio was determined by the proton NMR of crude products.
formation of the side product 11 in 60% yield.
2 mmol 5 was loaded.
Scheme 3.

Scope of the β-oriented nucleophilic attack with different nucleophiles.
Final maturation of 6aβ and 6aα to the desired frenolicin B and epi-frenolicin B, respectively, followed a slight modification of previously reported strategies (Scheme 4).6b Specifically, while boron tribromide-mediated demethylation led to product epimerization in prior reports,6b we found this could be avoided by simply replacing boron tribromide with boron trichloride.
Scheme 4.

Synthesis of (+)-frenolicin B 1 and epi-(+)-frenolicin B 2
While 6aβ was previously reported to display notable cancer cell line cytotoxicity,2d the cytotoxicity of 6aα was not previously reported. A comparison of the in vitro anticancer activities of 6aα and 6aβ (Figure S1) revealed similar potencies against the colon cancer cell line HCT116 (IC50 of 109±24 nM and 215±21 nM, respectively) compared to a slight reduction (~5-fold) in potency for 6aα against the non-small cell lung cancer cell line A549 (IC50 of 965±60 nM and 179±29 nM, respectively), suggesting the relative configuration of the C-ring to have a moderate influence upon cancer cell line cytotoxicity/specificity.
In summary, a diastereoselective oxa-Pictet-Spengler reaction has been established for the construction of naphthquinone analogs. The targeted strategy for the synthesis of α-configured congeners hinges upon the use of Cu(OTf)2, while desired β-configured analogs were assembled via nucleophilic substitution upon a suitable isochroman acetal precursor. The route has been successfully applied in the synthesis of various frenolicin derivatives with good to excellent diastereoselectivity/yields. Further anticancer structure-activity assessments are ongoing.
Supplementary Material
Acknowledgment
This work was supported, in part, by the University of Kentucky College of Pharmacy, the University of Kentucky Markey Cancer Center, National Institutes of Health grant CA175105 (to Q.-B. S.) and the National Center for Advancing Translational Sciences (UL1TR000117).
Footnotes
Supporting Information Available Complementary Table 1 for screening Lewis acids, experimental procedures for compounds 1–2, 5–14, and compounds characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Iwai Y, Kora A, Takahashi Y, Hayashi T, Awaya J, Masuma R, Oiwa R, Ōmura S. J. Antibiot. 1978;31:959. doi: 10.7164/antibiotics.31.959. [DOI] [PubMed] [Google Scholar]
- 2.(a) Ōmura S, Tsuzuki K, Iwai Y, Kishi M, Watanabe S, Shimizu H. J. Antibiot. 1985;38:1447. doi: 10.7164/antibiotics.38.1447. [DOI] [PubMed] [Google Scholar]; (b) Armer RE, Dutton CJ, Fenner BR, Greenwood SDW, Hall KT, Rudge AJ. Bioorg. Med. Chem. Lett. 1998;8:139. doi: 10.1016/s0960-894x(97)10200-1. [DOI] [PubMed] [Google Scholar]; (c) Brimble MA, Duncalf LJ, Nairn MR. Nat. Prod. Rep. 1999;16:267. doi: 10.1039/a804287j. [DOI] [PubMed] [Google Scholar]; (d) Salaski EJ, Krishnamurthy G, Ding W-D, Yu K, Insaf SS, Eid C, Shim J, Levin JI, Tabei K, Toral-Barza L, Zhang W-G, McDonald LA, Honores E, Hanna C, Yamashita A, Johnson B, Li Z, Laakso L, Powell D, Mansour TS. J. Med. Chem. 2009;52:2181. doi: 10.1021/jm900075g. [DOI] [PubMed] [Google Scholar]; (e) Fitzgerald JT, Henrich PP, O’Brien C, Krause M, Ekland EH, Mattheis C, Sa JM, Fidock D, Khosla C. J. Antibiot. 2011;64:799. doi: 10.1038/ja.2011.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Wang X, Shaaban KA, Elshahawi SI, Ponomareva LV, Sunkara M, Zhang Y, Copley GC, Hower JC, Kharel MK, Thorson JT. J. Nat. Prod. 2013;76:1441. doi: 10.1021/np400231r. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shaaban KA, Wang X, Elshahawi SI, Ponomareva LV, Sunkara M, Zhang Y, Copley GC, Hower JC, Kharel MK, Thorson JT. J. Nat. Prod. 2013;76:1441. doi: 10.1021/np400231r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Kraus GA, Li J, Gordon MS, Jensen JH. J. Am. Chem. Soc. 1993;115:5859. [Google Scholar]; (b) Kraus GA, Li J, Gordon MS, Jensen JH. J. Org. Chem. 1995;60:1154. [Google Scholar]
- 5.(a) Masquelin T, Hengartner U, Streith J. Helv. Chim. Acta. 1997;80:43. [Google Scholar]; (b) Contant P, Haess M, Riegl J, Scalone M. Synthesis. 1999:821. [Google Scholar]; (c) Donner CD. Synthesis. 2010:415. [Google Scholar]; (d) Donner CD, Casama MI. Tetrahedron Letters. 2012;53:1105. [Google Scholar]
- 6.(a) Fernandes RA, Chavan VP. T. Asy. 2011;22:1312. [Google Scholar]; (b) Fernandes RA, Chavan VP, Mulay SV, Manchoju A. J. Org. Chem. 2012;77:10455. doi: 10.1021/jo3019939. [DOI] [PubMed] [Google Scholar]
- 7.For reviews: Larghi EL, Kaufman TS. Eur. J. Org. Chem. 2011:5195. Larghi EL, Kaufman TS. Synthesis. 2006:187.
- 8.All the relative configurations were determined by key NOE correlations (see Supporting Information).
- 9.(a) Fernandes RA, Bruckner R. Synlett. 2005;8:1281. [Google Scholar]; (b) Eid CN, Shim J, Bikker J, Lin M. J. Org. Chem. 2009;74:423. doi: 10.1021/jo801945n. [DOI] [PubMed] [Google Scholar]
- 10.Enantioselectivity were determined by HPLC analysis [chiral IC column (Daciel Chemical Ind. Ltd.) 25.0 × 4.6 mm, 80/20 hexane/iPrOH, 0.6 mL/min, UV 254 nm, tmajor = 39.2 min, tminor = 41.8 min]. The observed ee value is consistent with the data reported in ref. 9a.
- 11.The epimers can be slightly isolated on a preparative reverse phase HPLC (Colunm: Supelco C18, 25 cm × 21.2 mm, 10 µm; eluent: gradient 35%–50% CH3CN in water; rate: 10 mL/min; tβ = 58.2 min, tα = 62.3 min; loading amount: 5 mg).
- 12.(a) Crimmins MT, Kirincich SJ, Wells AJ, Choy AL. Synth. Commun. 1998;28:3675. [Google Scholar]; (b) Airiau E, Spangenberg T, Girard N, Breit B, Mann A. Org. Lett. 2010;12:528. doi: 10.1021/ol902718q. [DOI] [PubMed] [Google Scholar]
- 13. Elmore SW, Coghlan MJ, Anderson DD, Pratt JK, Green BE, Wang AX, Stashko MA, Lin CW, Tyree CM, Miner JN, Jacobson PB, Wilcox DM, Lane BC. J. Med. Chem. 2001;44:4481. doi: 10.1021/jm010367u. Zhang Y, Li C-J. Angew. Chem. Int. Ed. 2006;45:1949. doi: 10.1002/anie.200503255. For a review: Li C-J. Acc. Chem. Res. 2009;42:335. doi: 10.1021/ar800164n.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.