

Abstract
Helicobacter pylori is a gram-negative bacterium that colonizes the human stomach, causes gastritis, and is associated with ulcers and gastric cancer. H. pylori is naturally competent for transformation. Natural genetic transformation is believed to be essential for the genetic plasticity observed in this species. While the relevance of horizontal gene transfer in H. pylori adaptiveness and antibiotic resistance is well documented, the DNA transformation machinery components are barely known. No enzymatic activity associated with the transformation process has been determined experimentally and described. We isolated, microsequenced, and cloned a major DNA nuclease from H. pylori. This protein, encoded by the open reading frame hp0323, was expressed in Escherichia coli. The purified protein, NucT, has a cation-independent thermostable nuclease activity that preferentially cleaves single-stranded DNA. NucT is associated with the membrane. NucT-deficient H. pylori strains are one or more orders of magnitude less efficient than the parental strain for transformation with either chromosomal or self-replicating plasmid DNA. To the best of our knowledge, NucT is the first nuclease identified in a gram-negative natural transformation system, and its existence suggests that there is a mechanism of DNA processing and uptake similar to the mechanisms in well-studied gram-positive systems.
Helicobacter pylori is a gram-negative bacterium that infects the gastric mucosa of more than one-half of the world's human population (10). It is the major cause of peptic ulcers and the main risk factor for the development of gastric cancer (10). Previous molecular genetic work and comparative analysis of the complete genomic sequences of two isolates of H. pylori revealed the highest degree of genetic diversity reported for any bacterial species (22, 36). The panmictic structure of H. pylori populations has been determined by mathematical and experimental approaches (11, 35). A panmictic structure is not common in haploids, and it is expected that sequential selective sweeps and/or bottlenecks remove genetic variants resulting from mutational events from populations (35, 39). However, extremely frequent horizontal genetic exchange could maintain most of the generated allelic variants in a population, in addition to creating new ones (11, 35).
H. pylori is naturally competent for transformation, and natural genetic transformation is believed to be essential for the genetic plasticity observed in this and other haploid species (11, 22). Genetic variability is expected to be relevant in H. pylori adaptiveness given the diverse conditions to which this persistent pathogen is exposed (37). It has been widely reported that more than one strain can simultaneously colonize a human host, and it has also been proven that genetic recombination occurs during natural mixed infections in humans, yielding better-adapted pathogen variants (4, 11, 16, 34). These experiments proved that horizontal genetic transfer occurs in vivo. Not only can the ability to transfer genes improve the adaptiveness to a human host, but it can also facilitate the spread of resistance to clinically useful antibiotics (18, 38).
For H. pylori, the molecular mechanisms of DNA uptake and integration are barely known. Genes of the com operon, which are homologues of Agrobacterium tumefaciens vir genes, have been implicated in DNA transfer in H. pylori (33). The com operon consists of four genes, designated comB7, comB8, comB9, and comB10 (14). Knockout of the individual genes in this operon leads to transformation efficiencies that are reduced by 10- to 100-fold (14). Other genes, such as comB4, comH, and dprA and the open reading frames hp0015, hp1089, hp1424, and hp1473, were identified as transformation genes because their absence impairs or eliminates the transformation process. However, their specific roles are still unknown (6, 15, 31, 32).
EndA and NucA are proteins that are required for Streptococcus pneumoniae and Bacillus subtilis DNA transport, respectively. Both EndA and NucA are membrane-localized proteins. EndA is a nuclease that acts on both DNA and RNA, and it is specifically involved in the degradation of the nontransported strand (3, 9). The fact that NucA is required for DNA cleavage during transformation of B. subtilis suggests that it probably acts as a nuclease. No protein with nuclease activity has been identified in any gram-negative competence system (9). In this paper we report biochemical and genetic characterization of the hp0323 protein product, which we designated NucT (for “nuclease involved in transformation”). Our results show that NucT is a vigorous membrane nuclease actively involved in H. pylori DNA transformation.
MATERIALS AND METHODS
H. pylori strains, plasmids, and media.
H. pylori strains and plasmids used in this study are listed in Table 1. Escherichia coli strains were grown in Luria-Bertani (LB) broth or agar. H. pylori strains were grown on brucella blood agar plates at 37°C in an atmosphere containing 5% CO2 with 95% humidity.
TABLE 1.
H. pylori strains and plasmids used in this study
| Strain or plasmid | Relevant genotype or phenotype | Reference or source |
|---|---|---|
| H. pylori strains | ||
| 26695 | Reference strain | 36 |
| X47-2AL | Parental strain | 20 |
| X47-2ALnucT-Km | nucT::kanamycin | This study |
| X47-2ALnucT-Gm | nucT::gentamicin | This study |
| N6 | Parental strain | A. Labigne collection |
| N6 nucT | nucT::kanamycin | This study |
| ADM1 | Clinical isolate | 25 |
| Plasmids | ||
| pMAL-p2 | N-terminal MBP exported fusion | New England Biolabs |
| pQE-30 | N-terminal His tag | Novagen, Inc. |
| pILL444 | ColE1, suicide vector for H. pylori, gentamicin resistance | 7 |
| pHEL3 | Self-replication in H. pylori, kanamycin resistance | 13 |
| pMAL-hp0323 | pMAL-p2 containing PCR-amplified fragment encoding NucT protein fused to an N-terminal MBP | This study |
| pQE-hp0323 | pQE30 containing PCR-amplified fragment encoding NucT protein fused to an N-terminal His tag | This study |
Nuclease assays.
Unless indicated otherwise, the buffer used in all assays contained 20 mM Tris-HCl, 200 mM NaCl, 10 mM β-mercaptoethanol, and 10 mM EDTA (pH 8.0). The different nuclease assays, performed for different purposes, are described below.
(i) Chromosomal DNA.
14C-labeled DNA was isolated from cultures of the thyA E. coli strain JS54 (2) supplemented with [methyl-14C]thymine as described previously (30); 1,000 to 50,000 cpm of preheated 14C-labeled DNA was used as the substrate in a 200-μl reaction mixture. The reaction was carried out at 70°C and was stopped by adding 1 volume of 10% trichloroacetic acid. After centrifugation at 20,000 × g for 15 min, the soluble radioactivity was measured by liquid scintillation counting. One unit was defined as the amount of protein required to degrade 1 μg of [14C]DNA per hour (specific activity of the DNA, 33,480 cpm/μg).
(ii) Plasmid DNA.
Reaction mixtures (20 μl) containing 250 ng of pUC18 supercoiled plasmid DNA and/or 1 μg of S. cerevisiae rRNA were incubated with 0.1 mM NucT at 37°C. At different times the reactions were terminated by addition of 3 μl of a stop solution (0.25% [wt/vol] bromophenol blue, 25% [vol/vol] glycerol). The reaction products were immediately separated on 0.8% (wt/vol) agarose gels. The gels were stained with 1 μg ethidium bromide per ml in 10 mM Tris-HCl buffer containing 1 mM EDTA at pH 8.0. Linear pUC18 DNA was prepared by cleavage with HindIII.
(iii) Oligonucleotides.
Oligonucleotides (Life Technologies, Inc.) were labeled with 32P at their 5′ ends with T4 polynucleotide kinase (New England Biolabs). Double-stranded substrates were prepared by annealing labeled oligonucleotides with the nonlabeled complementary strands. Reactions were performed with 1 μM NucT in activity buffer at 37°C. Products were separated from substrates in 20% polyacrylamide-7 M urea gels.
(iv) In situ gel assay.
Gels for Tricine-polyacrylamide gel electrophoresis (PAGE) consisting of 16% T-6% C, as described by Schagger and von Jagow (29), without sodium dodecyl sulfate (SDS) were prepared with 100 μg of heat-denatured salmon testis DNA per ml. Samples (2 μg) of the different nuclease-containing fractions were heated at 70°C in 10 mM Tris-HCl-10 mM EDTA-10 mM β-mercaptoethanol-10% glycerol (pH 6.8) and loaded onto the gel. The tank buffer contained 0.1% SDS. Electrophoresis was performed at 4°C. The gels were washed twice for 10 min in Tris-EDTA and incubated at 37°C for different times in 20 mM Tris-HCl-200 mM NaCl-10 mM EDTA-5 mM β-mercaptoethanol (pH 8.0) containing 0.5 μg of ethidium bromide per ml. Single-stranded DNA degradation was monitored by UV transillumination of the gel until dark bands of nuclease activity were visible against a fluorescent background.
Purification of NucT from H. pylori.
Bacteria from 20 saturated plates containing H. pylori ADM1 were harvested, washed in Tris-EDTA, and resuspended in 10 ml of lysis buffer (20 mM Tris-HCl, 500 mM NaCl, 5 mM dithiothreitol [DTT], 1 mM phenylmethylsulfonyl fluoride [PMSF], 0.1% Brij 35 [pH 8.0], 0.1 mg of lysozyme per ml). After incubation at 37°C for 30 min, the cells were disrupted with a French press and centrifuged at 20,000 × g and 4°C for 30 min. The supernatant was heated for 15 min at 70°C and centrifuged at 20,000 × g and 4°C for 30 min. The supernatant of the heated preparation was dialyzed against 20 mM Tris-HCl-50 mM NaCl-1 mM EDTA (pH 8.0) and passed through a 2-ml HyperD-heparin affinity column (BioSepra, Inc.) equilibrated with the same buffer. Elution was carried out with a 50 mM to 1 M NaCl gradient. Fractions that were positive for nuclease activity eluted at 250 mM NaCl. This affinity-purified pool was concentrated to obtain a final concentration of approximately 1 mg/ml with an Ultrafree 15 concentrator (Millipore). The concentrated pool was fractionated through a Superdex 75 gel filtration column that was equilibrated with 20 mM Tris-HCl-250 mM NaCl-1 mM EDTA-1 mM PMSF (pH 8.0). Active fractions were pooled, dialyzed against 500 volumes of 25 mM Tris-HCl-50 mM NaCl-1 mM EDTA (pH 7.0), and then fractionated by using a Pharmacia MonoS (5/5) ion-exchange column that was eluted with a linear gradient of 50 to 1,000 mM NaCl. The nuclease activity eluted at approximately 300 mM NaCl. The Mrof the enzyme was determined with a Superdex 75 column (1.5 by 20 cm) that was preequilibrated with 20 mM Tris-HCl-500 mM NaCl-1 mM EDTA (pH 8.0) by using ovalbumin (Mr, 43,000), carbonic anhydrase (Mr, 29,000), and cytochrome c (Mr, 12,400) as reference proteins.
N-terminal amino acid sequence analysis.
The pool of active fractions that were eluted from the MonoS column (better-purified nuclease fraction from H. pylori) was extensively dialyzed against MilliQ water and subjected to SDS-PAGE at pH 8.3 (29). The proteins were electroblotted onto a polyvinylidene difluoride (PVDF) membrane in 10 mM CAPS [3-(cyclohexylamino)-1-propane sulfonic acid] buffer (pH 11.0) containing 10% (vol/vol) methanol by using a constant current of 125 mA for 30 min. After the electrotransfer, the PVDF membrane was washed several times with MilliQ water and stained with Coomassie brilliant blue R-250 in 1% (vol/vol) high-quality glacial acetic acid. The N-terminal amino acid sequence was determined by subjecting the blot to Edman degradation with an automated protein sequencer.
Cloning of NucT.
For the constructions described below the hp0323 gene was PCR amplified from H. pylori reference strain 26695, and the cloned inserts were completely sequenced.
(i) Construction of an exportable MBP-NucT fusion.
The forward and reverse primers used for construction of an exportable maltose binding protein (MBP)-NucT fusion were 5′-GGAATTCAAAAACAGCTTATTTGTCTTAGG-3′ and 5′-GGGAAGCTTAAACCCAACGACTCCCTAAC-3′, respectively. The amplified 486-bp fragment was ligated between the EcoRI and HindIII sites of the pMAL-p2 vector, resulting in plasmid pMAL-hp0323.
(ii) Construction of His-tagged NucT.
For construction of His-tagged NucT, the forward primer was 5′-CGCGGATCCAAAAACAGCTTATTTGTCTTAGG-3′, and the reverse primer was the primer used for construction of pMAL-hp0323. The hp0323 gene was cloned by using BamHI and HindIII sites into the vector pQE30, which introduced a His tag at the N terminus of the recombinant protein, resulting in plasmid pQE-hp0323.
Amino acid sequence analyses.
Amino acid comparisons were performed by using BLAST at the National Center for Biotechnology Information website (21). The putative signal sequence and the most probable site of cleavage were determined by using SignalP V1.1 (24). Theoretical pI/Mw ratios were determined by using the Compute pI/Mw tool at ExPASy (41).
Overexpression and purification of recombinant NucT.
For recombinant overexpression and purification of the soluble and active NucT, E. coli/pMAL-hp0323 clones were constructed by transforming E. coli TB1 (New England Biolabs) with the pMAL-hp0323 plasmid. E. coli TB1 cells were also transformed with the pMAL-p2 vector as a control. Purification of MBP was carried out parallel to purification of MBP-NucT in order to obtain a control fraction that could be used to test the absence of E. coli contaminating nuclease activities in the purified fraction. A 10-ml overnight culture was inoculated into LB broth containing 1% glucose and 200 μg of ampicillin per ml. The cultures were incubated at 37°C with aeration until the optical density at 600 nm was 1.5, and cells were centrifuged and resuspended in LB broth containing 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG). After 3 h of induction, cells were harvested by centrifugation at 4,000 × g and 4°C for 10 min. Cell pellets were resuspended in 25 ml of 20 mM Tris-HCl-1 mM EDTA-1 mM β-mercaptoethanol-1 mM PMSF (pH 8.0) and treated by following the manufacturer's recommendations for periplasmic extraction. The MBP-NucT fusion present in the soluble fraction was absorbed onto amylose resin (New England Biolabs) by batch mixing at room temperature for 1 h and then eluted with 10 mM maltose. NucT was cleaved from the fusion protein by digestion with factor Xa for 4 h at room temperature. NucT was isolated from MBP and factor Xa by fast protein liquid chromatography by using a Pharmacia MonoS (5/5) ion-exchange column. NucT was concentrated to a concentration of 5 mg/ml, glycerol was added to a final concentration of 50%, and the protein solution was stored at −20°C.
To generate antiserum, plasmid pQE-hp0323 was transformed into E. coli JM109 (New England Biolabs) and grown in LB broth at 37°C until an A600 of 0.8 was reached. Cultures were induced with 1 mM IPTG and grown for a further 3 h before bacteria were harvested by centrifugation, washed, and resuspended in lysis buffer (50 mM Tris-HCl, 1 mM EDTA, 100 mM NaCl, 1 mM PMSF; pH 8.0). Lysozyme (1 mg/ml) was added to the cells, and the cells were incubated at 37°C for 15 min. The cells were sonicated and centrifuged at 4°C for 30 min. The pellet containing the inclusion bodies was washed by successive resuspension and centrifugation with lysis buffer containing 0.5% Triton X-100 and then with lysis buffer containing 2 mg of deoxycholate per ml and finally with lysis buffer containing 1 M urea. Washed inclusion bodies were resuspended in lysis buffer containing 4 M urea.
Antisera, cell fractionation, and immunoblots.
Antiserum was generated by using purified and solubilized inclusion bodies of His-NucT as the antigen. Five 8-week-old mice were challenged with 50 μg of protein. Three boosters consisting of 10 μg of protein were used. The specific antiserum was obtained by clarifying the sera with raw extracts of induced E. coli JM109 containing pQE30.
Cells from one plate grown for 24 h were harvested, washed, and resuspended in 30 mM Tris-HCl-20% sucrose-0.5 mM DTT-1 mM EDTA (pH 8.0). The cells were incubated for 10 min with shaking and then centrifuged at 8,000 × g and 4°C for 10 min. The supernatant was removed, and the pellet was resuspended in 10 ml of ice-cold 5 mM MgSO4. The suspension was incubated at 4°C for 10 min with shaking and then centrifuged as described above. The supernatant corresponded to the periplasmic fraction. Cells were treated with 0.1 mg of lysozyme per ml at 37°C for 30 min, disrupted with a French press, and ultracentrifuged at 100,000 × g and 4°C for 1 h. The supernatant and pellet corresponded to the cytoplasm and membrane fractions, respectively. Subcellular fractions were resuspended in identical volumes. Aliquots (10 μl) of each fraction (approximately 20 μg of protein for the cytoplasmic fraction) were loaded in the polyacrylamide gel. The samples were analyzed by Western blotting by using anti-NucT mouse antisera diluted 1:500. Anti-mouse immunoglobulin G alkaline phosphatase-conjugated antibodies (1:30,000) were used to detect anti-NucT by the colorimetric method; 5-bromo-4-chloro-3-indolyl-1-phosphate and nitroblue tetrazolium were the substrates. 2-Keto-3-deoxyoctanoic acid quantification and anti-OmpF Western blotting were used as fraction controls as described previously (12).
Construction and phenotype analysis of mutants of H. pylori.
H. pylori hp0323 mutants were created by allelic exchange and natural transformation with suicide constructs as reported previously (7). Disruption of the correct gene was confirmed by PCR and sequencing. The competence of the mutants for natural transformation was determined as described previously (40). Briefly, fresh 24-h cultures of H. pylori were harvested and transferred as thick patches onto fresh plates. After 5 h, 1 μg of chromosomal DNA from an H. pylori resistant strain or 1 μg of plasmid pILL444 or pHel3 isolated from E. coli strain MC1061 (5) was added to each patch. At 16 h posttransformation, the bacteria were harvested and suspended in brucella broth. All or 100 μl of this suspension was spread on selective plates containing metronidazole (15 μg/ml), streptomycin (50 μg/ml), or kanamycin (20 μg/ml), and the number of resistant colonies was determined by counting the colonies after 5 days of incubation. Viable cells were quantified by the serial dilution method. The frequency of spontaneous mutation to rifampin resistance was measured as previously described (26). The transformation frequency was defined as the number of resistant colonies divided by the total number of viable bacteria per microgram of DNA minus the spontaneous mutation frequency.
The abilities of the NucT-deficient and parental cells to colonize the Swiss model mouse strain were assessed by performing coinfection experiments as previously described (26).
RESULTS
hp0323 codes for a nuclease of H. pylori.
A strong nuclease activity can be detected in crude extracts of H. pylori (19). A specific assay for this nuclease activity was developed. Trichloroacetic acid-soluble products were analyzed after exposure of heat-denatured E. coli chromosomal [14C]DNA to H. pylori cell extracts or to the purified protein. Incubations that were 10 times longer were required to detect products in the soluble fraction when chromosomal DNA was not previously heated. Because of the thermostability and cation independence of nuclease activity (see below), the reaction was carried out at 70°C and in the presence of EDTA. The nuclease activity was monitored through different purification steps by this radioactive soluble method. The optimized protocol for purification of the nuclease activity included heat precipitation, heparin affinity, gel filtration, and anion exchange. The specific activity increased from 297 to 40,116 U/mg, and the overall yield was 16%. After purification, four discrete bands were detected on silver-stained SDS-PAGE gels. In order to distinguish the polypeptide responsible for the nuclease activity, an in situ gel assay was performed (see Materials and Methods). The activity was visualized as dark growing bands on a fluorescent background due to the ethidium bromide-stained DNA. After 24 h of incubation of the gel in activity buffer, the degradation zone reached the maximum size (Fig. 1A). Once the activity band was clearly observed and its position was marked by cutting its edges, the gel was stained with Coomassie blue, which revealed a protein band that overlapped the dark activity band (Fig. 1B). The molecular weight markers provided a negative control, showing that DNA-free regions (dark bands) were not artifacts caused by exclusion of DNA from areas in the gel occupied by any protein. This conclusion was also supported by the fact that the dark bands grew over time. By using this method we were able to assign the nuclease activity to a single band at an apparent molecular mass of 17 kDa (Fig. 1). The active band was electrotransferred to PVDF membranes and microsequenced by the Edman method. The amino-terminal sequence obtained was KNSLFVLPY. This sequence corresponds to the amino-terminal end of a putative membrane nuclease, without the predicted signal peptide, encoded by the hp0323 open reading frame from H. pylori (36).
FIG. 1.

Assignment of the DNase activity to a protein band on an in situ activity gel. (A) DNase activity with preheated chromosomal DNA in 16% T-6% C Tricine-PAGE, as revealed by ethidium bromide staining after incubation at 37°C for 24 h in activity buffer. Lane 1, better-purified DNase fraction; lane 2, nuclease S1 (positive control). (B) Molecular mass markers (lane MW) and the same samples that were used for panel A. The gel was stained with Coomassie blue. The numbers on the left indicate the sizes of the molecular mass markers (in kilodaltons).
Analysis of the hp0323 nucleotide and amino acid sequences.
Based on the genome sequence of H. pylori 26695, it is predicted that the HP0323 gene is transcribed monocistronically since the upstream and downstream genes are in opposite orientations. Analysis of the sequence upstream of the predicted ATG revealed a perfect TATAAT −10 promoter element; however, no typical −35 E. coli consensus sequence was found, which is consistent with previous analyses of transcription in H. pylori (8, 23). A ribosomal binding site was identified upstream of the hp0323 open reading frame, which has the coding capacity for a protein consisting of 180 amino acids. A 23-residue sequence consistent with a signal peptide was found at the N terminus. The predicted molecular mass of the mature protein (amino acids 24 to 180) is 17.75 kDa. The experimentally determined N terminus of HP0323 is consistent with removal of the predicted 23-amino-acid signal peptide. The protein is highly conserved in H. pylori J99 (accession no. NP_223026), exhibiting 96% identity and 99% similarity. BLASTp analysis of the NucT amino acid sequence showed that it has significant similarity to a large number of nucleases and putative nucleases from different organisms. The highest levels of similarity were with the proteins from Helicobacter hepaticus strain ATCC 51449 (accession no. NP_860236; 55% identity and 72% similarity) and Wolinella succinogenes strain DSMZ 1740 (accession no. NP_906451; 48% identity and 69% similarity). The closest relative of NucT in the BLASTp list whose biological role has been determined is a nuclease from Proteus vulgaris. This enzyme has been characterized as a hydroxymethylcytosine-specific restriction enzyme (PvuRts1I) (17). NucT and PvuRts1I exhibit only 29% identity in a region consisting of 119 residues. Other characterized homologs are also involved in providing or preventing restriction barriers to DNA invasion.
NucT can be overexpressed and purified as an exported MBP fusion from E. coli.
To obtain purified NucT, hp0323 was PCR amplified from the H. pylori 26695 reference strain, purified, and cloned into the pMAL-p2 vector to be expressed as an MBP-NucT fusion. Upon IPTG induction, E. coli TB1 cells transformed with the pMAL-hp0323 plasmid produced the expected ∼58-kDa MBP-NucT fusion protein (Fig. 2). The MBP-NucT fusion was released from cells by osmotic shock, and the fusion was isolated from the periplasmic fluid by amylose-resin affinity purification. Between 1 and 2 mg of MBP-NucT was obtained from 3-liter cultures. Since E. coli chromosomal DNA is efficiently degraded by NucT, the low yield could have been a consequence of a potential NucT toxic effect. In fact, the only construction that gave detectable quantities of active NucT was the fusion to periplasmic MBP, whereas other constructions, such as glutathione S-transferase-MBP, His tag-MBP, S tag-MBP, and cytoplasmic MBP fusions, yielded an inactive protein and/or undetectable levels. Purification of NucT from MBP after factor Xa treatment was performed by anion-exchange chromatography. NucT eluted from the MonoS column at 250 mM NaCl as a pure single band (Fig. 2). The purified enzyme was stored at a concentration of 5 mg/ml in 50 mM Tris-HCl-10 mM β-mercaptoethanol-100 mM NaCl-10 mM EDTA-50% glycerol (pH 8.0) at −20°C. Under these conditions, the enzyme was stable for several months with a minimal loss of activity.
FIG. 2.

NucT purification from E. coli: SDS-PAGE of preparations after the different steps used for purification of NucT from an MBP-NucT fusion overexpressed in E. coli TB1. Lane 1, total induced cells; lane 2, periplasmic fraction; lane 3, amylase-purified fusion; lane 4, fusion after Xa treatment; lane 5, NucT after MonoS purification. Lane MW contained molecular mass markers, whose sizes (in kilodaltons) are indicated on the right.
The same purification protocol was carried out by using E. coli TB1 cells transformed with pMALp2 without any inserted gene. The purified MBP did not have the ability to degrade DNA or RNA. The results served as control for the specificity of the products observed in the following experiments. Furthermore, no nonspecific nuclease activity able to degrade single- or double-stranded DNA was present in the wild-type E. coli raw extracts under the conditions assayed.
NucT is a thermostable cation-independent nuclease.
The purified 17-kDa nuclease was characterized by the radioactive soluble method, in which E. coli heated chromosomal [14C]DNA was used as the substrate. The optimal pH for NucT activity was 8.0, and the optimal temperature was 80°C. The activity was 20 times higher at 70°C than at 37°C. After incubation at 70°C for 30 min, this enzyme was fully active. NucT was not inhibited by 10 mM EDTA, and its activity was not stimulated by the presence of the divalent cations Ca, Mg, and Mn. NucT was more active in the presence of reducing agents, such as DTT or β-mercaptoethanol, and it was inactivated by 50 mM oxidized glutathione but not by 1 mM ZnCl2, PbCl2, or HgCl2 (data not shown). The Mr of the enzyme, as determined by gel filtration, was approximately 32,000, suggesting that NucT acts as a dimer.
NucT acts preferentially on single-stranded DNA with weak sequence selectivity.
The substrate preference and the products of NucT were analyzed by using the recombinant protein. When pUC18 DNA, including supercoiled covalently closed circular, open circular, and linear forms, was treated with 0.1 mM purified NucT, all the DNA was degraded (Fig. 3). Since supercoiled DNA has no free ends, this result showed that NucT possesses an endonucleolytic activity. Complete degradation could result from repeated rounds of endonucleolysis or by subsequent exonucleolytic degradation from the free ends produced by the initial cleavage. The specificity of cleavage of NucT was analyzed further by using 32P-labeled oligonucleotides as substrates.
FIG. 3.
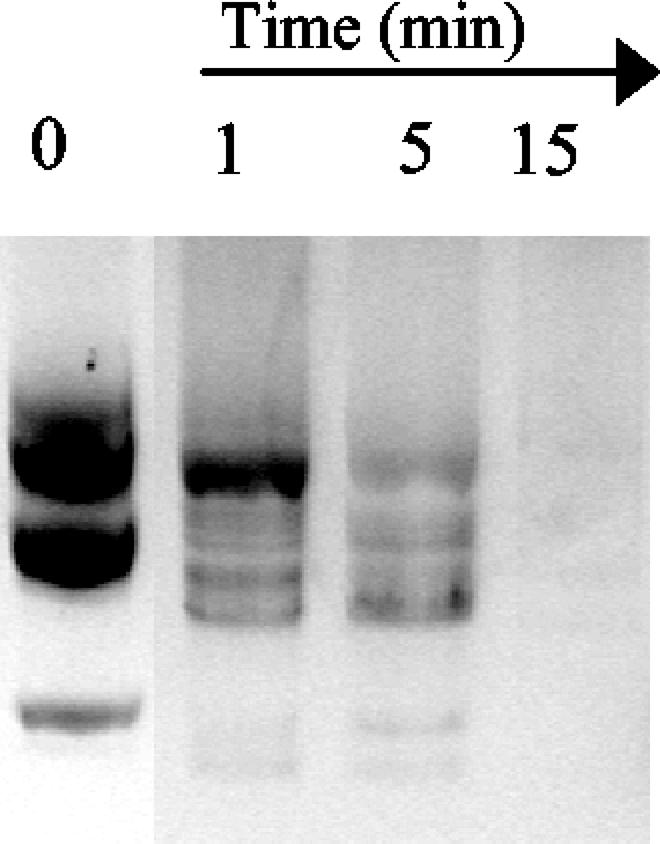
Endonuclease activity of NucT: degradation of pUC18 at 37°C when it was exposed to 0.1 mM NucT in activity buffer for several different times. No degradation was observed after 1 h of incubation with 50 μg of MBP purified in parallel with MBP-NucT from E. coli TB1/pMAL-p2 (data not shown).
Figure 4 shows the products obtained when a 34-mer that was labeled with 32P at the 5′ end was incubated with the purified enzyme at a concentration of 1 μM for various lengths of time at 37°C. This experiment was carried out with either single- or double-stranded 34-mer. The different intermediates produced did not permit us to unambiguously determine if NucT possesses an exonucleolytic activity in addition to its endonucleolytic activity or if it fully degrades DNA by successive rounds of endonucleolysis. The heterogeneity in the intensities of some of the bands produced by NucT, in conjunction with the fact that two oligonucleotides of the same length did not give the same pattern of products, suggests that there is some type of weak selectivity that is not strictly sequence dependent as it occurs for DNase I (1).
FIG. 4.

Exonuclease activity of NucT: degradative activity of 1 μM NucT with 100 fmol of a 34-base oligonucleotide that was labeled a the 5′ end with 32P. Block 1, hydrolysis of a single-stranded 34-mer for several different times; block 2, 5-min reaction with 100 fmol of the single-stranded 34-mer shown in block 1 preincubated with 1 μg of S. cerevisiae rRNA; block 3, activity with a second single-stranded 34-mer with a different sequence; block 4, hydrolysis of the double-stranded form of the 34-mer shown in block 1 for several different times. The sequences of the two oligonucleotides used as substrates and molecular weight markers are shown. Lanes MW contained molecular mass markers, whose sizes (in kilodaltons) are indicated on the right.
When the same oligonucleotide in its single- and double-stranded forms was used as the substrate, it was observed that NucT degraded the single-stranded form more efficiently than it degraded the double-stranded form (Fig. 4, compare blocks 1 and 4). It is worth noting that under these conditions the products were different, suggesting that the structure of the substrate has an effect on NucT activity.
Hydrolysis of single-stranded DNA could be prevented by addition of a 1,000:1 excess of eukaryotic rRNA (Fig. 4, block 2), but a 100:1 excess of RNA did not inhibit degradation of the oligonucleotide. Figure 5 shows specifically the degradation of RNA by 0.1 mM NucT. No RNase activity was detected in the purified MBP fraction under the conditions assayed.
FIG. 5.
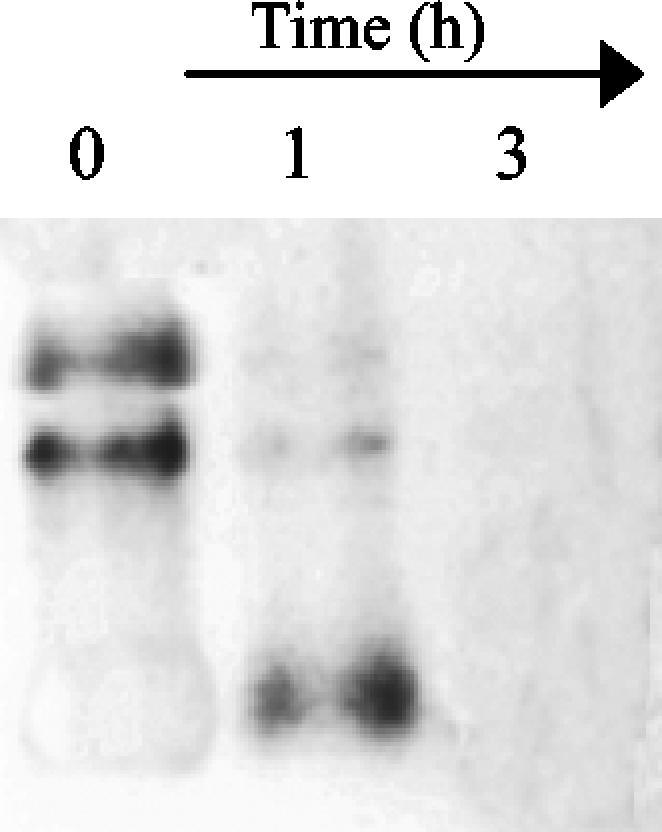
RNase activity of NucT: degradation rRNA from S. cerevisiae when it was exposed to 0.1 mM NucT at 37°C for several different times. No degradation was observed after 5 h of incubation with 50 μg of MBP purified in parallel with MBP-NucT from E. coli TB1/pMAL-p2 (data not shown).
Taken together, the results show that NucT efficiently degrades nucleic acids endonucleolytically and probably exonucleolytically, that single-stranded DNA is a better substrate than double-stranded DNA, and that DNA is hydrolyzed more efficiently than RNA is hydrolyzed.
NucT is associated with the H. pylori membrane.
The anti-NucT serum was used to examine the subcellular localization of NucT. Periplasmic, cytoplasmic, and membrane fractions were prepared sequentially by osmotic shock, treatment with a French press, and ultracentrifugation. The fractions were resuspended in equal volumes, and identical aliquots were loaded on SDS-polyacrylamide gels. The fraction contents were verified by using 2-keto-3-deoxyoctanoic acid quantification and anti-OmpF antisera as controls (data not shown). Figure 6 shows that NucT was located mainly in the membrane fraction. Washing of the membranes with 1 M NaCl released the enzyme into the soluble fraction, suggesting that the protein is associated with but not anchored to the membranes of H. pylori.
FIG. 6.

NucT subcellular localization: immunoblot analysis of whole-cell lysates and subcellular fractions of the H. pylori wild type and its nucT derivative (see Materials and Methods). Lane 1, wild-type whole-cell lysate; lane 2, mutant whole-cell lysate; lane 3, periplasm; lane 4, membranes; lane5, cytoplasm; lane 6, NaCl wash of the membrane fraction; lane 7, membrane fraction after salt wash.
NucT-deficient H. pylori mutants are deficient in transformation capability.
In order to elucidate the role of NucT in the DNA metabolism of H. pylori, nucT insertional mutants with different backgrounds were constructed. Extracts from the mutant cells were not able to hydrolyze DNA in the presence of EDTA at 70°C. Some activity (200-fold lower) remained at 37°C. The results show that hp0323 codes for the main nuclease activity of H. pylori.
There were no differences in in vitro growth between the parental and nucT derivative cells (data not shown). Furthermore, coinfection experiments showed that NucT-deficient cells colonized the mouse stomach at the same rate as the wild-type strain (approximately 5 × 105 CFU/g of tissue15 and 30 days after infection). Taken together, the results provide evidence that NucT is not required for cell duplication and does not act as a virulence factor, at least in the mouse model (Swiss mice) and under the conditions assayed.
The frequency of spontaneous mutation was analyzed as previously described (26). The absence of NucT did not change the mutation rate of the deficient H. pylori derivatives.
By contrast, the competence of the hp0323 mutants for transformation with chromosomal DNA was between 10- and 100-fold lower in the NucT-deficient derivatives than in the parental strain. While transformation with chromosomal DNA depends on allelic exchange through recombination, transformation with self-replicating plasmids does not involve integration of the DNA into the chromosome. To further elucidate the role that NucT plays in H. pylori transformation, experiments were carried out in which a self-replicating plasmid was used as the donor DNA. The parental strain X47-2AL (20) and derivative mutants of this strain were tested for their natural transformation competence with pHel3. In this case, a 10-fold reduction in the transformation capacity was observed in the mutant strain (Table 2). A plasmid of the expected size was purified from the transformants, which indicates that pHel3 was indeed maintained as a plasmid. This result indicates that NucT is not involved in DNA integration but is involved in DNA binding and/or uptake by the cell.
TABLE 2.
Transformation rates of H. pylori wild-type and NucT-deficient strains
| H. pylori strain | Transformation ratesa
|
||||
|---|---|---|---|---|---|
| Chromosomal DNA (gentamicin resistant)b | pILL444 (gentamicin resistant) | Chromosomal DNA (streptomycin resistant)b | Chromosomal DNA (kanamycin resistant)b | pHEL3 (kanamycin resistant) | |
| N6 | 5.6 × 10−6 | 1.23 × 10−6 | NDc | ||
| N6 nucT | 1.3 × 10−9 | <1 × 10−9 | ND | ||
| X47-2AL | ND | ND | 1.19 × 10−7 | 5.75 × 10−7 | 5.26 × 10−9 |
| X47-2ALNucT-Km | ND | ND | 6.70 × 10−9 | ||
| X47-2ALNucT-Gm | ND | 2.29 × 10−8 | 4.67 × 10−10 | ||
The transformation rates are expressed as the number of transformants per microgram of DNA per viable cell.
Chromosomal DNA from streptomycin-, gentamicin-, and kanamycin-resistant strains were used in the transformation experiments.
ND, not done.
DISCUSSION
The strong DNase activity found in H. pylori has been used as a species marker for years (19). In this work we identified the gene coding for the major nuclease from H. pylori, which we designated NucT. The product of this gene was characterized by functional and biochemical approaches. NucT has endonuclease and possibly 3′-to-5′ exonuclease activities that exhibit weak structural selectivity. Single-stranded DNA is a better substrate for NucT than other nucleic acids are, although the enzyme recognizes and hydrolyzes double-stranded DNA, as well as RNA. NucT seems to be structurally very stable. It is thermostable, it recovers from SDS treatment, and it resists multiple freeze-thaw cycles. NucT stability could be a by-product of its function, localization, and/or interaction with some partners. We are performing site-directed mutagenesis in order to investigate the nature of NucT stability further.
The results of a computer analysis of the amino acid sequence of NucT were consistent with a protein that is exported from the cytosol and undergoes cleavage of the signal peptide. Furthermore, the sequence showed high levels of similarity and identity with a large number of nucleases and putative endonucleases. However, the level of homology can lead to misinterpretation of the H. pylori metabolism, as we have previously demonstrated (25). Here we experimentally demonstrated that the product of the hp0323 open reading frame actually encodes a nuclease activity, NucT. Furthermore, the biological role of NucT was established.
Since disruption of hp0323 resulted in a significant deficiency for transformation, the gene was designated nucT (nuclease involved in transformation). By contrast, the absence of NucT, which resulted in a complete loss of the promiscuous thermoresistant and cation-independent nuclease activity present in wild-type H. pylori, did not affect the viability, colonization efficiency, or mutation frequency of the mutant cells. The absence of NucT resulted in at least a 1-log reduction in the transformation efficiency with a chromosomal marker. The fact that NucT-deficient cells also exhibited a reduction in the transformation efficiency when a self-replicating plasmid was used as the donor DNA strongly suggests that NucT is not involved in chromosomal integration but is involved in a previous step in the transformation process, ranging from DNA binding to the cell surface to DNA translocation through the membrane. We observed transformation frequencies lower than the values reported previously with all kind of donor DNAs. The strains assayed might be less competent than other strains, or our culture conditions may have disfavored competence.
It seems unlikely that DNA binding to the cell surface requires a protein with a potent nuclease activity. In spite of the fact that we cannot rule out this hypothesis, in a simpler model NucT is actively involved in DNA transfer through the H. pylori membrane. Two other proteins located in the membrane, EndA and NucA, have been found to be involved in bacterial transformation (9, 27). Like NucT, EndA is a promiscuous membrane nuclease that is capable of degrading DNA and RNA endonucleolytically and exonucleolytically (9). By contrast, NucA seems to possess only endo-DNase activity. However, since the experiments described by Provvedi et al. (27) were in vivo experiments, the specific nature of the NucA substrates and products remains uncertain.
The deduced amino acid sequence of NucT includes a signal peptide in the N-terminal region (36), which is not present in the mature protein; this supports the hypothesis that the peptide is cleaved during translocation from the cytoplasm to the membrane. We were unable to clone an active form of the nuclease that harbored the signal peptide sequence, suggesting that the presence of the signal peptide could inhibit NucT activity, perhaps by affecting protein folding. The mature form of NucT could not be actively expressed in the cytoplasm, and the purified protein was activated by reducing agents and inhibited by oxidized glutathione.
Taken together, the biochemical activity, the subcellular localization of the enzyme, and the phenotype of the NucT-deficient cells support the hypothesis that NucT is actively involved in the intermediate steps of the transformation process.
Given the association of NucT with the membrane and its role in transformation, it is tempting to suggest that NucT is a component of the Com transformation complex. The two-hybrid protein-protein interaction map of H. pylori did not reveal any particular interaction between NucT and other H. pylori proteins (28). However, other Com-Com interactions, such as those that involve ComB9, have been detected, although they were not found by the two-hybrid analysis (14, 28, 33). Elucidation of this question requires further experiments. Smeets and Kusters originally proposed that H. pylori possesses a nuclease function as part of the transformation machinery (33). Furthermore, these authors suggested that comH, a gene required for transformation whose product shows no homology to other known enzymes involved in DNA metabolism and whose localization and interaction with other Com proteins were not experimentally proved, could encode a nuclease responsible for DNA translocation through the membrane (31, 33). Although we cannot rule out this hypothesis, we can state that NucT is a good candidate for fulfilling such a DNA transfer function in H. pylori. Although many questions arose in the present study, the results described here portray the first reported nuclease involved in natural competence in gram-negative bacteria and represent a significant step forward in our understanding of the mechanism of natural competence in H. pylori.
Acknowledgments
We thank Ana Di Martino (Mitre Clinic, Buenos Aires, Argentina) for providing H. pylori clinical isolates and Rainer Haas (Max von Pettenkofer-Institut für Hygiene und Medizinische Mikrobiologie, LMU München, Munich, Germany) for providing pHel 3. We acknowledge Agnès Labigne (Institut Pasteur) and members of her lab, particularly Catherine Chevalier and Chantal Ecobichon, for providing materials, help, and advice. We especially thank J. Pablo Radicella (Département de Radiobiologie et Radiopathologie, CEA) for his invaluable collaboration. We thank to Gonzalo Prat Gay (Fundación Instituto Leloir) and members of his lab for helpful discussions and Marta Bravo and Jimena Ortega (Fundación Instituto Leloir) for their assistance with fast protein liquid chromatography and the sequencing experiment, respectively. We are grateful to Veronica Ielmini (Fundación Instituto Leloir) for providing anti-OmpF sera, S. cerevisiae RNA, suggestions, and help.
This work was supported by grants from Laboratorios Bagó SA and the Argentine Federal Government (grant ANPCyT PICT 01-09016) (to L.I.) and from CSUF (to M.E.T.). E.O. was the recipient of a doctoral fellowship from the University of Buenos Aires (FOMEC).
REFERENCES
- 1.Adams, R. L. P., J. T. Knowler, and D. P. Leader. 1992. The biochemistry of the nucleic acids, 11th ed. Chapman & Hall, London, United Kingdom.
- 2.Bejar, S., and J. P. Bouche. 1985. A new dispensable genetic locus of the terminus region involved in control of cell division in Escherichia coli. Mol. Gen. Genet. 201:146-150. [DOI] [PubMed] [Google Scholar]
- 3.Berge, M., M. Moscoso, M. Prudhomme, B. Martin, and J. P. Claverys. 2002. Uptake of transforming DNA in Gram-positive bacteria: a view from Streptococcus pneumoniae. Mol. Microbiol. 45:411-421. [DOI] [PubMed] [Google Scholar]
- 4.Bjorkholm, B., A. Lundin, A. Sillen, K. Guillemin, N. Salama, C. Rubio, J. I. Gordon, P. Falk, and L. Engstrand. 2001. Comparison of genetic divergence and fitness between two subclones of Helicobacter pylori. Infect. Immun. 69:7832-7838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Casadaban, M. J., and S. N. Cohen. 1980. Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J. Mol. Biol. 138:179-207. [DOI] [PubMed] [Google Scholar]
- 6.Chang, K. C., Y. C. Yeh, T. L. Lin, and J. T. Wang. 2001. Identification of genes associated with natural competence in Helicobacter pylori by transposon shuttle random mutagenesis. Biochem. Biophys. Res. Commun. 288:961-968. [DOI] [PubMed] [Google Scholar]
- 7.Clayton, C. L., and H. L. T. Mobley. 1997. Helicobacter pylori protocols. Humana Press, Totowa, N.J.
- 8.Delany, I., G. Spohn, R. Rappuoli, and V. Scarlato. 2002. Growth phase-dependent regulation of target gene promoters for binding of the essential orphan response regulator HP1043 of Helicobacter pylori. J. Bacteriol. 184:4800-4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dubnau, D. 1999. DNA uptake in bacteria. Annu. Rev. Microbiol. 53:217-244. [DOI] [PubMed] [Google Scholar]
- 10.Ernst, P. B., and B. D. Gold. 2000. The disease spectrum of Helicobacter pylori: the immunopathogenesis of gastroduodenal ulcer and gastric cancer. Annu. Rev. Microbiol. 54:615-640. [DOI] [PubMed] [Google Scholar]
- 11.Falush, D., C. Kraft, N. S. Taylor, P. Correa, J. G. Fox, M. Achtman, and S. Suerbaum. 2001. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: estimates of clock rates, recombination size, and minimal age. Proc. Natl. Acad. Sci. 98:15056-15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerhardt, P., R. G. E. Murray, W. A. Wood, and N. R. Krieg (ed.). 1994. Methods for general and molecular bacteriology. American Society for Microbiology, Washington, D.C.
- 13.Heuermann, D., and R. Haas. 1998. A stable shuttle vector system for efficient genetic complementation of Helicobacter pylori strains by transformation and conjugation. Mol. Gen. Genet. 257:519-528. [DOI] [PubMed] [Google Scholar]
- 14.Hofreuter, D., S. Odenbreit, and R. Haas. 2001. Natural transformation competence in Helicobacter pylori is mediated by the basic components of a type IV secretion system. Mol. Microbiol. 41:379-391. [DOI] [PubMed] [Google Scholar]
- 15.Hofreuter, D., S. Odenbreit, G. Henke, and R. Haas. 1998. Natural competence for DNA transformation in Helicobacter pylori: identification and genetic characterization of the comB locus. Mol. Microbiol. 28:1027-1038. [DOI] [PubMed] [Google Scholar]
- 16.Israel, D. A., N. Salama, U. Krishna, U. M. Rieger, J. C. Atherton, S. Falkow, and R. M. Peek, Jr. 2001. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proc. Natl. Acad. Sci. 98:14625-14630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Janosi, L., H. Yonemitsu, H. Hong, and A. Kaji. 1994. Molecular cloning and expression of a novel hydroxymethylcytosine-specific restriction enzyme (PvuRts1I) modulated by glucosylation of DNA. J. Mol. Biol. 242:45-61. [DOI] [PubMed] [Google Scholar]
- 18.Kersulyte, D., H. Chalkauskas, and D. E. Berg. 1999. Emergence of recombinant strains of Helicobacter pylori during human infection. Mol. Microbiol. 31:31-43. [DOI] [PubMed] [Google Scholar]
- 19.Lior, H., and A. Patel. 1987. Improved toluidine blue-DNA agar for detection of DNA hydrolysis by campylobacters. J. Clin. Microbiol. 25:2030-2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Londono-Arcila, P., D. Freeman, H. Kleanthous, A. M. O'Dowd, S. Lewis, A. K. Turner, E. L. Rees, T. J. Tibbitts, J. Greenwood, T. P. Monath, and M. J. Darsley. 2002. Attenuated Salmonella enterica serovar Typhi expressing urease effectively immunizes mice against Helicobacter pylori challenge as part of a heterologous mucosal priming-parenteral boosting vaccination regimen. Infect. Immun. 70:5096-5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Madden, T. L., R. L. Tatusov, and J. Zhang. 1996. Applications of network BLAST server. Methods Enzymol. 266:131-141. [DOI] [PubMed] [Google Scholar]
- 22.Marshall, D. G., W. G. Dundon, S. M. Beesley, and C. J. Smyth. 1998. Helicobacter pylori—a conundrum of genetic diversity. Microbiology 144:2925-2939. [DOI] [PubMed] [Google Scholar]
- 23.McGowan, C. C., A. S. Necheva, M. H. Forsyth, T. L. Cover, and M. J. Blaser. 2003. Promoter analysis of Helicobacter pylori genes with enhanced expression at low pH. Mol. Microbiol. 48:1225-1239. [DOI] [PubMed] [Google Scholar]
- 24.Nielsen, H., J. Engelbrecht, S. Brunak, and G. von Heijne. 1997. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10:1-6. [DOI] [PubMed] [Google Scholar]
- 25.O'Rourke, E. J., C. Chevalier, S. Boiteux, A. Labigne, L. Ielpi, and J. P. Radicella. 2000. A novel 3-methyladenine DNA glycosylase from Helicobacter pylori defines a new class within the endonuclease III family of base excision repair glycosylases. J. Biol. Chem. 275:20077-20083. [DOI] [PubMed] [Google Scholar]
- 26.O'Rourke, E. J., C. Chevalier, A. V. Pinto, J. M. Thiberge, L. Ielpi, A. Labigne, and J. P. Radicella. 2003. Pathogen DNA as target for host-generated oxidative stress: role for repair of bacterial DNA damage in Helicobacter pylori colonization. Proc. Natl. Acad. Sci. 100:2789-2794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Provvedi, R., I. Chen, and D. Dubnau. 2001. NucA is required for DNA cleavage during transformation of Bacillus subtilis. Mol. Microbiol. 40:634-644. [DOI] [PubMed] [Google Scholar]
- 28.Rain, J. C., L. Selig, H. De Reuse, V. Battaglia, C. Reverdy, S. Simon, G. Lenzen, F. Petel, J. Wojcik, V. Schachter, Y. Chemama, A. Labigne, and P. Legrain. 2001. The protein-protein interaction map of Helicobacter pylori. Nature 409:211-215. [DOI] [PubMed] [Google Scholar]
- 29.Schagger, H., and G. von Jagow. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368-379. [DOI] [PubMed] [Google Scholar]
- 30.Sekiguchi, M., S. Yasuda, S. Okubo, H. Nakayama, K. Shimada, and Y. Takagi. 1970. Mechanism of repair of DNA in bacteriophage. I. Excision of pyrimidine dimers from ultraviolet-irradiated DNA by an extract of T4-infected cells. J. Mol. Biol. 47:231-242. [DOI] [PubMed] [Google Scholar]
- 31.Smeets, L. C., J. J. Bijlsma, S. Y. Boomkens, C. M. Vandenbroucke-Grauls, and J. G. Kusters. 2000. comH, a novel gene essential for natural transformation of Helicobacter pylori. J. Bacteriol. 182:3948-3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Smeets, L. C., J. J. Bijlsma, E. J. Kuipers, C. M. Vandenbroucke-Grauls, and J. G. Kusters. 2000. The dprA gene is required for natural transformation of Helicobacter pylori. FEMS Immunol. Med. Microbiol. 27:99-102. [DOI] [PubMed] [Google Scholar]
- 33.Smeets, L. C., and J. G. Kusters. 2002. Natural transformation in Helicobacter pylori: DNA transport in an unexpected way. Trends Microbiol. 10:159-162. [DOI] [PubMed] [Google Scholar]
- 34.Suerbaum, S., and M. Achtman. 1999. Evolution of Helicobacter pylori: the role of recombination. Trends Microbiol. 7:182-184. [DOI] [PubMed] [Google Scholar]
- 35.Suerbaum, S., J. M. Smith, K. Bapumia, G. Morelli, N. H. Smith, E. Kunstmann, I. Dyrek, and M. Achtman. 1998. Free recombination within Helicobacter pylori. Proc. Natl. Acad. Sci. 95:12619-12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tomb, J. F., O. White, A. R. Kerlavage, R. A. Clayton, G. G. Sutton, R. D. Fleischmann, K. A. Ketchum, H. P. Klenk, S. Gill, B. A. Dougherty, K. Nelson, J. Quackenbush, L. Zhou, E. F. Kirkness, S. Peterson, B. Loftus, D. Richardson, R. Dodson, H. G. Khalak, A. Glodek, K. McKenney, L. M. Fitzegerald, N. Lee, M. D. Adams, J. C. Venter, et al. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539-547. [DOI] [PubMed] [Google Scholar]
- 37.Wang, G., M. Z. Humayun, and D. E. Taylor. 1999. Mutation as an origin of genetic variability in Helicobacter pylori. Trends Microbiol. 7:488-493. [DOI] [PubMed] [Google Scholar]
- 38.Wang, G., T. J. Wilson, Q. Jiang, and D. E. Taylor. 2001. Spontaneous mutations that confer antibiotic resistance in Helicobacter pylori. Antimicrob. Agents Chemother. 45:727-733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang, I. I., D. E. Taylor, and M. Z. Humayun. 2000. Response from Wang, Humayun and Taylor. Trends Microbiol. 8:58. [DOI] [PubMed] [Google Scholar]
- 40.Wang, Y., K. P. Roos, and D. E. Taylor. 1993. Transformation of Helicobacter pylori by chromosomal metronidazole resistance and by a plasmid with a selectable chloramphenicol resistance marker. J. Gen. Microbiol. 139:2485-2493. [DOI] [PubMed] [Google Scholar]
- 41.Wilkins, M. R., E. Gasteiger, A. Bairoch, J. C. Sanchez, K. L. Williams, R. D. Appel, and D. F. Hochstrasser. 1999. Protein identification and analysis tools in the ExPASy server. Methods Mol. Biol. 112:531-552. [DOI] [PubMed] [Google Scholar]