

Abstract
In the process of human hematopoiesis, precise regulation of the expression of lineage-specific gene products is critical for multiple cell-fate decisions that govern cell differentiation, proliferation, and self-renewal. Given the important role of microRNAs (miRNAs) in development and differentiation, we examined the global expression of miRNA in CD34+ cells during lineage specific hematopoiesis and found 49 miRNAs to be differentially expressed, with functional roles in cellular growth and proliferation, and apoptosis. miR-18a was upregulated during erythropoiesis and downregulated during megakaryopoiesis. miR-145 was upregulated during granulopoiesis and down regulated during erythropoiesis. Megakaryopoitic differentiation resulted in significant alteration in the expression of many miRNAs that are believed to play critical roles in the regulation of B and T cell differentiation. Target prediction analyses on three different miRNA databases indicated that TargetScan outperformed microCosm and miRDB in identifying potential miRNA targets associated with hematopoietic differentiation process. An integrated analysis of the observed miRNAs and messenger RNAs (mRNAs) resulted in 87 highly correlated miRNA-mRNA pairs that have major functional roles in cellular growth and proliferation, hematopoietic system development, and Wnt/B-catenin and Flt 3 signaling pathways. We believe that this study will enhance our understanding on the regulatory roles of miRNA in hematopoiesis by providing a library of mRNA-miRNA networks.
The phenotype of a cell is controlled by regulation of gene expression, which is the basis for cell differentiation, morphogenesis, and the adaptability of cells. Modification of gene expression can occur at different levels. Apart from epigenetic mechanisms (cytosine methylation, histone acetylation), regulation can be observed at the level of transcription initiation (transcription factors), heteronucleic transcript processing (RNA splicing), messenger (mRNA) transport from the nucleus into the cytoplasm (nucleocytoplasmatic transport factors, such as exportin-5), and translation and post-translational modifications [1–5]. It has recently become evident that non–protein-coding genes play an important role in the control of gene expression [5].
For example, regulation of gene expression through mechanisms that involve microRNAs (miRNAs) has attracted much attention. miRNAs are small noncoding RNAs that suppress gene expression by binding to partially complementary sequences mostly in the 3′UTR of mRNAs and inhibiting their translation into protein or accelerating their degradation. miRNAs regulate at least 30% of the protein-encoding genes and are involved in the regulation of a broad range of cellular aspects such as differentiation, function, proliferation, survival, metabolism, and response to changes in its environment. It is thought that miRNAs make an important contribution to the regulation of gene expression and that their dysregulation is implicated in disease pathophysiology [6–9]. Cumulative evidence now suggests that specific miRNAs and genetic variations interfering with miRNA function (miRNA polymorphisms) are involved in the prognosis and progression of a variety of diseases [10].
Hematopoietic lineage differentiation is known to be controlled by complex molecular events that regulate the self-renewal, commitment, proliferation, apoptosis, and maturation of stem and progenitor cells. Traditionally, the major focus of research has been to study the role of transcription factors in regulating hematopoiesis. Lineage-specific transcription factors are key regulators of gene expression in multiple cell-fate decisions that govern hematopoietic differentiation. Given the important role of miRNAs in development and differentiation, it is not surprising that these regulatory RNAs also play crucial roles in hematopoiesis [11–13]. It is believed that transcription factors and miRNAs act in concert to regulate gene expression during hematopoietic differentiation [14].
Because of the wealth of information available about the transcriptional and cellular networks involved in hematopoietic differentiation, and well-characterized processes for in vitro lineage-specific differentiation, the hematopoietic system is ideal for studying cell lineage specification and its regulation by microRNA. The integration of miRNA and mRNA expression data have been shown to be a good method for filtering sequence-based putative predictions [15]. Thus, we undertook a systematic approach to integrate analysis of miRNA and mRNA expression during hematopoietic differentiation.
Methods
Human CD34+ peripheral blood cells
Human CD34+ peripheral blood cells (PBCs) were collected by apheresis from healthy volunteers who were given G-CSF for 5 days (10 μg/kg per day). After CD34 antigen-mediated selection with immunomagnetic beads (ISOLEX300i system; Baxter Healthcare, Deerfield, IL, USA), purified CD34+ PBCs were collected and cryopreserved in liquid nitrogen until use.
Suspension cultures and growth factors
CD34+ PBCs were cultured in X-VIVO10 (BioWhittaker, Walkersville, MD, USA) supplemented with 1% human serum albumin. At least 1 × 106 CD34+ cells were seeded in six-well plates and incubated at 37°C and 5% CO2 in a fully humidified atmosphere. To induce lineage-specific differentiation, growth factors (R&D Systems, Irvine, CA, USA), were added to each well as follows: for erythropoietic differentiation (designated E), stem cell factor (SCF; 50 ng/mL), Flt3-ligand (50 ng/mL), IL-3 (10 ng/ml), and EPO (10 U/mL); for granulopoietic differentiation (designated G), SCF (50 ng/mL), Flt3-ligand (50 ng/mL), IL-3 (10 ng/mL), G-CSF, and GM-CSF (each, 10 ng/mL); for megakaryopoietic differentiation (designated M), SCF (50 ng/mL), Flt3-ligand (50 ng/mL), and TPO (20 ng/mL). All growth factors were added at the beginning of the culture period. The cells were cultured in triplicate in a final volume of 2 mL, in separate wells for each of the conditions and each of the time points. The cells were inspected daily for 11 days and harvested on days 0 and 11. To increase the purity of cells belonging to specific lineages, lineage-specific cells were enriched at the end of culture by immunomagnetic bead selection via the MACS system [16]. Cells of the erythropoietic lineage were selected using CD71+ microbeads (BD Biosciences, Pharmingen); cells of granulopoietic linage were selected using CD15+ microbeads (BD Biosciences, Pharmingen); and cells of the megakaryopoietic lineage were selected using CD61+ microbeads (BD Biosciences, Pharmingen). Purity of the differentiated cells were calculated based on the cells with the specific marker relative to percentage of stained cells.
Flow cytometry
Starting and cultured CD34+ cells (n = 3) were characterized by immunofluorescence using a BD FACSCanto flow cytometer. Erythropoietic cells (n = 3) were characterized by staining with an anti-CD71 FITC antibody (Miltenyi Biotec, Auburn, CA, USA). Megakaryocytic cells (n = 3) were determined with an anti-CD61 FITC antibody (Miltenyi Biotec, Auburn, CA, USA) and granulopoietic cells (n = 3) were analyzed with anti-CD15 FITC antibodies (Miltenyi Biotec, Auburn, CA, USA). Isotype-matched nonspecific antibodies were used a control group. Analysis gates were set to exclude dead cells and debris, with 10,000 viable cells analyzed per sample.
Total RNA with miRNA isolation
Total RNA enriched for microRNAs was extracted from all the 12 samples using the RNeasy mini kit (Qiagen, Valencia, CA, USA) following the manufacturer’s directions. The concentration of the isolated RNA was determined using the Nanodrop ND-100 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). Quality and integrity of the total RNA isolated was assessed on the Agilent 2100 bioanalyzer (Agilent Technologies, Palo Alto, CA, USA).
Affymetrix miRNA labeling, array hybridization, data preprocessing, and statistical analysis
Total RNA containing low-molecular-weight RNA was labeled using the Flashtag RNA labeling kit (Genisphere, Hatfield, PA, USA) according to the manufacturer’s instructions. For each sample, 2 μg total RNA was subjected to a tailing reaction (2.5 mmol/L MnCl2, adenosine triphosphate, Poly(A) Polymerase; incubation for 15 min at 37°C), followed by ligation of the biotinylated signal molecule to the target RNA sample (1 × Flash Tag ligation mix biotin, T4 DNA ligase; incubation for 30 min at room temperature) and the addition of stop solution.
Each sample was hybridized to a GeneChip miRNA Array version 2.0 (Affymetrix, Santa Clara, CA, USA) at 48°C and 60 rpm for 16 hours and then washed and stained on Fluidics Station 450 and scanned on a GeneChip Scanner 3000 7G (Affymetrix). The image data were analyzed with the miRNA QC Tool software for quality control (http://www.affymetrix.com/products_services/arrays/specific/mi_rna.affx#1_4). The raw data were preprocessed by Affymetrix miRNA QC Tool (Version 1.1.1.0) with the work-flow of RMA global background correction, quantile normalization, and median polish summarization. We selected 4592 unique mature human miRNA probes for differentially expressed miRNA detection analysis. Differential expression between CD34+ and E, M, and G was assessed using one-way ANOVA implemented in the MSCL Analyst’s Toolbox developed by two of the authors (J.B. and P.J.M.; http://abs.cit.nih.gov/MSCLtoolbox/) written in the JMP statistical software package (SAS Institute, Cary, NC, USA). The differentially expressed miRNAs were selected when the false discovery rate was less than 20% and the absolute fold change between any of E, G, or M cell groups compared with CD34+ was greater than 2. Unsupervised hierarchical clustering analysis was performed for selected differentially expressed miRNAs. The data have been deposited in Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, accession number: GSE35532).
MicroRNA putative target selection
TARGETSCAN 5.1 (www.targetscan.com), MicroCosm (http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/), and miRDB (http://mirdb.org/miRDB/) miRNA target databases were used to identify putative miRNA targets. The 49 differentially expressed miRNAs were uploaded to each of the three databases, and a list of their potential targets was obtained. The three target prediction tools were evaluated and compared against each other using NcNemar test [17] with Bonferroni correction for multiple comparisons and post hoc analysis for enrichment in predicted mRNA targets.
Integrated analysis of microRNA and mRNA expression profiles
Integrated analysis was performed on 172 mRNAs that were significantly modulated during lineage specific differentiation of CD34+ cells as reported in our published study [18] and the miRNAs identified from the current study. Individual miRNAs determined to be differentially expressed were submitted to each of the three databases (TargetScan, MicroCosm, mirDB) to determine the predicted mRNA targets. We selected only mRNA targets that were shown previously to be differentially expressed in these same samples. Next, the correlation between the expression levels of each miRNA and any of the predicted, differentially expressed target mRNA from any of the three databases was computed, and significant correlations were determined at p < 0.05. mRNAs that were highly correlated with any miRNA were further investigated by Ingenuity pathway analysis (IPA; Ingenuity Systems, Redwood City, CA, USA) to assign biological functions to putative target genes of the differentially expressed miRNAs.
Micro RNA quantitative polymerase chain reaction assays
Validation of expression of selected miRNAs was performed using commercially available predesigned TaqMan RT-qPCR assays (Applied Biosystems, Foster City, CA, USA) using the same RNA samples as used for the microarray profiling. Total RNA (500 ng) isolated from cell lysate was reverse transcribed using Human MegaPlex RT Primer Pools. Amplified samples were injected into TaqMan Array Human (version 2.0) MicroRNA Cards and analyzed using a 7900 Real-Time PCR System (Applied Biosystems). This platform consists of two arrays: TLDA panel A (377 functionally defined microRNAs) and TLDA panel B (289 microRNAs, whose function is not yet completely defined) for a total of 666 microRNA assays—all TaqMan MicroRNA. Each array or panel includes, among other endogenous controls, the mammalian U6 assay that is repeated four times on each card; this was used for data normalization. TLDA cards were run in the 7900 HT Sequence Detection system. The ABI TaqMan SDS version 2.3 software was used to obtain raw CT values. To review results, the raw CT data (SDS file format) was exported from the Plate Centric View into the ABI TaqMan RQ manager software. Automatic baseline and manual CT were set to 0.2 for all samples. Fold changes in the expression of selected significantly differentially expressed miRNAs (based on the microarray data) were compared with the fold changes obtained from microarrays to determine the correlation coefficient.
Results
In vitro differentiation of CD34+ cells
Lineage-specific cells were characterized by immunophenotyping as shown in Figure 1. The histograms illustrate the expression of lineage-specific cell surface markers that define the undifferentiated CD34+ and differentiated erythropoietic, granulopoietic, and megakaryopoietic cells. Figure 1A shows the unstained CD34+ cells on day 11 as a control group for panels in Figure 1B, 1C, and 1D following culture of the cells under erythroid (E), granulocytic (G) and megakaryocytic (M) conditions. After 11 days in E culture, out of the stained cells, 98% stained positive for CD71 representing erythropoietic differentiation, 99% of the G culture stained positive for CD15, and 78% of the M culture stained positive for CD61 representing megakaryopoietic differentiation. These numbers are shown in red in Figure 1.
Figure 1.

Flow cytometry characterization of CD34+ and differentiated E, G, and M cells by dual-color immunofluorescence using a BD FACSCanto flow cytometer. (A) Unstained CD34+ cells. (B) Erythropoietic cells stained with anti-CD71 FITC antibody. (C) Granulopoietic cells stained with anti-CD15 FITC antibody. (D) Megakaryocytic cells stained with anti-CD61 FITC antibody. The numbers in red show the actual percentage of cells positively stained with the specific CD surface marker out of the total number of stained cells analyzed in the flow cytometer.
Differential expression of microRNA during lineage specific differentiation of CD34+ cells
In an effort to identify differentially expressed microRNAs during differentiation of CD34+ cell into E, M, and G cells, hierarchical cluster analysis was performed showing the differential expression pattern across the lineage-specific stem cell types (Fig. 2). We observed differential expression on 49 miRNAs (Table 1) during hematopoietic differentiation. Analysis of these significantly differentially expressed miRNAs under the E, G, and M conditions revealed an overlap in the expression levels in the same direction for miR-181a, miR-493, miR-150, and miR-221 in all three groups of differentiated cells. miR-1975, miR-886, and miR-1308 showed significant downregulation during megakaryopoiesis, but upregulation during granulopoiesis and erythropoiesis. During granulopoiesis and megakarypoiesis, a group of 6 miRNAs (miR-181b, miR-92a, miR-17, miR-423, miR-1275, miR-106a) showed significant downregulation. Conversely, a group of six miRNAs that include, miR-1825, miR-1281, miR-198, miR-551b, miR-500, and miR-501 showed significant upregulation. For groups E and M, miR-320b and miR-222 showed downregulated expression, whereas miR-103, miR-107, and miR-191 showed upregulation in M and downregulation in E. Interestingly, miR-18a was found to be significantly unregulated only during erythropoiesis, whereas miR-145 showed significant upregulation only during granuolopoiesis. Megakaryopoitic differentiation resulted in unique alteration in miR-7f-star, miR-1228, miR-4306, miR-409, miR-190, miR-198, miR-191, miR-3138, miR-588, miR-204, and miR-500 (Table 1).
Figure 2.

Hierarchical cluster analysis performed on the 49 differentially expressed miRNA. miRNAs were chosen to be differentially expressed between four lineage-specific cell types, megakaryopoietic (M), granulopoietic (G), erythropoietic (E), and CD34+ stem cells are shown here. The cluster shows cell type fold change expression in E, G, and M groups compared with CD34+.
Table 1.
Differentially expressed miRNA during CD34+ differentiation
| miRNA title | (Fold changes –Log2)
|
||
|---|---|---|---|
| E vs CD34 | G vs CD34 | M vs CD34 | |
| hsa-let-7b | −0.68 | −0.63 | −1.82 |
| hsa-let-7f-1-star | 0.13 | 0.05 | 2.21 |
| hsa-let-7i | 0.25 | −0.01 | −5.79 |
| hsa-miR-103 | 1.39 | 0.22 | −4.14 |
| hsa-miR-106a | 0.86 | −1.59 | −5.76 |
| hsa-miR-107 | 1.56 | 0.23 | −3.27 |
| hsa-miR-1228 | 0.16 | 0.08 | 3.42 |
| hsa-miR-126 | 1.18 | −0.46 | 3.42 |
| hsa-miR-1268 | 1.00 | 1.72 | −2.89 |
| hsa-miR-1275 | −0.96 | −1.28 | −5.19 |
| hsa-miR-1281 | 0.51 | 1.48 | 5.51 |
| hsa-miR-1308 | 2.22 | 2.02 | −5.30 |
| hsa-miR-1308 | 1.23 | 1.42 | 0.09 |
| hsa-miR-145 | −0.25 | 3.90 | 0.32 |
| hsa-miR-150 | −3.97 | −3.94 | −3.07 |
| hsa-miR-15b | 0.70 | −0.15 | −6.74 |
| hsa-miR-17 | 0.76 | −1.33 | −5.69 |
| hsa-miR-181a | −1.32 | −2.49 | −7.52 |
| hsa-miR-181b | −0.86 | −3.17 | −7.04 |
| hsa-miR-1825 | 0.78 | 1.20 | 5.79 |
| hsa-miR-18a | 2.02 | 0.08 | −0.18 |
| hsa-miR-190 | 0.16 | −0.11 | 1.14 |
| hsa-miR-191 | 1.35 | 0.27 | −3.52 |
| hsa-miR-191 | 0.82 | 0.80 | 4.59 |
| hsa-miR-193b | 0.76 | 1.13 | 3.34 |
| hsa-miR-1975 | 2.20 | 2.96 | −3.76 |
| hsa-miR-198 | 0.55 | 1.85 | 5.94 |
| hsa-miR-198 | 0.30 | 0.38 | 3.33 |
| hsa-miR-204 | 0.35 | 0.12 | 1.53 |
| hsa-miR-210 | 2.76 | 3.29 | −0.12 |
| hsa-miR-221 | −2.11 | −1.09 | −7.11 |
| hsa-miR-222 | −1.82 | −0.45 | −6.12 |
| hsa-miR-3138 | 0.34 | 0.48 | 1.36 |
| hsa-miR-320b | −1.13 | −0.73 | −4.98 |
| hsa-miR-409 | 0.15 | 0.50 | 1.81 |
| hsa-miR-423 | −0.96 | −1.64 | −3.71 |
| hsa-miR-4258 | −0.05 | 1.04 | 4.38 |
| hsa-miR-4306 | 0.86 | 0.80 | 2.38 |
| hsa-miR-486 | 5.95 | −1.41 | −2.45 |
| hsa-miR-493 | 1.18 | 1.58 | 5.29 |
| hsa-miR-500 | 0.92 | 1.61 | 5.49 |
| hsa-miR-500 | 0.58 | 0.56 | 1.46 |
| hsa-miR-501 | 0.30 | 1.42 | 5.31 |
| hsa-miR-551b | −0.06 | 1.60 | 5.10 |
| hsa-miR-588 | 0.10 | 0.21 | 2.55 |
| hsa-miR-720 | 3.55 | 4.51 | −0.98 |
| hsa-miR-886 | 4.80 | 1.36 | −0.38 |
| hsa-miR-92a | −0.90 | −2.26 | −3.88 |
| hsa-miR-93 | 1.20 | 1.20 | −6.05 |
Validation of differentially expressed miRNAs by quantitative polymerase chain reaction analysis
We performed quantitative polymerase chain reaction (qPCR) analysis on selected miRNAs that were significantly upregulated and downregulated on the Affymetrix miRNA genechips and correlated the relative fold changes from these two platforms. We observed a strong correlation (R = 0.85 0.68, and 0.87) for the E, G, and M groups between the two platforms for the 49 miRNAs relative to CD34+ as shown in Figure 3. All three comparisons showed concordance in the same direction, thereby validating the microarray data.
Figure 3.
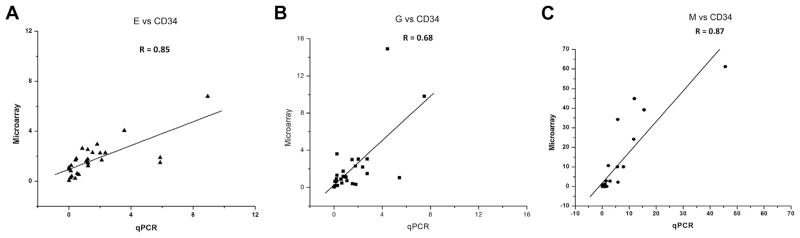
Validation by qPCR. Log2 expression fold change (A) E versus CD34; (B) G versus CD34; (C) M versus CD34 measured by microarray and qPCR on selected genes.
Selection of putative gene targets regulated by the differentially expressed miRNAs
miRNAs can regulate a large number of target genes, and several databases based on various algorithms are currently available for predicting miRNA gene targets. We chose to use Target Scan version 5.1, MicroCosm, and mirDB to predict gene targets of the 49 differentially expressed miRNAs identified in this study. This resulted in the identification of 12,440 unique gene targets from TargetScan, 11,621 from MicroCosm and 6072 from miRDB for the differentially expressed miRNA during lineage-specific CD34+ differentiation (Supplementary Table E1, online only, available at www.exphem.org).
Integrated analysis of miRNA and mRNA expression profiles
In an effort to verify that the predicted targets of differentially expressed miRNAs do regulate the mRNA expression levels during CD34+ differentiation, we performed an integrated analysis of miRNA predicted target genes and the experimentally observed target genes. After comparing the target predictions from the three different databases, TargetScan was found to give the largest number of miRNA-mRNA pairs (n = 428) and significantly correlated pairs (n = 87; Supplementary Table E2, online only, available at www.exphem.org). In addition, TargetScan demonstrated more negatively correlated miRNA-mRNA pairs (n = 54), which seems to be the more accepted mode of action between miRNA and mRNA. Of the three target prediction tools, mirDB overlaps to a large extent (72.6%) with TargetScan, and this overlap was more pronounced for the negatively correlated pairs (80%). Microcosm seems to be an outlier by showing poor overlap with both mirDB (4.2%) and TargetScan (23.2%). The McNemar tests with post hoc analyses revealed an enrichment in negatively correlated mRNA-miRNA pairs for TargetScan versus MicroCosm and TargetScan versus mirDB (p < 2.33 × 10−7 and p < 1.80 × 10−10, respectively), and no enrichment for MicroCosm versus mirDB (p > 0.22, NS). These findings provided the rationale for moving forward with using only the TargetScan prediction results in the current study (Supplementary Table E3, online only, available at www.exphem.org).
Table 2 lists the top predicted targets (TargetScan) with the high-context percentile score at and above 91 that overlap with the experimentally observed targets during CD34+ differentiation. Supplementary Tables E2 and E4 (online only, available at www.exphem.org) show the complete list of conserved and nonconserved predicted targets derived from the differentially expressed miRNAs from the current study; this list also includes the TargetScan high-context percentile score for the corresponding miRNA as described in the methods section.
Table 2.
List of predicted miRNA target genes selected and ranked based on high context percentile score by Target Scan analysis
| miRNA title | Target gene symbol | Context % score | Context score | UTR-end | UTR-start |
|---|---|---|---|---|---|
| hsa-miR-1205 | EGLN3 | 99 | −0.411 | 235 | 229 |
| hsa-miR-545 | CTTN | 99 | −0.494 | 168 | 162 |
| hsa-miR-576-3p | CXCL3 | 99 | −0.51 | 430 | 424 |
| hsa-miR-23a | ADRA2A | 98 | −0.495 | 1396 | 1390 |
| hsa-miR-23b | ADRA2A | 98 | −0.495 | 1396 | 1390 |
| hsa-miR-498 | EGLN3 | 98 | −0.44 | 1287 | 1281 |
| hsa-miR-516b | EGLN3 | 98 | −0.421 | 224 | 218 |
| hsa-miR-665 | ABCC3 | 98 | −0.384 | 383 | 377 |
| hsa-miR-873 | EGLN3 | 98 | −0.397 | 47 | 41 |
| hsa-miR-1324 | C20orf112 | 97 | −0.385 | 489 | 483 |
| hsa-miR-138 | CCND3 | 97 | −0.4 | 298 | 292 |
| hsa-miR-182 | CTTN | 97 | −0.457 | 284 | 278 |
| hsa-miR-488 | C20orf112 | 97 | −0.418 | 355 | 349 |
| hsa-miR-491-5p | ASB13 | 97 | −0.339 | 662 | 656 |
| hsa-miR-876-3p | CEBPA | 97 | −0.429 | 1023 | 1017 |
| hsa-miR-1 | C20orf112 | 96 | −0.408 | 522 | 516 |
| hsa-miR-106a | EGLN3 | 96 | −0.408 | 333 | 327 |
| hsa-miR-106b | EGLN3 | 96 | −0.408 | 333 | 327 |
| hsa-miR-1271 | ARHGAP6 | 96 | −0.406 | 1026 | 1020 |
| hsa-miR-17 | EGLN3 | 96 | −0.408 | 333 | 327 |
| hsa-miR-182 | CTTN | 96 | −0.435 | 980 | 974 |
| hsa-miR-206 | C20orf112 | 96 | −0.402 | 522 | 516 |
| hsa-miR-20a | EGLN3 | 96 | −0.408 | 333 | 327 |
| hsa-miR-20b | EGLN3 | 96 | −0.408 | 333 | 327 |
| hsa-miR-218 | EGLN3 | 96 | −0.421 | 1544 | 1538 |
| hsa-miR-330-3p | CCND3 | 96 | −0.431 | 650 | 644 |
| hsa-miR-519d | EGLN3 | 96 | −0.406 | 333 | 327 |
| hsa-miR-520d-5p | ADCY6 | 96 | −0.438 | 2391 | 2385 |
| hsa-miR-524-5p | ADCY6 | 96 | −0.438 | 2391 | 2385 |
| hsa-miR-613 | C20orf112 | 96 | −0.4 | 522 | 516 |
| hsa-miR-656 | ARHGAP6 | 96 | −0.478 | 814 | 808 |
| hsa-miR-7 | CKAP4 | 96 | −0.38 | 469 | 463 |
| hsa-miR-93 | EGLN3 | 96 | −0.406 | 333 | 327 |
| hsa-miR-96 | ARHGAP6 | 96 | −0.4 | 1026 | 1020 |
| hsa-miR-145 | ARHGAP6 | 95 | −0.422 | 863 | 857 |
| hsa-miR-302a | ARHGEF17 | 95 | −0.342 | 427 | 421 |
| hsa-miR-302b | ARHGEF17 | 95 | −0.342 | 427 | 421 |
| hsa-miR-302c | ARHGEF17 | 95 | −0.342 | 427 | 421 |
| hsa-miR-302d | ARHGEF17 | 95 | −0.342 | 427 | 421 |
| hsa-miR-302e | ARHGEF17 | 95 | −0.338 | 427 | 421 |
| hsa-miR-338-5p | CTTN | 95 | −0.468 | 213 | 207 |
| hsa-miR-339-5p | ADRA2A | 95 | −0.331 | 718 | 712 |
| hsa-miR-448 | CTTN | 95 | −0.447 | 1327 | 1321 |
| hsa-miR-450b-5p | ENDOD1 | 95 | −0.415 | 135 | 129 |
| hsa-miR-513b | BCL2L1 | 95 | −0.395 | 1216 | 1210 |
| hsa-miR-520d-5p | ASB13 | 95 | −0.413 | 1724 | 1718 |
| hsa-miR-875-3p | EGLN3 | 95 | −0.388 | 465 | 459 |
| hsa-miR-1271 | ADCY6 | 94 | −0.344 | 609 | 603 |
| hsa-miR-153 | CKAP4 | 94 | −0.363 | 633 | 627 |
| hsa-miR-342-5p | BCL2L1 | 94 | −0.27 | 684 | 678 |
| hsa-miR-372 | ARHGEF17 | 94 | −0.338 | 427 | 421 |
| hsa-miR-373 | ARHGEF17 | 94 | −0.338 | 427 | 421 |
| hsa-miR-512-5p | BCL2L2 | 94 | −0.298 | 2362 | 2356 |
| hsa-miR-520a-3p | ARHGEF17 | 94 | −0.34 | 427 | 421 |
| hsa-miR-520b | ARHGEF17 | 94 | −0.342 | 427 | 421 |
| hsa-miR-520c-3p | ARHGEF17 | 94 | −0.342 | 427 | 421 |
| hsa-miR-520d-3p | ARHGEF17 | 94 | −0.342 | 427 | 421 |
| hsa-miR-520e | ARHGEF17 | 94 | −0.336 | 427 | 421 |
| hsa-miR-524-5p | ASB13 | 94 | −0.413 | 1724 | 1718 |
| hsa-miR-548p | ENDOD1 | 94 | −0.394 | 626 | 620 |
| hsa-miR-767-3p | C20orf112 | 94 | −0.333 | 371 | 365 |
| hsa-miR-96 | ADCY6 | 94 | −0.354 | 609 | 603 |
| hsa-miR-122 | C20orf112 | 93 | −0.263 | 135 | 129 |
| hsa-miR-138 | ADRA2A | 93 | −0.32 | 666 | 660 |
| hsa-miR-345 | CDKN1A | 93 | −0.318 | 1179 | 1173 |
| hsa-miR-1254 | C5orf4 | 92 | −0.248 | 1038 | 1032 |
| hsa-miR-135a | ARHGAP6 | 92 | −0.339 | 583 | 577 |
| hsa-miR-135b | ARHGAP6 | 92 | −0.337 | 583 | 577 |
| hsa-miR-146b-3p | CECR1 | 92 | −0.2452 | 1671 | 1665 |
| hsa-miR-361-5p | CD177 | 92 | −0.365 | 237 | 231 |
| hsa-miR-491-3p | FLJ11151 | 92 | −0.4091 | 134 | 128 |
| hsa-miR-506 | FLJ11151 | 92 | −0.3012 | 2703 | 2697 |
| hsa-miR-124 | FLJ11151 | 91 | −0.2992 | 2703 | 2697 |
| hsa-miR-1274a | ADRA2A | 91 | −0.259 | 719 | 713 |
| hsa-miR-220c | ADRA2A | 91 | −0.254 | 840 | 834 |
| hsa-miR-326 | BCL2L2 | 91 | −0.261 | 2279 | 2273 |
| hsa-miR-338-5p | CD9 | 91 | −0.406 | 321 | 315 |
| hsa-miR-377 | BCL2L1 | 91 | −0.353 | 1446 | 1440 |
| hsa-miR-409-3p | CXCL3 | 91 | −0.305 | 508 | 502 |
| hsa-miR-511 | ADRA2A | 91 | −0.376 | 1108 | 1102 |
| hsa-miR-519b-5p | EGLN3 | 91 | −0.308 | 535 | 529 |
| hsa-miR-519c-5p | EGLN3 | 91 | −0.308 | 535 | 529 |
| hsa-miR-520g | CDKN1A | 91 | −0.295 | 1155 | 1149 |
| hsa-miR-520h | CDKN1A | 91 | −0.295 | 1155 | 1149 |
| hsa-miR-548b-5p | BCL2L2 | 91 | −0.361 | 2596 | 2590 |
| hsa-miR-548c-3p | CTTN | 91 | −0.328 | 1375 | 1369 |
| hsa-miR-548c-5p | BCL2L2 | 91 | −0.361 | 2596 | 2590 |
| hsa-miR-548d-5p | BCL2L2 | 91 | −0.361 | 2596 | 2590 |
| hsa-miR-548h | BCL2L2 | 91 | −0.361 | 2596 | 2590 |
| hsa-miR-23a | C5orf4 | 90 | −0.402 | 1575 | 1569 |
| hsa-miR-23b | C5orf4 | 90 | −0.402 | 1575 | 1569 |
| hsa-miR-330-5p | BCL2L2 | 90 | −0.255 | 2279 | 2273 |
| hsa-miR-518d-5p | CD9 | 90 | −0.31 | 310 | 304 |
| hsa-miR-518d-5p | EGLN3 | 90 | −0.308 | 535 | 529 |
| hsa-miR-520c-5p | CD9 | 90 | −0.31 | 310 | 304 |
| hsa-miR-520c-5p | EGLN3 | 90 | −0.308 | 535 | 529 |
| hsa-miR-526a | CD9 | 90 | −0.31 | 310 | 304 |
| hsa-miR-526a | EGLN3 | 90 | −0.308 | 535 | 529 |
| hsa-miR-548a-5p | BCL2L2 | 90 | −0.345 | 2596 | 2590 |
| hsa-miR-944 | BCL2L2 | 90 | −0.368 | 2694 | 2688 |
Predicted targets with contect % score at and above 91 are shown in this table.
Of the TargetScan predicted targets, 105 targets were found on the previously experimentally identified list of 172 differentially expressed genes [19]. Correlation analysis of mRNA average gene expression and miRNA average gene expression for each cell type revealed a total of 87 pairs (p < 0.05; 33 positive and 54 negative) from a list of 50 mRNA and 20 miRNA (Fig. 4). The negatively correlated pairs are shown in Table 3.
Figure 4.

Correlation of selected differentially expressed miRNAs with potential differentially expressed targets. Twenty miRNAs had significant correlation with at least 1 of 50 being differentially expressed. Potential target mRNAs are shown. Blue indicates a negative, and red indicates a positive correlation. There were 87 significant correlations at p ≤ 0.05. All correlation coefficients have an absolute value of 0.95 or higher.
Table 3.
Highly negatively correlated miRNA and mRNA pairs
| miRNA targeted gene title | Gene symbol | miRNA | Correlation |
|---|---|---|---|
| Phosphodiesterase 5A, cGMP-specific | PDE5A | let-7i | −0.999 |
| Regulator of G-protein signaling 10 | RGS10 | mir-320b | −0.999 |
| Ubiquitin-conjugating enzyme E2L 6 | UBE2L6 | mir-1825 | −0.998 |
| Potassium voltage-gated channel, shaker-related subfamily, member 3 | KCNA3 | mir-1308 | −0.998 |
| Hexamethylene bis-acetamide inducible 1 | HEXIM1 | let-7i | −0.998 |
| Integrin, beta 3 (platelet glycoprotein IIIa, antigen CD61) | ITGB3 | mir-222 | −0.997 |
| Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | CDKN1A | mir-93 | −0.996 |
| SEC14-like 5 (S. cerevisiae) | SEC14L5 | let-7i | −0.994 |
| Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | CDKN1A | let-7i | −0.994 |
| Pannexin 1 | PANX1 | mir-320b | −0.993 |
| Lipase, hepatic | LIPC | let-7i | −0.992 |
| Integrin, beta 3 (platelet glycoprotein IIIa, antigen CD61) | ITGB3 | mir-221 | −0.991 |
| Hypothetical protein FLJ11151 | FLJ11151 | mir-204 | −0.990 |
| Chemokine (C-X-C motif) ligand 3 | CXCL3 | mir-320b | −0.988 |
| Zinc finger, X-linked, duplicated B | ZXDB | mir-500 | −0.988 |
| Regulator of G-protein signaling 6 | RGS6 | mir-222 | −0.988 |
| Proprotein convertase subtilisin/kexin type 6 | PCSK6 | mir-320b | −0.987 |
| Phosphodiesterase 3A, cGMP-inhibited | PDE3A | mir-320b | −0.987 |
| BCL2-like 2 | BCL2L2 | mir-15b | −0.987 |
| Regulator of G-protein signaling 6 | RGS6 | mir-221 | −0.985 |
| SEC14-like 5 (S. cerevisiae) | SEC14L5 | mir-1275 | −0.983 |
| Rho GTPase activating protein 6 | ARHGAP6 | mir-320b | −0.982 |
| Gamma-aminobutyric acid (GABA) A receptor, epsilon | GABRE | mir-1275 | −0.981 |
| Regulator of G-protein signaling 6 | RGS6 | mir-1275 | −0.981 |
| Proprotein convertase subtilisin/kexin type 6 | PCSK6 | mir-15b | −0.980 |
| Potassium voltage-gated channel, shaker-related subfamily, member 3 | KCNA3 | mir-1268 | −0.980 |
| Thrombospondin 1 | THBS1 | mir-221 | −0.980 |
| 5-Hydroxytryptamine (serotonin) receptor 2A | HTR2A | mir-15b | −0.979 |
| Translocase of inner mitochondrial membrane 13 homolog (yeast) | TIMM13 | mir-198 | −0.978 |
| Tumor necrosis factor receptor superfamily, member 1B | TNFRSF1B | mir-193b | −0.978 |
| Microfibrillar-associated protein 3-like | MFAP3L | mir-15b | −0.977 |
| Serpin peptidase inhibitor, clade E | SERPINE1 | mir-1275 | −0.976 |
| Phosphodiesterase 5A, cGMP-specific | PDE5A | mir-93 | −0.976 |
| KIAA0513 | KIAA0513 | mir-1268 | −0.974 |
| Tubulin, alpha 8 | TUBA8 | mir-1275 | −0.972 |
| 5-Hydroxytryptamine (serotonin) receptor 2A | HTR2A | mir-93 | −0.972 |
| Microfibrillar-associated protein 3-like | MFAP3L | mir-93 | −0.972 |
| Phospholipase A2, group IVC (cytosolic, calcium-independent) | PLA2G4C | mir-181a | −0.970 |
| Glycine-N-acyltransferase-like 1 | GLYATL1 | mir-181a | −0.970 |
| Cyclin D3 | CCND3 | mir-320b | −0.969 |
| SEC14-like 5 (S. cerevisiae) | SEC14L5 | mir-181a | −0.969 |
| Insulin receptor substrate 2 | IRS2 | mir-107 | −0.969 |
| KIAA0513 | KIAA0513 | mir-93 | −0.968 |
| Platelet-derived growth factor beta polypeptide | PDGFB | mir-93 | −0.966 |
| Regulator of G-protein signaling 6 | RGS6 | let-7i | −0.965 |
| Insulin receptor substrate 2 | IRS2 | mir-103 | −0.964 |
| Thrombospondin 1 | THBS1 | mir-222 | −0.962 |
| Gamma-aminobutyric acid (GABA) A receptor, epsilon | GABRE | mir-93 | −0.961 |
| Insulin receptor substrate 2 | IRS2 | mir-15b | −0.961 |
| Stannin | SNN | let-7i | −0.958 |
| Tetraspanin 9 | TSPAN9 | mir-93 | −0.957 |
| 5-Hydroxytryptamine (serotonin) receptor 2A | HTR2A | mir-103 | −0.956 |
| Proprotein convertase subtilisin/kexin type 6 | PCSK6 | mir-103 | −0.955 |
| Coagulation factor II (thrombin) receptor-like 2 | F2RL2 | mir-93 | −0.953 |
Ingenuity pathway analysis (Supplementary Figs. 1, 2, and 3)—on the functions of 12,440 predicted miRNA targets 87 highly correlated miRNA-mRNA pairs from the integrated analysis and 53 inversely correlated mRNA-miRNA pairs with a p value cut off of p < 0.001 (a measure of the association between a set of functional analysis genes derived from the experiment and a given process or pathway is due to random chance)—revealed the top functional categories to be cell growth and proliferation, cell development, hematologic system development, cell death and survival, and embryonic development. These results suggest that although the predicted genes from miRNA fall into the category of hematopoiesis and hematological system development at p < 0.001, the inversely correlated genes appear to be more tightly associated with hematopoietic process based on its ranking in the top five functional categories.
Analysis of the negatively correlated miRNA-mRNA pairs by IPA as shown in Figure 5 revealed miR-1275, miR-221, miR-320b, miR-1268, miR-1308, and miR-193b to be potentially specifically involved in erythropoietic differentiation. During granulopoiesis miR-1268, miR-204 and miR-93 showed a possible regulatory role in the expression of lineage specific genes (Fig. 5B). Differentiation of CD34+ cells to megakaryocytes demonstrated possible regulatory roles of miR-138, miR-1285, miR-500, miR-103, miR-320b, miR-1275, miR-7i, miR-93, miR-181a, and miR-15b in the differential expression of lineage specific genes as depicted in Figure 5C. We also observed 12 miRNAs showing direct positive correlation as shown in Table 4. In particular, we observed that 15 of the significantly correlated microRNAs play a significant role in Wnt/B-catenin and Flt3 signaling pathways. As a result, this study has generated a library of mRNA-miRNA networks that are important candidates for control of the cellular processes of hematopoietic differentiation.
Figure 5.

Inversely correlated miRNA-mRNA pairs with significant correlation (R = 0.95). The x axis shows the log2-fold changes in the expression of miRNA and mRNA. The green bars depict the miRNA, and the red bars depict the mRNA expression. (A) E versus CD34. (B) G versus CD34. (C) M versus CD34.
Table 4.
Highly positively correlated miRNA-mRNA pairs
| Targeted mRNA title | Gene symbol | miRNA | Correlation |
|---|---|---|---|
| Serum deprivation response (phosphatidylserine binding protein) | SDPR | mir-1825 | 0.951 |
| Cortactin | CTTN | mir-588 | 0.953 |
| Protein O-fucosyltransferase 1 | POFUT1 | mir-103 | 0.955 |
| Protocadherin 21 | PCDH21 | mir-1825 | 0.959 |
| Regulator of G-protein signaling 6 | RGS6 | mir-588 | 0.961 |
| RAB3D, member RAS oncogene family | RAB3D | mir-1275 | 0.963 |
| RNA pseudouridylate synthase domain containing 1 | RPUSD1 | mir-1268 | 0.965 |
| Transient receptor potential cation channel, subfamily C, member 6 | TRPC6 | mir-198 | 0.966 |
| Rho GTPase activating protein 6 | ARHGAP6 | mir-500 | 0.966 |
| Mitochondrial ribosomal protein S16 | MRPS16 | mir-103 | 0.967 |
| Regulator of G-protein signaling 10 | RGS10 | mir-198 | 0.968 |
| Paralemmin | PALM | mir-1268 | 0.970 |
| Integrin, beta 5 | ITGB5 | mir-493 | 0.970 |
| Guanine nucleotide binding protein (G protein), alpha z polypeptide | GNAZ | mir-198 | 0.972 |
| Phosphodiesterase 5A, cGMP-specific | PDE5A | mir-198 | 0.973 |
| Leucine rich repeat containing 32 | LRRC32 | mir-1825 | 0.973 |
| Carcinoembryonic antigen-related cell adhesion molecule 6 | CEACAM6 | mir-1228 | 0.974 |
| Tripartite motif-containing 14 | TRIM14 | mir-222 | 0.976 |
| Regulator of G-protein signaling 10 | RGS10 | mir-1825 | 0.982 |
| Pannexin 1 | PANX1 | mir-1825 | 0.982 |
| Rho GTPase activating protein 6 | ARHGAP6 | mir-204 | 0.983 |
| Tripartite motif-containing 14 | TRIM14 | mir-221 | 0.984 |
| Hexamethylene bis-acetamide inducible 1 | HEXIM1 | mir-1825 | 0.984 |
| Serpin peptidase inhibitor, clade E | SERPINE1 | mir-1825 | 0.986 |
| SEC14-like 5 (S. cerevisiae) | SEC14L5 | mir-198 | 0.986 |
| Glycoprotein V (platelet) | GP5 | mir-1825 | 0.989 |
| Guanine nucleotide binding protein (G protein), alpha z polypeptide | GNAZ | mir-588 | 0.990 |
| Hexamethylene bis-acetamide inducible 1 | HEXIM1 | mir-588 | 0.990 |
| Glycoprotein V (platelet) | GP5 | mir-588 | 0.991 |
| Mitochondrial ribosomal protein S16 | MRPS16 | mir-93 | 0.994 |
| Proprotein convertase subtilisin/kexin type 6 | PCSK6 | mir-588 | 0.995 |
| Gamma-aminobutyric acid (GABA) A receptor, epsilon | GABRE | mir-1825 | 0.998 |
| Platelet-derived growth factor beta polypeptide | PDGFB | mir-588 | 0.999 |
Discussion
Hematopoietic lineage differentiaton, the process of continuous development of hematopoietic stem cells into at least eight different blood lineages is known to be controlled by complex molecular events that work in unison to regulate the commitment, proliferation, apoptosis, and maturation of stem cells and progenitor cells [20–23]. In the past, gene expression analyses have provided information on important transcripts that dictate the fate of cells; however, it is still a puzzle as to why so many genes are involved in the cell differentiation process, and why stem cells and the respective progenitor cells express mRNA for some genes but do not express the encoded protein.
Recently, miRNAs have emerged as essential factors to regulate a broad range of cellular events such as development, differentiation, proliferation, morphogenesis, apoptosis, and metabolism [24–26]. The few studies to date that have addressed miRNAs in hematopoiesis have focused on few selected genes and pathways to demonstrate the regulatory roles of miRNA in hematopoiesis [27–36]. However, a global analysis of miRNA during hematopoiesis would be beneficial to delineate novel miRNA-mediated regulatory mechanisms during cellular differentiation.
We undertook this study to gain insight into the regulatory roles of miRNAs in human hematopoietic process by using an in vitro human model system that permits microarray based analysis of CD34+ differentiation into major blood cell lineages. Importantly, by integrating the experimentally observed expression levels of mRNA and miRNA in this model system, we have attempted to deepen the understanding of the regulatory roles of miRNA targeting genes that are involved in cell differentiation process.
We observed significant modulation in the expression of several miRNAs, including the previously identified miR-15b, miR-146, miR-150, miR-181, and miR-223 during lineage-specific differentiation of CD34+ cells. As observed in previous studies, these miRNAs appear to fall within the functional categories of cellular growth and proliferation, cell cycle, and signaling and play an important role in hematopoietic system development [37–45]. We also identified several novel differentially expressed miRNAs that have not been reported previously. A major finding of this study is that there are characteristic expression patterns of transcripts (mRNAs and miRNAs) during lineage-specific differentiation of CD34+ cells. We also corroborated the results obtained using microarrays with real-time qPCR analysis on few selected miRNAs.
Although multiple computational approaches have been developed recently, a challenge still remains to accurately predict miRNA targets. In an effort to identify potential targets of the miRNA associated with hematopoiesis, we applied and compared three different prediction databases to identify potential mRNA targets. Predicted targets identified by the three databases were evaluated further to rule out false positives by correlating to experimentally observed targets that were manipulated during CD34+ differentiation process. We observed poor overlap between the prediction tools that were used in the current study. TargetScan outperformed miRDB and microcosm by providing the largest number of significantly correlated pairs and more negatively correlated miRNA-mRNA pairs. TargetScan and miRDB algorithms both emphasize on pairing in the seed region (7-mer and 8-mer) for target prediction. Microcosm, however, does not consider the seed region per se, which might explain its poor performance against the other two prediction tools. The lack of poor overlap between the three target prediction tools has also been reported previously by others [46–48] as a major challenge because of very limited sequence complimentarity with the use of only 7–8 bases out of 21–22 nucleotides and the scarcity of experimentally validated gene targets. This limitation warrants future functional studies on identified miRNA targets for confirmation.
The mechanisms by which miRNAs degrade their targets are still not well characterized. There may be cooperation between translation inhibition and mRNA degradation involving deadenylase and decapping complexes. The expression profiles of both mRNAs and miRNAs from our studies allowed us to ask whether there was a correlation between the expression levels of miRNAs and the predicted mRNA targets of each miRNA. MicroRNAs bind to mRNAs and negatively regulate their expression, either by repression of translation or degradation of the mRNA sequence [49]. Increased expression levels of miRNAs can also result in upregulation of previously suppressed target genes either directly, by decreasing the expression of inhibitory proteins or transcription factors, or indirectly, by inhibiting the expression levels of inhibitory miRNAs [50]. In a few cases, miRNAs have also been observed to activate the translation of target mRNA by regulation of their stability. miRNAs that do not show inverse correlation to mRNA might not be direct transcriptional regulators, but could have an indirect mechanism in the regulation of target gene expression. The detection of miRNAs with a positive correlation between the miRNA and target mRNA (either both downregulated or both upregulated) suggests some alternative indirect mechanisms or a positive regulatory role for miRNAs, as has been reported for some genes involved in cell cycle regulation.
Ingenuity pathway analysis on the highly correlated genes and miRNA from the current study revealed the genes to be involved in top functional categories of hematologic system development, Wnt/B-catenin and FLT3 signaling, hematopoietic tissue development, and cellular growth and proliferation. Wnt proteins control cell fate and differentiation during development in hematopoietic stem cells [51]. Alterations of the Wnt/β-catenin signaling pathway are known to be associated with the tumorigenesis of tissues with a high renewal potential, such as that of bone marrow hematopoietic tissue. FLT3 is normally expressed in hemotopoietic progenitor cells in the bone marrow, thymus, and lymph nodes, and interaction of FLT3 with its ligand results in receptor dimerization, autophosphorylation, and the subsequent phosphorylation of cytoplasmic substrates that are involved in signaling pathways regulating the proliferation of pluripotent stem cells, early progenitor cells and immature lymphocytes.
Our results demonstrate interesting correlations between differentially expressed miRNAs and their corresponding target genes during lineage differentiation. These results also provide solid evidence that miRNAs represent a class of molecules that regulate hematopoietic process. However, besides miRNA negative regulation, there are other modes of gene regulation (e.g., miRNA positive regulation, DNA methylation, chromatin remodeling) that are involved in different potentials for differentiation and division. We also believe that the data available from this study would lead discovery of “hubs” of miRNAs that together regulate lineage fate/gene expression. Deeper analysis of the differentially expressed miRNAs by motif enrichment analysis might provide insights into how groups of miRNA and mRNAs might be regulated together at a transcriptional level during the differentiation process. Future studies are warranted to study how these different regulatory interactions are synchronized to maintain the phenotype in the process of lineage specific cell differentiation.
Supplementary Material
Supplementary Table E1. Number of miRNA-mRNA pairs predicted by each algorithm (column headers), out of a total 8428 possible pairs (49 miRNAs × 172 mRNAs)
Supplementary Table E3. Number of overlaping predicted miRNA-mRNA pairs (predicted by 2 algorithms)
Supplementary Figure E1. Pathway analysis on predicted miRNA targets.
Supplementary Figure E2. Pathway analysis on 87 highly correlated miRNA-mRNA pairs.
Supplementary Figure E3. Pathway analysis on inversely correlated miRNA-mRNA pairs.
Acknowledgments
This work was funded by Intramural Research, National Heart, Lung, and Blood Institute. We thank Harry L. Malech and Uimook Choi, in Genetic Immunotherapy Section, Laboratory of Host Defenses, National Institute of Allergy and Infectious Diseases, National Institutes of Health, for providing the CD34+ cells; Leigh Samsel in the NHLBI Flow Cytometry Core for help with the flow characterization of cells; Cuhur Yusuf Demirkale for his helpful suggestions in data analysis; and Ms. Bandana at Strand Life Sciences, Bangalore (Agilent Technologies’ partner) for help with TargetScan analysis.
Footnotes
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.exphem.2013.10.003.
Author contributions: N.R. designed the study, collected data, and prepared the manuscript; P.L. and R.W. collected data; Y.Y., J.B., Q.N., and P.J.M. performed data analysis; P.J.M. and J.B. edited the manuscript.
Conflict of interest disclosure
No financial interest/relationships with financial interest relating to the topic of this article have been declared.
References
- 1.Huang S, He X. microRNAs: tiny RNA molecules, huge driving forces to move the cell. Protein Cell. 2010;1:916–926. doi: 10.1007/s13238-010-0116-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rager JE, Smeester L, Jaspers I, Sexton KG, Fry RC. Epigenetic changes induced by air toxics: formaldehyde exposure alters miRNA expression profiles in human lung cells. Environ Health Perspect. 2011;119:494–500. doi: 10.1289/ehp.1002614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thorne JL, Maguire O, Doig CL, et al. Epigenetic control of a VDR-governed feed-forward loop that regulates p21(waf1/cip1) expression and function in non-malignant prostate cells. Nucleic Acids Res. 2011;39:2045–2056. doi: 10.1093/nar/gkq875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gotte M. MicroRNAs in breast cancer pathogenesis. Minerva Ginecol. 2010;62:559–571. [PubMed] [Google Scholar]
- 5.Rearick D, Prakash A, McSweeny A, Shepard SS, Fedorova L, Fedorov A. Critical association of ncRNA with introns. Nucleic Acids Res. 2011;39:2357–2366. doi: 10.1093/nar/gkq1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shi Y, Zhao X, Hsieh J, et al. MicroRNA regulation of neural stem cells and neurogenesis. J Neurosci. 2010;30:14931–14936. doi: 10.1523/JNEUROSCI.4280-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong W. Focus issue: signals for gene expression. Sci Signal. 2010;3:eg10. doi: 10.1126/scisignal.3146eg10. [DOI] [PubMed] [Google Scholar]
- 8.Schanen BC, Li X. Transcriptional regulation of mammalian miRNA genes. Genomics. 2011;97:1–6. doi: 10.1016/j.ygeno.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.David M. Interferons and microRNAs. J Interferon Cytokine Res. 2010;30:825–828. doi: 10.1089/jir.2010.0080. [DOI] [PubMed] [Google Scholar]
- 10.Mishra PJ. MicroRNA polymorphisms: a giant leap towards personalized medicine. Per Med. 2009;6:119–125. doi: 10.2217/17410541.6.2.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuchen S, Resch W, Yamane A, et al. Regulation of microRNA expression and abundance during lymphopoiesis. Immunity. 2010;32:828–839. doi: 10.1016/j.immuni.2010.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Slavov SN, Gimenes Teixeira HL, Rego EM. The role of micro-ribonucleic acids in normal hematopoiesis and leukemic T-lymphogenesis. Braz J Med Biol Res. 2010;43:619–626. doi: 10.1590/s0100-879x2010007500057. [DOI] [PubMed] [Google Scholar]
- 13.Zhao G, Yu D, Weiss MJ. MicroRNAs in erythropoiesis. Curr Opin Hematol. 2010;17:155–162. doi: 10.1097/MOH.0b013e328337ba6c. [DOI] [PubMed] [Google Scholar]
- 14.Vasilatou D, Papageorgiou S, Pappa V, Papageorgiou E, Dervenoulas J. The role of microRNAs in normal and malignant hematopoiesis. Eur J Haematol. 2010;84:1–16. doi: 10.1111/j.1600-0609.2009.01348.x. [DOI] [PubMed] [Google Scholar]
- 15.Muniategui A, Nogales-Cadenas R, Vazquez M, et al. Quantification of miRNA-mRNA interactions. PLoS One. 2012;7:e30766. doi: 10.1371/journal.pone.0030766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grutzkau A, Radbruch A. Small but mighty: how the MACS-technology based on nanosized superparamagnetic particles has helped to analyze the immune system within the last 20 years. Cytometry A. 2010;77:643–647. doi: 10.1002/cyto.a.20918. [DOI] [PubMed] [Google Scholar]
- 17.McNemar Q. Statistical theory and research design. Annu Rev Psychol. 1952;3:409–418. doi: 10.1146/annurev.ps.03.020152.002205. [DOI] [PubMed] [Google Scholar]
- 18.Liu P, Barb J, Woodhouse K, Taylor JG, Munson PJ, Raghavachari N. Transcriptome profiling and sequencing of differentiated human hematopoietic stem cells reveal lineage-specific expression and alternative splicing of genes. Physiol Genomics. 2011;43:1117–1134. doi: 10.1152/physiolgenomics.00099.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu P, Barb J, Woodhouse K, Taylor JG, Munson PJ, Raghavachari N. Transcriptome profiling and sequencing of differentiated human hematopoietic stem cells reveal lineage-specific expression and alternative splicing of genes. Physiol Genomics. 2011;43:1117–1134. doi: 10.1152/physiolgenomics.00099.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santos P, Arumemi F, Park KS, Borghesi L, Milcarek C. Transcriptional and epigenetic regulation of B cell development. Immunol Res. 2011;50:105–112. doi: 10.1007/s12026-011-8225-y. [DOI] [PubMed] [Google Scholar]
- 21.Welch JS, Yuan W, Ley TJ. PML-RARA can increase hematopoietic self-renewal without causing a myeloproliferative disease in mice. J Clin Invest. 2011;121:1636–1645. doi: 10.1172/JCI42953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geissmann F, Manz MG, Jung S, Sieweke MH, Merad M, Ley K. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327:656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher AG. Cellular identity and lineage choice. Nat Rev Immunol. 2002;2:977–982. doi: 10.1038/nri958. [DOI] [PubMed] [Google Scholar]
- 24.Fatica A, Rosa A, Fazi F, et al. MicroRNAs and hematopoietic differentiation. Cold Spring Harb Symp Quant Biol. 2006;71:205–210. doi: 10.1101/sqb.2006.71.014. [DOI] [PubMed] [Google Scholar]
- 25.Pasquinelli AE, Hunter S, Bracht J. MicroRNAs: a developing story. Curr Opin Genet Dev. 2005;15:200–205. doi: 10.1016/j.gde.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Chen CZ, Lodish HF. MicroRNAs as regulators of mammalian hematopoiesis. Semin Immunol. 2005;17:155–165. doi: 10.1016/j.smim.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 27.Hua Z, Chun W, Fang-Yuan C. MicroRNA-146a and hemopoietic disorders. Int J Hematol. 2011;94:224–229. doi: 10.1007/s12185-011-0923-7. [DOI] [PubMed] [Google Scholar]
- 28.Guglielmelli P, Tozzi L, Bogani C, et al. Overexpression of microRNA-16-2 contributes to the abnormal erythropoiesis in polycythemia vera. Blood. 2011;117:6923–6927. doi: 10.1182/blood-2010-09-306506. [DOI] [PubMed] [Google Scholar]
- 29.Byon JC, Papayannopoulou T. MicroRNAs: Allies or foes in erythropoiesis? J Cell Physiol. 2012;227:7–13. doi: 10.1002/jcp.22729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tenedini E, Roncaglia E, Ferrari F, et al. Integrated analysis of microRNA and mRNA expression profiles in physiological myelopoiesis: role of hsa-mir-299-5p in CD34+ progenitor cells commitment. Cell Death Dis. 2010;1:e28. doi: 10.1038/cddis.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castoldi M, Vujic Spasic M, Altamura S, et al. The liver-specific microRNA miR-122 controls systemic iron homeostasis in mice. J Clin Invest. 2011;121:1386–1396. doi: 10.1172/JCI44883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pillai MM, Yang X, Balakrishnan I, Bemis L, Torok-Storb B. MiR-886-3p down regulates CXCL12 (SDF1) expression in human marrow stromal cells. PLoS One. 2010;5:e14304. doi: 10.1371/journal.pone.0014304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li G, Wu Z, Li X, Ning X, Li Y, Yang G. Biological role of microRNA-103 based on expression profile and target genes analysis in pigs. Mol Biol Rep. 2011;38:4777–4786. doi: 10.1007/s11033-010-0615-z. [DOI] [PubMed] [Google Scholar]
- 34.Rasmussen KD, Simmini S, Abreu-Goodger C, et al. The miR-144/451 locus is required for erythroid homeostasis. J Exp Med. 2010;207:1351–1358. doi: 10.1084/jem.20100458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kong KY, Owens KS, Rogers JH, et al. MIR-23A microRNA cluster inhibits B-cell development. Exp Hematol. 2010;38:629–640. e621. doi: 10.1016/j.exphem.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Georgantas RW, 3rd, Hildreth R, Morisot S, et al. CD34+ hematopoietic stem-progenitor cell microRNA expression and function: a circuit diagram of differentiation control. Proc Natl Acad Sci U S A. 2007;104:2750–2755. doi: 10.1073/pnas.0610983104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bruchova-Votavova H, Yoon D, Prchal JT. miR-451 enhances erythroid differentiation in K562 cells. Leuk Lymphoma. 2010;51:686–693. doi: 10.3109/10428191003629362. [DOI] [PubMed] [Google Scholar]
- 38.Han YC, Park CY, Bhagat G, et al. microRNA-29a induces aberrant self-renewal capacity in hematopoietic progenitors, biased myeloid development, and acute myeloid leukemia. J Exp Med. 2010;207:475–489. doi: 10.1084/jem.20090831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Felli N, Pedini F, Romania P, et al. MicroRNA 223-dependent expression of LMO2 regulates normal erythropoiesis. Haematologica. 2009;94:479–486. doi: 10.3324/haematol.2008.002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaar DG, Medina DJ, Moore DF, Strair RK, Ting Y. miR-320 targets transferrin receptor 1 (CD71) and inhibits cell proliferation. Exp Hematol. 2009;37:245–255. doi: 10.1016/j.exphem.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 41.Ben-Ami O, Pencovich N, Lotem J, Levanon D, Groner Y. A regulatory interplay between miR-27a and Runx1 during megakaryopoiesis. Proc Natl Acad Sci U S A. 2009;106:238–243. doi: 10.1073/pnas.0811466106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Romania P, Lulli V, Pelosi E, Biffoni M, Peschle C, Marziali G. MicroRNA 155 modulates megakaryopoiesis at progenitor and precursor level by targeting Ets-1 and Meis1 transcription factors. Br J Haematol. 2008;143:570–580. doi: 10.1111/j.1365-2141.2008.07382.x. [DOI] [PubMed] [Google Scholar]
- 43.Fu YF, Du TT, Dong M, et al. Mir-144 selectively regulates embryonic alpha-hemoglobin synthesis during primitive erythropoiesis. Blood. 2009;113:1340–1349. doi: 10.1182/blood-2008-08-174854. [DOI] [PubMed] [Google Scholar]
- 44.Zhao H, Kalota A, Jin S, Gewirtz AM. The c-myb proto-oncogene and microRNA-15a comprise an active autoregulatory feedback loop in human hematopoietic cells. Blood. 2009;113:505–516. doi: 10.1182/blood-2008-01-136218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barroga CF, Pham H, Kaushansky K. Thrombopoietin regulates c-Myb expression by modulating micro RNA 150 expression. Exp Hematol. 2008;36:1585–1592. doi: 10.1016/j.exphem.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rajewsky N. microRNA target predictions in animals. Nat Genet. 2006;38(Suppl):S8–13. doi: 10.1038/ng1798. [DOI] [PubMed] [Google Scholar]
- 47.Wang X. miRDB: a microRNA target prediction and functional annotation database with a wiki interface. RNA. 2008;14:1012–1017. doi: 10.1261/rna.965408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang X, El Naqa IM. Prediction of both conserved and nonconserved microRNA targets in animals. Bioinformatics. 2008;24:325–332. doi: 10.1093/bioinformatics/btm595. [DOI] [PubMed] [Google Scholar]
- 49.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 50.van Rooij E. The art of microRNA research. Circ Res. 108:219–234. doi: 10.1161/CIRCRESAHA.110.227496. [DOI] [PubMed] [Google Scholar]
- 51.Hoang T. The origin of hematopoietic cell type diversity. Oncogene. 2004;23:7188–7198. doi: 10.1038/sj.onc.1207937. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table E1. Number of miRNA-mRNA pairs predicted by each algorithm (column headers), out of a total 8428 possible pairs (49 miRNAs × 172 mRNAs)
Supplementary Table E3. Number of overlaping predicted miRNA-mRNA pairs (predicted by 2 algorithms)
Supplementary Figure E1. Pathway analysis on predicted miRNA targets.
Supplementary Figure E2. Pathway analysis on 87 highly correlated miRNA-mRNA pairs.
Supplementary Figure E3. Pathway analysis on inversely correlated miRNA-mRNA pairs.