

Abstract
Controlled cell death, or apoptosis, occurs in response to many different environmental stimuli. The apoptotic cascade that occurs within the cell in response to these cues leads to morphological and biochemical changes that trigger the dismantling and packaging of the cell. Caspases are a family of cysteine-dependent aspartate-directed proteases that play an integral role in the cascade that leads to apoptosis. Caspases are grouped as either initiators or effectors of apoptosis, depending on where they enter the cell death process. Prior to activation, initiator caspases are present as monomers that must dimerize for full activation whereas effector caspases are present as dimeric zymogens that must be processed for full activation. The stability of the dimer may be due predominately to the interactions in the dimer interface as each caspase has unique properties in this region that lend to its specific mode of activation. Moreover, dimerization is responsible for active site formation because both monomers contribute residues that enable the formation of a fully functional active site. Overall, dimerization plays a key role in the ability of caspases to form fully functional proteases.
INTRODUCTION: APOPTOSIS LEADS TO CELL DEATH
Apoptosis is a type of cell death in which a cell uses specialized machinery to dismantle itself. Under normal growth and developmental conditions, apoptosis is a cell suicide mechanism that enables eumetazoans to control cell number, that is, to maintain homeostasis and to eliminate damaged cells. A healthy adult human produces approximately ten billion cells each day by mitosis and a similar number are removed by apoptosis.1 Disregulation of the cell death mechanism results in a loss of homeostasis. Indeed, alterations in the cell death program have been implicated in several diseases, including neurodegenerative disorders, inflammatory diseases and cancer.2 Cancer cells, in particular, are known to have a decreased sensitivity to proapoptotic signals when compared to normal cells. It is well established, however, that anticancer drugs are effective at inducing the cell death program by a variety of mechanisms.3–5
Apoptosis is a highly regulated process that may be triggered by a variety of stimuli including, but not limited to, virus infection, toxic stress, environmental insults and hormones.6 The morphology of the cell changes during apoptosis due to cytoplasmic shrinkage, active membrane blebbing, chromatin condensation and fragmentation of membrane-enclosed vesicles.7 In addition, the nuclear DNA is degraded, the cytoskeleton in dismantled and cell cycle progression is halted.8,9 In short, every aspect of the cell is disrupted so that the contents are dismantled and packaged into vesicles, called apoptotic bodies, which are phagocytozed by macrophages or surrounding tissue.10
CASPASES ARE PART OF THE CELL DEATH MACHINERY
A family of cysteine-dependent aspartate-directed proteases, known as caspases, is intimately involved in apoptosis. The cleavage of key proteins in the cell by caspases leads to the morphological and biochemical changes observed in apoptosis. For example, the cleavage of ICAD (inhibitor of CAD) by caspases releases the DNase CAD (caspase activated DNase) from an inactive complex and ultimately results in the cleavage of nuclear DNA by CAD.11 To date, fourteen caspases have been identified, with eleven caspases present in humans.6,12
Depending on their involvement in the life and death of a cell, caspases are broadly classified either as apoptotic or inflammatory caspases (Fig. 1). Those involved in the inflammatory response, namely caspases-1, -4 and -5, are cytokine activators.13–15 The apoptotic caspases are further divided into two groups, the initiators and the effectors, depending on their time of entry into the apoptotic cascade (Fig. 1). Initiator caspases such as caspases-2, -8, -9 and -10 have an early entry into the cascade and are responsible for activating the effector caspases (-3, -6, or -7). They are themselves activated either by so-called extrinsic or intrinsic mechanisms.
Figure 1.

Human caspase organization. Caspases are grouped on the left according to function and on the right according to the recognition sequence of the substrate. Each caspase has an N-terminal prodomain, where some contain either a CARD (caspase recruitment domain) or DED (death effector domain) motif, followed by the large subunit (LARGE), an intersubunit linker and the small subunit (SMALL). The numbers on each caspase molecule refers to the length of each specific domain, which was determined using the NCBI domain organization database (http://www.ncbi.nlm.nih.gov/).
The extrinsic pathway for initiator caspase activation ultimately is responsible for the elimination of unwanted cells that are produced during development or that have tumorogenic qualities.16 This pathway is initiated by ligation of a transmembrane death receptor in response to an extracellular signal, followed by recruitment and activation of initiator caspases as a part of a multiprotein complex (Fig. 2). Caspases-8 and -10 are the initiator caspases that are activated by way of the extrinsic pathway. In contrast, the intrinsic pathway primarily is responsible for the removal of cells in response to cytotoxic stress, chemotherapeutic drugs, mitochondrial damage and certain developmental cues.17 The mitochondria release cytochrome c into the cytoplasm in response to one or more of these cues (Fig. 2). The increase in the cytoplasmic concentration of cytochrome c is sensed by the protein Apaf-1 (apoptosis activating factor 1), which leads to recruitment of caspase-9 to a multiprotein complex, called the apoptosome, followed by activation of the caspase in a cofactor-dependent manner. The end result of initiator caspase activation is the downstream activation of the effector caspases-3, -6 and -7, which ultimately are responsible for cleavage of intracellular proteins that lead to the dismantling of the cell.
Figure 2.

The caspase cascade. A) Inflammatory caspase activation: a ligand binds to a toll-like receptor (TLR), which signals a NALP protein to bind to the TLR. ASC interacts with the pyrin domain (PYD) of NALP via PYD:PYD interactions. The CARD domain of ASC interacts with the CARD domain of procaspase-1 and the CARD domain of NALP interacts with the CARD domain of procaspase-5, forming the inflammasome. The inflammasome complex promotes dimerization of caspases-1 and -5, leading to their activation and the inflammatory response. B) The extrinsic apoptotic pathway: a death ligand binds to a death receptor, which signals an adaptor molecule to bind to the receptor via death domain (DD) interactions. The DED motif of the adaptor molecule interacts with the DED of procaspases-8 and -10, forming a DISC complex. Dimerization (mechanism unknown) results in maturation and full activity. Caspases-8 and -10 then process executioner caspases. C) Procaspase-2, a unique caspase, is activated when a ligand binds to a death receptor, which signals an adaptor molecule to bind via interactions with the death domain. The CARD of the adaptor molecule interacts with the CARD of procaspase-2 to promote dimerization in a DISC-like complex. Upon removal of the prodomain, caspase-2 cleaves Bid, a protein responsible for the increased permeability of the mitochondria. D) The intrinsic apoptotic pathway: an increase in the cytosolic concentration of cytochrome c leads to the formation of the apoptosome. The apoptosome is composed of Apaf-1 monomers that form a heptameric structure when cytochrome c binds to the WD40 motifs of Apaf-1, in an ATP-dependent manner, leading to interactions of the CARDs. The CARD of procaspase-9 then interacts with the CARD of Apaf-1, increasing the local concentration of procaspase-9 monomers and thereby promoting dimerization and activation. Caspase-9 then processes effector caspases, which leads to apoptosis. Effector caspases are activated by cleavage of their prodomain and intersubunit linker.
CASPASES ALSO ARE PART OF THE INFLAMMATORY RESPONSE
Some members of the caspase family are involved in processes within the cell that are not lethal. Caspases-1, -4 and -5 function in the regulation of inflammatory processes, for example. Caspase-1, which was known originally as interleukin-1β converting enzyme (ICE), is responsible for the activation of interleukin-1β (IL-1β) in macrophages13 as well as interleukin-18 (IL-18), a member of the IL-1 cytokine superfamily.18 Caspase-5 interacts with caspase-1 in a multiprotein complex known as the inflammasome, where caspase-1 is activated19 (Fig. 2). The function of caspase-4 is more obscure, although it has been shown that its transcription is induced by interferons.15
CASPASES CLEAVE WITH HIGH SPECIFICITY
Caspases show a high degree of specificity for substrates, with an absolute requirement for an aspartic acid residue and a recognition sequence of four amino acids N-terminal to the cleavage site. These residues generally are referred to as positions 1–4, or P1-P4, where the aspartate occupies the P1 position and cleavage of the substrate occurs C-terminal to the P1 aspartate. In addition to the functional groupings described above, caspases also have been grouped into three sub-families based on consensus recognition sequences.20 Group I caspases (caspases-1, -4, -5 and -14) recognize Trp-Glu-His-Asp; Group II caspases (caspases-2, -3 and -7) recognize Asp-Glu-X-Asp, where X is any amino acid; and Group III (caspases-6, -8, -9 and -10) recognize (Leu/Val)-Glu-X-Asp (Fig. 1). The specificity of the sub-families is crucial to the apoptotic mechanism because it prevents indiscriminate proteolysis of proteins that are not destined to be destroyed in the cell death process.21
CASPASES ARE PRODUCED INITIALLY AS INACTIVE ZYMOGENS
Many of the events that occur during apoptosis are posttranslational in nature. Caspases are synthesized initially as zymogens (procaspases) and the pool of inactive zymogen is converted rapidly to active protease upon induction of apoptosis. While some procaspases are monomers and some are dimers, as described below, all procaspases have a common organization in that each monomeric unit is composed of an N-terminal prodomain, a large subunit and a small subunit, as shown schematically in Figure 1. In all procaspases, the large and small subunits are covalently connected by a sequence of amino acids referred to as the intersubunit linker. The linker is cleaved and in some cases completely excised, upon maturation. At the N-terminus, the caspase prodomain ranges in length from 15 amino acids for caspase-14 to 219 amino acids for caspase-10. In general, initiator caspases contain a long prodomain and effector caspases contain a short prodomain.
The prodomains of caspases have been shown to have a variety of functions, from facilitating dimerization,22–24 to assisting in the folding of the caspase as an intramolecular chaperone,25 to sequestering the protein in the cytoplasm,26 to silencing the zymogen in vivo.27,28 The long prodomain of initiator caspases contains recognition motifs that are involved in homo- or heterotypic interactions that lead to zymogen activation. For example, the death effector domain (DED) is found within the prodomain of caspases-8 and -10 and interacts with an adaptor molecule that is recruited upon ligation of a death receptor, as shown schematically in Figure 2. Utilizing a similar activation protocol, the caspase recruitment domain (CARD) is found within the prodomain of caspases-1, -2, -4, -5 and -9 and is involved in recognition of the caspase by the particular multiprotein complex that facilitates activation.
An active caspase molecule is derived from the processing and association of two procaspase monomers. Structurally, mature caspases are dimers of heterodimers that contain two copies each of the large and small subunits. The dimer is arranged in an LSSL configuration, where L represents the large subunit and S represents the small subunit, with two active sites at nearly opposite ends of the molecule. The dimer is composed of a 12-stranded β-sheet core surrounded by a network of α-helices (Fig. 3A) and the substrate-binding region is formed on the protein surface by five loops (L1, L2, L3, L4 and L2’). L1-L4 are contributed by one heterodimer and L2’ is contributed by the second heterodimer.
Figure 3.

Comparison of caspase dimer interfaces. A) Structure of caspase-3 (PDB entry 2J30). α-helices 5 and 5’ and active site loops L1, L2, L2’, L3 and L4 are labeled. The prime (‘) indicates residues from the second heterodimer. Residues in β-strands 8 and 8’ are shown below the structure and ionic interactions between residues in helices 5 and 5’ are shown above the structure rotated by 180° to view sidechains. Amino acids in the dimer interface (β-strands 8 and 8’) of caspase-1 (B) (PDB entry 2HBQ), caspase-8 (C) (PDB entry 1QTN), caspase-2 (D) (PDB entry 1PYO) and of caspase-9 (E) (PDB entry 1JXQ) are shown. For A-E, the dashed lines indicate main chain hydrogen bonds between β-strands 8 and 8’. Structures were generated using Pymol (Delano Scientific LLC, Palo Alto, CA).
DIMERIZATION OF CASPASE ZYMOGENS
For all caspases, dimerization is critical for proper active site formation. Since the active site loops are contributed by both heterodimers in the dimeric structure (Fig. 3), caspase zymogens demonstrate little or no activity in the monomeric state. Overall, the initiator procaspases are thought to dimerize by an induced proximity model where oligomeric activation complexes mediate clustering of zymogens and increase the local concentration of procaspase monomers, which in turn facilitates dimerization.29,30 For example, procaspases-8, -9 and -10, the initiator caspases, are found as monomers at physiological concentrations and must undergo dimerization to become active. Once an extracellular ligand binds to the death receptor, the intracellular region of the receptor binds to the death domain (DD) of an adaptor molecule (Fig. 2). The adaptor molecule then recruits procaspase-8 or -10 by interacting with the death effector domain (DED) located in the long prodomain. The resulting assembly is known as the death-inducing signaling complex (DISC) (Fig. 2).31 Likewise, Apaf-1 assembles into a heptameric structure in the presence of cytochrome c and ATP to recruit procaspase-9 through CARD-CARD interactions. The resulting assembly is referred to as the apoptosome (Fig. 2). For initiator caspases, the intersubunit linker is cleaved following dimerization and while this stabilizes the dimer, cleavage of the linker is not necessary for the gain of enzymatic activity since a functional active site can form without this event.32
A unique initiator caspase, caspase-2, is found as a dimer at physiological concentrations and is activated by proteolytic cleavage. This caspase functions as an initiator of both the intrinsic and extrinsic pathways and in some cases, as in neuronal cells, it acts as an effector caspase.33,34 In the extrinsic pathway, caspase-2 initiates apoptosis in response to ligation of a death receptor utilizing the CARD of caspase-2. In a DISC-like complex, caspase-2 is proteolytically cleaved in the intersubunit linker and within the prodomain, which releases the active protease into the cytoplasm. Unlike other initiator caspases, caspase-2 is completely inactive toward the effector caspase zymogens. Caspase-2, instead, cleaves Bid (BH3 interacting death agonist), which leads to an increase in the permeability of the outer membrane of the mitochondria. This event elevates the cytosolic levels of cytochrome c, resulting in the formation of the apoptosome and the subsequent activation of caspase-9. The mechanism of caspase-2 activation in the intrinsic pathway remains obscure; however, it is known that caspase-2 plays an important role in initiating chemotherapy-induced cell death.21
Procaspases-1, -4 and -5, those involved in the regulation of inflammatory processes, are zymogens that exist in the cell as monomers and require dimerization to become active. Procaspases-1 and -5 can be activated by several different inflammasomes that contain a variety of proteins such as Ipaf, NALP1, NALP2, NALP3 and ASC. The inflammasomes are formed similarly to the multi-protein complexes of the extrinsic apoptotic pathway. That is, an interaction with a ligand and its cognate receptor results in the recruitment of an adaptor molecule that in turn binds the CARD of the inflammatory caspases (Fig. 2). The induced proximity effect, described above, apparently is utilized by these procaspases as well. The binding of the procaspases to the inflammasome increases the local concentration of monomers and thereby facilitates oligomerization.35
CASPASE DIMERIZATION AFFECTS STABILITY
In contrast to the initiator caspases, procaspases-3, -6 and -7, the effectors, are found as stable, but inactive, dimers at physiological concentration. As described below, the procaspases are inactive due to misaligned active sites that prevent efficient catalysis. Activation of these caspases occurs after proteolytic cleavage of the intersubunit linker, which allows the active site loops to rearrange.
The stability of caspases is due largely to dimerization. Studies of procaspase-3 showed that the protein unfolds by an unusual mechanism that includes two partially folded intermediates in equilibrium with the native dimer and the unfolded monomer.36,37 One intermediate is an inactive dimer, the second is a partially folded monomer. An examination of the free energy change for the formation of each species in the unfolding process showed that dimerization had a significant contribution to the conformational free energy of the protein. While the monomer has a conformational free energy of ~7 kcal/mol (25°C and pH 7.2), the protein gains an additional ~18 kcal/mol upon dimerization and subsequent isomerization of the inactive dimer.36 Dimerization, therefore, results in a very stable protein under physiological conditions. Indeed, the equilibrium dissociation constant of the procaspase-3 dimer is estimated to be in the low nanomolar range or lower.36
Dimerization of procaspase-3 is a pH-dependent event. While procaspase-3 is a dimer at neutral pH, the dimer is destabilized in favor of the monomer at pH 4. Titration studies have shown that two events occur as the pH is lowered.37 First, binding of the pro-domain to the protease domain is reduced between pH 7 and 5, although the protein remains dimeric. Second, the dimer dissociates between pH ~6 and 4. The dimeric state of procaspase-3 is unlikely to be affected during apoptosis because the pH of a cell decreases from 7.4 to ~6.8.38 This may not be true for the mature caspase-3, however, since the dimer dissociates between pH ~7 and ~5.39 Small fluctuations around pH 6.8 are predicted to have larger effects on the dimeric structure of caspase-3 than on that of procaspase-3. In contrast to procaspase-3, folding studies of monomeric caspases have not been done, so currently it is not known whether these principles hold true for other caspases.
A COMPARISON OF CASPASE DIMER INTERFACES
As described above, mature caspases are dimers of heterodimers, i.e., each heterodimer comprises a newly cleaved large and small subunit. The caspase structure includes a water-filled central cavity on one side and α-helices on the other side. Interactions across the dimer interface among charged amino acids in the α-helices are predicted to stabilize the structure (Fig. 3A). Approximately 2000 Å is buried in the dimer interface.40,41
Most of the interface interactions occur in β-strands 8 and 8’ in the small subunits since they form the lining of the central cavity (Fig. 3A). In general, for initiator procaspases, the dimerization rather than the processing of the zymogen is the key event in activation. That is, the acquisition of enzymatic activity is coupled to dimer formation rather than to chain cleavage. This mechanism is fundamentally different in the effector procaspases since those proteins are stable dimers that have low activity.
An understanding of the molecular interactions at the dimer interface may help to explain why some caspase zymogens are monomers in the cell while others form stable dimers under physiological conditions. Although the three-dimensional structures are quite similar among the caspases, the dimer interfaces are very different. The network of hydrogen bonds, the presence or absence of salt bridges or other polar interactions and the hydrophobic nature of the dimer interface likely influence the stability of the dimer. The combination of these features could explain why caspase-8, for instance, remains associated with the DISC once activated. The multi-protein complex could help to stabilize the dimeric form of caspase-8 since the monomer appears to be the preferred structure in solution. The same scenario holds true for caspase-9. It remains associated with the apoptosome to carry out catalytic functions apparently because of the weak interactions at the dimer interface. On the other hand, caspase-3 forms a stable dimer in solution and thus does not require activation complexes to facilitate dimerization.
In addition to stabilizing the protein, dimerization also is critical to active site formation. All caspases contain five loops that form the active site. Four loops (L1, L2, L3 and L4) are contributed by one heterodimer and the fifth loop, L2’, is contributed by the second heterodimer. As described below, the loops rearrange upon maturation of the protein, enabling the substrate to bind productively in the active site. The resulting conformation is stabilized by interactions in the dimer interface, although the specific stabilizing interactions are unique to each caspase as follows.
Caspase-3 contains four buried hydrogen bonds that are contributed by main chain atoms in β-strands 8 and 8’ and span the dimer interface (see Fig. 3A). In addition, residues from helices 5 and 5’ (Glu231, His234, Arg238 and Glu272, see Fig. 3A) create a network of salt-bridges on the surface of the protein that are predicted to stabilize the dimer. The region of the caspase-3 interface composed of β-strands 8 and 8’ has, in general, a hydrophobic character, possibly explaining why procaspase-3 is found as a stable dimer in the cell. That is, the protein forms a stable dimer as a mechanism to shield the amino acids from solvent. Otherwise, the hydrophobic β-strand 8 would be solvent-exposed in the monomer.
In contrast, the dimer interface of caspase-1 is more hydrophilic and presents a different hydrogen bonding network. The addition of Glu390 and Arg391 on β-strand 8 and Glu390’ and Arg391’ on β-strand 8’ provides four additional charged residues in the center of the dimer (Fig. 3B). Arg391 forms a β-bulge that perturbs the pattern of hydrogen bonds in β-strand 8 and results in a so-called “negative design element”.42 The element prevents the formation of indiscriminate oligomers from an exposed β-strand by requiring a precise hydrogen bonding pattern. Overall, this is consistent with the presence of a procaspase-1 monomer in solution because the exposed β-strand 8 in the procaspase-1 monomer would hydrogen bond only with another monomer that presents a complementary surface. The side chain of Glu390 hydrogen bonds with the amide backbone nitrogen of Arg391’ across the dimer interface and also forms a salt-bridge with Arg286, from the active site of the same heterodimer. This salt bridge is predicted to stabilize active site loop 2 (see also Fig. 5).43
Figure 5.

Active site rearrangements of caspases-1 and -3. A) Movements in L2 and L3 of caspase-1 upon substrate binding. The following colors are used. Green: apo-caspase-1 (PDB entry 1SC1), blue: holo-caspase-1 with malonate bound (PDB entry 1SC3). B) Different rotamer configurations for Gln283 in the ligand-free (blue) or ligand-bound (green) conformations of caspase-1 and loop movements in L2. Ala285 refers to the catalytically inactive mutant of caspase-1. C) Movements of Arg286 upon substrate binding result in intercalation of the side chain between Cys331 and Pro335, from active site loop 3, forming a new salt bridge with Glu390 from the dimer interface. The red sphere indicates a water molecule between Glu390 and Glu390’. D) In caspase-3 (PDB entry 2J30), the charge of Arg164 (equivalent to Arg286 in caspase-1) is neutralized by Glu124, which is situated above the dimer interface. For B and C, the secondary structure and active site loops are colored the same as those in Figures 3A and 4A and side chains are colored using the cpk color mode. Structures were generated using Pymol (Delano Scientific LLC, Palo Alto, CA). A color version of this image is available at www.landesbioscience.com/curie.
The dimer interface of caspase-8 contains only two main-chain hydrogen bonds, between Phe468 and Pro466’ as well as the symmetry-related Phe468’ and Pro466 interaction (Fig. 3C). The interface of caspase-8 is hydrophilic in nature and is striking for the absence of an arginine residue next to the catalytic cysteine, C360. Glutamine occupies this position in caspase-8 (Table 1). Currently, it is not known how the polar interface might contribute to the formation of a stable monomer rather than a stable dimer in solution. Current models suggest that the protein-protein interactions between the pro-domain and the DISC result in dimerization of caspase-8 by increasing the local concentration of interacting species, although the mechanistic details of initiator caspase dimerization are not known.
Table 1.
Caspase sequence alignment
 |
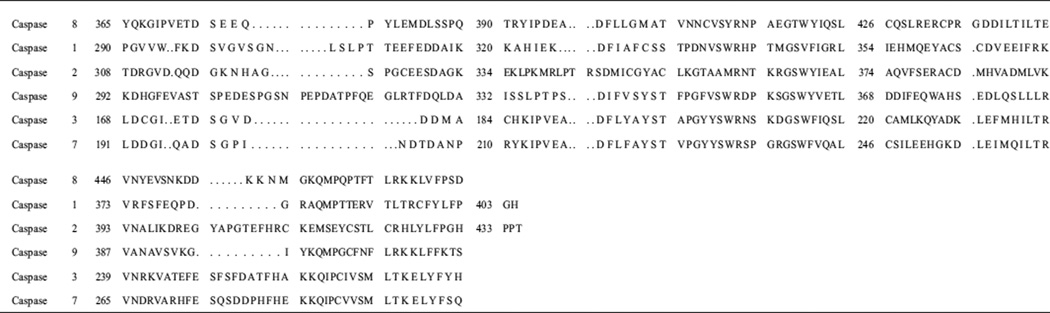 |
Sequence data were obtained using NCBI (http://www.ncbi.nlm.nih.gov/)
Caspase-2 is unique among the caspases because it has a covalent linkage that spans the dimer interface. The central cysteine pair, Cys419 and Cys419’, forms a disulfide bridge (Fig 3D and Table 1).21 Interestingly, the residue at this position in many caspases is implicated in interactions across the dimer interface, such as Glu390 described above for caspase-1. However, the disulfide linkage is not required for dimerization of caspase-2,34 so the role of the disulfide bond is not known at present.
The interface of caspase-9 has three primary differences at the dimer interface that are distinct from those of other caspases, resulting in the formation of only one functional active site. First, caspase-9 contains a phenylalanine residue at position 404 (Fig. 3D and Table 1) rather than valine, glutamate, or cysteine at this position in caspases-3, -1, or -2, respectively (Fig. 3A, 3B and 3D). As a result, caspase-9 cannot simultaneous form two productive active sites due to steric clashes in the dimer interface. Second, a seven-residue loop is inserted between strands 3 and 4 of the large subunit (see Fig. 6A). Several residues in the insertion fill the central cavity on the surface of the protein and form polar contacts across the dimer interface.32 Third, the intersubunit linker is about 20 residues longer than that of procaspase-3. Together, these features somehow allow for a zymogen that has a relatively high activity when compared to that of the activated caspase.
Figure 6.

Active site rearrangements of caspase-9 upon inhibitor binding. A) Structure of caspase-9 (PDB entry 1JXQ) with inhibitor bound to one active site. The structure is colored as in Figure 3 except the loop insertion between β-strands 3 and 4 is shown in green. B) Upon inhibitor binding to one active site, loop rearrangements result in movement of the Tyr345 side chain, on β-strand 7, away from the active site and toward the protein interior, causing the side chain of Phe404 to move toward the dimer interface. Due to steric constraints in the interface, these movements can occur only in one heterodimer, so the second active site remains disorganized. C) Interactions among amino acids in the elbow loop (Phe348-Phe351) and the second heterodimer (Phe246’, Pro338’ and Phe406’). For A-C, the secondary structure and active site loops are colored the same as those in Figures 3A and 4A and side chains are colored using the cpk color mode. Structures were generated using Pymol (Delano Scientific LLC, Palo Alto, CA). A color version of this figure is available at www.landesbioscience.com/curie.
ACTIVE SITE FORMATION AFTER CHAIN CLEAVAGE
Structural studies of caspase-7,45,46 an effector caspase and of caspases-147,48 and -932 provide clues to conformational changes that occur during maturation. In general, the active site is misaligned in the zymogen and cleavage of the intersubunit linker allows for realignment of the active site loops. While the mechanistic details differ for caspases-7 and -1, both proteins follow the general scheme that the chain cleavage results in proper formation of the substrate binding pocket as well as formation of the loop bundle among L2, L4 and L2’ (Fig. 4A). Formation of the loop bundle stabilizes the active site configuration and moves the catalytic cysteine into the S1 subsite where it is positioned to attack the substrate at the P1 site.
Figure 4.

Active site loop movements upon maturation of caspase-7. A) Active site loops 1–4 and 2’. B) Movements in L2 and L2’ upon maturation and substrate binding. Holo-caspase-7 (PDB entry 1F1J), apo-caspase-7 (PDB entry 1K86), procaspase-7 (PDB entry 1GQF). Note that in procaspase-7 the intersubunit linker encompasses both L2 and L2’. C) Movements in L3 upon maturation. D) Movements in L4 upon maturation. For A-D, the inhibitor bound to holo-caspase-7 is indicated. Structures were generated using Pymol (Delano Scientific LLC, Palo Alto, CA).
For caspase-7, cleavage of the intersubunit linker occurs at Asp198. Prior to the cleavage event, one of the two intersubunit linkers binds in the central cavity whereas the other is positioned on the outside of the cavity (Fig. 4B). Following cleavage, residues in active site loop 3 move into the central cavity and toward the dimer interface, resulting in formation of the substrate-binding groove (Fig. 4C). This insertion is prevented in the zymogen by the blocking segment of the intersubunit linker. More specifically, amino acids 212–215 of one monomer occupy this position in the central cavity and prevent movement of the so-called elbow loop of the second monomer into the central cavity (Fig. 4C). The elbow loop contains residues 224–231 and, in the activated caspase, forms several backbone hydrogen bonds with L2 as well as stacking interactions with other side chains. Thus, preventing insertion of the elbow loop destabilizes the substrate-binding pocket as well as the catalytic loop (L2). As a result, L3 in the procaspase occupies the substrate-binding pocket (Fig. 4C). Proteolytic cleavage of the intersubunit linker allows the formation of a new N-terminus of the small subunit (L2’) and C-terminus of the large subunit (L2). L2’ leaves the central cavity and rotates by ~180° to form new contacts in the loop bundle with L2, L3 and L4 from the opposing monomer (Fig. 4A,B).
The structure of apo-caspase-7, determined in the absence of inhibitor, reveals an intermediate form in which the active site is neither fully active nor fully misaligned. For example, one observes that L2’ remains bound in the central cavity and retains the misaligned conformation (Fig. 4B); that is, it resembles the configuration of the zymogen rather than that of the holo-enzyme. As a consequence, the loop bundle (L2, L4, L2’) is not properly assembled. In contrast, L3 (Fig. 4C) and L4 (Fig. 4D) are positioned closer to their productive conformations, although they are not fully stabilized until L2’ provides the additional interactions. Once L2’ rotates out of the central cavity upon substrate binding, the elbow loops from each heterodimer can insert into the cavity and a functional active site forms. More specifically, the arginine positioned next to the catalytic cysteine rotates from a solvent exposed orientation in the substrate binding pocket toward the dimer interface and intercalates between tyrosine 223 on β-strand 7 and proline 227 on the elbow loop, to form an ordered lining to the central cavity (Fig. 5D). The reorientation of the arginine on L2 toward the dimer interface and the accompanying loop movements lock the S1-S4 subsites into place and position the backbone amide of the catalytic cysteine into the correct orientation to form part of the oxyanion hole.45,46 These findings suggest that substrate binding plays a key role in the full activation of caspases and that proteolytic cleavage of the intersubunit linker is not sufficient for activation. The caspase active site is dynamic in that it fluctuates between the misaligned conformation, in which L2’ occupies the central cavity and the substrate binding pocket is not formed and the active conformation, in which formation of the loop bundle allows the active site to align fully. The binding of substrate traps the protein in the active conformation.
The formation of the active site of caspase-1 is somewhat different than that of caspase-7. As described above, β-strand 8 contains a bulge in the region that accommodates the blocking segment in (pro)caspase-7 due to the insertion of Arg391. In order for the formation of the active site of caspase-1 to mimic that of caspase-7, Arg391 would have to interact with another residue so that the blocking segment can fit into the central cavity. Structural data for caspase-1 reveal that in the ligand-free form the active site loops exhibit a misaligned conformation that is catalytically inactive prior to substrate binding.48 Three segments (L2, L3 and L4) undergo conformational changes upon substrate/inhibitor binding, resulting in catalytically active caspase-1. In particular, Arg341 in L3 rotates from a solvent exposed position on the surface of the enzyme into the S1 pocket of the active site (Fig. 5A). Upon substrate binding, Arg341 forms hydrogen bonds with amino acids in L1 and residues in the P1 and P3 positions of the substrate. The movement of Arg341 creates movement of the entire loop to place other key residues in their catalytically active form (e.g., Trp340 rotates from the surface of the protein into the S2 region of the active site, where it hydrogen bonds with the substrate; Fig. 5A).
The ligand-free form of caspase-1 lacks the salt bridge between Arg286 and Glu390 that stabilizes the active site and the dimer interface in the ligand-bound form (Fig. 5C). Arg286 is surface-exposed in the ligand-free form but is completely internalized upon ligand binding (Fig. 5A) and is stabilized by formation of the salt bridge with Glu390 (Fig. 5C).
As described above caspase-9 is unique from all other caspases because only one active site has the correct conformation to bind substrate. The other active site is unable to form due to steric constraints of residues in the dimer interface, largely from interactions involving Phe404, Tyr345 and Phe404’/Tyr345’ (Fig. 6A and 6B).32 In order for a functional active site to form, the side chains of Ser344-Ser353, the so-called “elbow loop”, from one heterodimer, contact residues in the dimer interface and insert into the opposing monomer (Fig. 6C) and allow the S2 and S1 binding pockets to assume an active conformation. The movement of the active site loops of caspase-9 upon ligand-binding is similar to those of the loops in caspase-1.48
ACTIVE SITE COOPERATIVITY
As described above, the interactions at the dimer interface lend to the overall stability of mature caspases. The integrity of the dimer interface also could be responsible for cooperativity between the two active sites. Given that interactions at the dimer interface affect the conformations of the active site loops, the question remained as to whether the binding of substrate to one active site enhanced the binding of substrate to the other active site. This was examined using wild-type caspase-1 and a mutant containing a Glu390 to Ala substitution.43 The rationale was that Glu390 had a dual role in dimer stabilization and communication across the dimer interface. The results showed that wild-type caspase-1 exhibits limited positive cooperativity with a Hill coefficient of 1.5 and replacing Glu390 with alanine removes the cooperativity (Hill coefficient of 1.0). Current models suggest that Glu390 and Glu390’ interact through a central water molecule in the dimer interface (Fig. 5C) and play a key role in communication between the two active sites. At present, very little is known about the mechanisms of cooperativity in caspase-1 or whether it occurs in other caspases.
In contrast to communications across the dimer interface, the interactions at the dimer interface of one heterodimer are tightly coupled to the productive binding of the substrate to the active site of the same heterodimer. To further illustrate this point, tethering experiments were performed to find allosteric sites for inhibitor binding.43,49 Tethering uses a library of small thiol-containing compounds that make disulfide bonds with naturally occurring cysteine residues in a protein. This method was used to determine allosteric sites in caspases-3, -7 and -1. Two classes of compounds were identified after screening thousands of molecules. Representatives of these classes are FICA (5-fluoro-1H-indole-2-carboxylic acid (2-mercapto-ethyl) amide) and DICA (2-(2,4-dichlorophenoxy-N-(2-mercapto-ethyl)-acetamide). Both compounds bound to a single cysteine in the small subunit on β-strand 8, Cys264 in caspase-3, which sits close to the dimer interface and is 14 Å from the catalytic cysteine. Binding of the inhibitors did not cause dissociation of the dimer but did prevent binding of the substrate at the active site. Structural data showed that in caspase-7, FICA interacts with Tyr223’ (β-strand 7) on the opposing heterodimer to occupy the central cavity (Fig. 7), whereas DICA interacts with Tyr223 in the same heterodimer. Regardless, the same conformational changes occur at the active site in the presence of either FICA or DICA. By displacing Tyr223, Arg187 is pushed out of the central cavity and placed into a position that occludes substrate binding to the active site and Cys186 is moved by 3.7 Å away from the S1 subsite. In addition, L2’ remains bound over the allosteric site and away from the substrate binding site (Fig. 7). This precludes formation of the loop bundle. As a result, L3 adopts a disordered structure and becomes completely destabilized. In fact, the large conformational changes that occur as a result of inhibitor binding to the interface resemble the conformation of the zymogen rather than that of the active enzyme.
Figure 7.

Structure of caspase-7 with FICA bound in the dimer interface (PDB entry 1SHL). Binding of inhibitor in the dimer interface displaces the side chain of Tyr223 and results in disorganized active site loops L2, L3 and L4. L2’ occupies the central cavity, as observed for apo-caspase-7 (see Fig. 4B). The secondary structure and active site loops are colored the same as those in Figures 3A and 4A and side chains are colored using the cpk color mode. Structures were generated using Pymol (Delano Scientific LLC, Palo Alto, CA).
In the case of caspase-1, it was found that a thienopyrazole bound to Cys331, which is near the dimer interface on β-strand 7 (Fig. 5C). Like FICA in caspase-7, the caspase-1 inhibitor interacted with residues on the neighboring heterodimer. Overall, the binding of the tethered inhibitor resulted in a disorganized active site by causing the catalytic cysteine to be rotated 5 Å away from the S1 subsite, the substrate-binding loop to be collapsed so that it could not interact productively with substrate and the side chain of Arg286 to be rotated away from the dimer interface and toward the substrate-binding cleft. As with caspase-7, caspase-1 adopted a conformation closely resembling its ligand-free form when bound to the allosteric inhibitor. Current models suggest that binding of the allosteric inhibitor to the dimer interface shifts the equilibrium of species found in solution from the active form to the inactive form,43 underlining the coupling of the dimer interface to the active site.
CONCLUSION
In caspases, the composition of the dimer interface is a key determinant of the mode of activation taken by an individual enzyme, in that interfaces that are mostly hydrophobic, like caspase-3, are found as stable, yet inactive, dimers prior to activation and only require processing to achieve a productively active conformation. Dimer interfaces that are more hydrophilic, like those of caspases-1 and -8, tend to result in monomeric caspases in physiological conditions. In those cases, activation requires an increased local concentration to promote dimerization. Overall, dimerization of caspases is a critical event in apoptotic initiation because it ultimately leads to rearrangement of the active site loops, resulting in the formation of the active site and it increases the stability of the caspase. These features ultimately lead to cell death.
ACKNOWLEDGEMENT
This work was supported by a grant from the National Institutes of Health (GM065970).
Footnotes
REFERENCES
- 1.Jacobson MD, Weill M, Raff MC. Programmed cell death in animal development. Cell. 1997;88:347–354. doi: 10.1016/s0092-8674(00)81873-5. [DOI] [PubMed] [Google Scholar]
- 2.Fadeel B, Orrenius S. Apoptosis: a basic biological phenomenon with wide-ranging implications in human disease. J Internal Med. 2005;258:479–517. doi: 10.1111/j.1365-2796.2005.01570.x. [DOI] [PubMed] [Google Scholar]
- 3.Kabore AF, Johnston JB, Gibson SB. Changes in the apoptotic and survival signaling in cancer cells and their potential therapeutic implications. Current Cancer Drug Targets. 2004;4:147–163. doi: 10.2174/1568009043481551. [DOI] [PubMed] [Google Scholar]
- 4.Fulda S, Debatin K-M. Targeting apoptosis pathways in cancer therapy. Current Cancer Drug Targets. 2004;4:569–576. doi: 10.2174/1568009043332763. [DOI] [PubMed] [Google Scholar]
- 5.Meng XW, Lee S-H, Kaufmann SH. Apoptosis in the treatment of cancer: a promise kept? Curr Op Cell Biol. 2006;18:668–676. doi: 10.1016/j.ceb.2006.10.008. [DOI] [PubMed] [Google Scholar]
- 6.Earnshaw WC, Martins LM, Kaufmann SH. Mammalian caspases: structure, activation, substrates and functions during apoptosis. Ann Rev Biochem. 1999;68:383–424. doi: 10.1146/annurev.biochem.68.1.383. [DOI] [PubMed] [Google Scholar]
- 7.Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Intern Rev Cytol. 1980;68:251–306. doi: 10.1016/s0074-7696(08)62312-8. [DOI] [PubMed] [Google Scholar]
- 8.Kaufmann SH. Induction of endonucleolytic DNA cleavage in human acute myelogenous leukemia cells by etoposide, camptothecin and other cytotoxic anticancer drugs: a cautionary note. Cancer Res. 1989;49:5870–5878. [PubMed] [Google Scholar]
- 9.Canman CE, Tange H-Y, Normolle DP, et al. Variations in patterns of DNA damage induced in human colorectal tumor cells by 5-fluorodeoxyuridine: implications for mechanisms of resistance and cytotoxicity. Proc Natl Acad Sci. 1992;89:10474–10478. doi: 10.1073/pnas.89.21.10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin Z, El-Deiry W. Overview of cell death signaling pathways. Cancer Biology and Therapy. 2005;4:139–163. doi: 10.4161/cbt.4.2.1508. [DOI] [PubMed] [Google Scholar]
- 11.Enari M, Sakahira H, Yokoyama H, et al. A caspase-activated DNase that degrades DNA during apoptosis and its inhibitor ICAD. Nature. 1998;391:43–50. doi: 10.1038/34112. [DOI] [PubMed] [Google Scholar]
- 12.Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity activation and inhibition. Biochem J. 2004;384:201–232. doi: 10.1042/BJ20041142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–774. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- 14.Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561–574. doi: 10.1016/j.cell.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Ahn E-Y, Pan G, Vickers SM, et al. IFN-γ upregulates apoptosis-related molecules and enhances FAS-mediated apoptosis in human cholangiocarcinoma. Intern J Cancer. 2002;100:445–451. doi: 10.1002/ijc.10516. [DOI] [PubMed] [Google Scholar]
- 16.Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Op Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 17.Boatright KM, Renatus M, Scott FL, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 18.Leung BP, Culshaw S, Gracie JA, et al. A role for IL-18 in neutrophil activation. J Immunol. 2001;167:2879–2886. doi: 10.4049/jimmunol.167.5.2879. [DOI] [PubMed] [Google Scholar]
- 19.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 20.Thornberry NA, Rano TA, Peterson EP, et al. A combinatorial approach defines specificities of members of the caspase family and granzyme B. J Biol Chem. 1997;272:17907–17911. doi: 10.1074/jbc.272.29.17907. [DOI] [PubMed] [Google Scholar]
- 21.Schweizer A, Briand C, Grutter MG. Crystal structure of caspase-2, apical initiator of the intrinsic apoptotic pathway. J Biol Chem. 2003;278:42441–42447. doi: 10.1074/jbc.M304895200. [DOI] [PubMed] [Google Scholar]
- 22.Hofmann K, Bucher P, Tschopp J. The CARD domain: a new apoptotic signalling motif. Trends Biochem Sci. 1997;22:155–156. doi: 10.1016/s0968-0004(97)01043-8. [DOI] [PubMed] [Google Scholar]
- 23.Thome M, Hofmann K, Burns K, et al. Identification of CARDIAK, a RIP-like kinase that associates with caspase-1. Curr Biol. 1998;8:885–888. doi: 10.1016/s0960-9822(07)00352-1. [DOI] [PubMed] [Google Scholar]
- 24.Weber CH, Vincenz C. The death domain superfamily: a tale of two interfaces? Trends Biochem Sci. 2001;26:475–481. doi: 10.1016/s0968-0004(01)01905-3. [DOI] [PubMed] [Google Scholar]
- 25.Feeney B, Clark AC. Reassembly of active caspase-3 is facilitated by the propeptide. J Biol Chem. 2005;280:39772–39785. doi: 10.1074/jbc.M505834200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Denault J-B, Salvesen GS. Human caspase-7 activity and regulation by its N-terminal peptide. J Biol Chem. 2003;278:34042–34050. doi: 10.1074/jbc.M305110200. [DOI] [PubMed] [Google Scholar]
- 27.Meergans T, Hildebrandt A-K, Horak D, et al. The short prodomain influences caspase-3 activation in HeLa cells. Biochem J. 2000;349:135–140. doi: 10.1042/0264-6021:3490135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cowling V, Downward J. Caspase-6 is the direct activator of caspase-8 in the cytochrome c-induced apoptosis pathway: absolute requirement for removal of caspase-6 prodomain. Cell Death and Differentiation. 2002;9:1046–1056. doi: 10.1038/sj.cdd.4401065. [DOI] [PubMed] [Google Scholar]
- 29.Muzio M, Stockwell BR, Stennicke H, et al. An induced proximity model for capase-8 activation. J Biol Chem. 1998;273:2926–2930. doi: 10.1074/jbc.273.5.2926. [DOI] [PubMed] [Google Scholar]
- 30.Pop C, Timmer J, Sperandio S, et al. The apoptosome activates caspase-9 by dimerization. Mol Cell. 2006;22:269–275. doi: 10.1016/j.molcel.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 31.Boatright KM, Renatus M, Scott FL, et al. A unified model for apical caspase activation. Mol Cell. 2003;11:529–541. doi: 10.1016/s1097-2765(03)00051-0. [DOI] [PubMed] [Google Scholar]
- 32.Renatus M, Stennicke HR, Scott FL, et al. Dimer formation drives the activation of the cell death protease caspase 9. Proc Natl Acad Sci. 2001;98:14250–14255. doi: 10.1073/pnas.231465798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H, Bergeron L, Cryns V, et al. Activation of caspase-2 in apoptosis. J Biol Chem. 1997;272:21010–21017. doi: 10.1074/jbc.272.34.21010. [DOI] [PubMed] [Google Scholar]
- 34.Baliga BC, Read SH, Kumar S. The biochemical mechanism of caspase-2 activation. Cell Death and Differentiation. 2004;11:1234–1241. doi: 10.1038/sj.cdd.4401492. [DOI] [PubMed] [Google Scholar]
- 35.Launay S, Hermine O, Fontenay M, et al. Vital functions for lethal caspases. Oncogene. 2005;24:5137–5148. doi: 10.1038/sj.onc.1208524. [DOI] [PubMed] [Google Scholar]
- 36.Bose K, Clark AC. Dimeric procaspase-3 unfolds via a four-state equilibrium process. Biochemistry. 2001;40:14236–14242. doi: 10.1021/bi0110387. [DOI] [PubMed] [Google Scholar]
- 37.Bose K, Clark AC. pH effects on the stability and dimerization of procaspase-3. Protein Sci. 2005;14:24–36. doi: 10.1110/ps.041003305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsuyama S, Llopis J, Deveraux QL, et al. Changes in intramitochondrial and cytosolic pH: early events that modulate caspase activation during apoptosis. Nat Cell Biol. 2000;2:318–325. doi: 10.1038/35014006. [DOI] [PubMed] [Google Scholar]
- 39.Bose K, Pop C, Feeney B, et al. An uncleavable procaspase-3 mutant has a lower catalytic efficiency but an active site similar to that of mature caspase-3. Biochemistry. 2003;42:12298–12310. doi: 10.1021/bi034998x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mittl PRE, DiMarco S, Krebs JF, et al. Structure of recombinant human CPP32 in complex with the tetrapeptide acetyl-asp-val-ala-asp fluoromethyl ketone. J Biol Chem. 1997;272:6539–6547. doi: 10.1074/jbc.272.10.6539. [DOI] [PubMed] [Google Scholar]
- 41.Ganesan R, Mittl PRE, Jelakovic S, et al. Extended substrate recognition in caspase-3 revealed by high resolution X-ray structure analysis. J Mol Biol. 2006;359:1378–1388. doi: 10.1016/j.jmb.2006.04.051. [DOI] [PubMed] [Google Scholar]
- 42.Richardson JS, Richardson DC. Natural β—sheet proteins use negative design to avoid edge-to-edge aggregation. Proc Natl Acad Sci. 2002;99:2754–2759. doi: 10.1073/pnas.052706099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Scheer JM, Romanowski MJ, Wells JA. A common allosteric site and mechanism in caspases. Proc Natl Acad Sci. 2006;103:7595–7600. doi: 10.1073/pnas.0602571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blanchard H, Kodandapani L, Mittl PRE, et al. The three-dimensional structure of caspase-8: an initiator enzyme in apoptosis. Structure. 1999;7:1125–1133. doi: 10.1016/s0969-2126(99)80179-8. [DOI] [PubMed] [Google Scholar]
- 45.Chai J, Wu Q, Shiozaki E, et al. Crystal structure of a procaspase-7 zymogen: Mechanisms of activation and substrate binding. Cell. 2001;107:399–407. doi: 10.1016/s0092-8674(01)00544-x. [DOI] [PubMed] [Google Scholar]
- 46.Riedl SJ, Fuentes-Prior P, Renatus M, et al. Structural basis for the activation of human procaspase-7. Proc Natl Acad Sci. 2001;98:14790–14795. doi: 10.1073/pnas.221580098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilson KP, Black J-AF, Thomson JA, et al. Structure and mechanism of interleukin-1β converting enzyme. Nature. 1994;370:270–275. doi: 10.1038/370270a0. [DOI] [PubMed] [Google Scholar]
- 48.Romanowski MJ, Scheer JM, O’Brien T, et al. Crystal structures of ligand-free and malonate-bound human caspase-1: implications for the mechanism of substrate binding. Structure. 2004;12:1361–1371. doi: 10.1016/j.str.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 49.Hardy JA, Lam J, Nguyen JT, et al. Discovery of an allosteric site in the caspases. Proc Natl Acad Sci. 2004;101:12461–12466. doi: 10.1073/pnas.0404781101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walters J, Pop C, Scott FL, et al. A constitutively active and uninhibitable caspase-3 zymogen efficiently induces apoptosis. Biophys J. 2009;424:335–345. doi: 10.1042/BJ20090825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schipper JL, MacKenzie SH, Sharma A, Clark AC. A bifunctional allosteric site in the dimer interface of procaspase-3. Biophys Chem. 2011;159:100–109. doi: 10.1016/j.bpc.2011.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolan DW, Zorn JA, Gray DC, Wells JA. Small molecule activators of a proenzyme. Science. 2009;326:853–858. doi: 10.1126/science.1177585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peterson QP, Goode DR, West DC, Ramsey KN, Lee JJ, Hergenrother PJ. PAC-1 activates procaspase-3 in vitro through relief of zinc-mediated inhibition. J Mol Biol. 2009;388:144–158. doi: 10.1016/j.jmb.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]