

Abstract
We developed a series of interrelated locus-specific databases to store all published and unpublished genetic variation related to these disorders, and then implemented microattribution to encourage submission of unpublished observations of genetic variation to these public repositories 1. A total of 1,941 unique genetic variants in 37 genes, encoding globins (HBA2, HBA1, HBG2, HBG1, HBD, HBB) and other erythroid proteins (ALOX5AP, AQP9, ARG2, ASS1, ATRX, BCL11A, CNTNAP2, CSNK2A1, EPAS1, ERCC2, FLT1, GATA1, GPM6B, HAO2, HBS1L, KDR, KL, KLF1, MAP2K1, MAP3K5, MAP3K7, MYB, NOS1, NOS2, NOS3, NOX3, NUP133, PDE7B, SMAD3, SMAD6, and TOX) are currently documented in these databases with reciprocal attribution of microcitations to data contributors. Our project provides the first example of implementing microattribution to incentivise submission of all known genetic variation in a defined system. It has demonstrably increased the reporting of human variants and now provides a comprehensive online resource for systematically describing human genetic variation in the globin genes and other genes contributing to hemoglobinopathies and thalassemias. The large repository of previously reported data, together with more recent data, acquired by microattribution, demonstrates how the comprehensive documentation of human variation will provide key insights into normal biological processes and how these are perturbed in human genetic disease. Using the microattribution process set out here, datasets which took decades to accumulate for the globin genes could be assembled rapidly for other genes and disease systems. The principles established here for the globin gene system will serve as a model for other systems and the analysis of other common and/or complex human genetic diseases.
Keywords: Globin genes, microattribution, locus-specific databases, genetic variation, hemoglobinopathies, thalassemia, fetal hemoglobin
Since completion of the human genome project, a major aim has been to determine how individual genomes differ from each other and how these differences explain variation in phenotype. However, it often remains unclear which variants cause changes in phenotype and which are neutral; furthermore, in many instances the mechanisms by which variants cause changes in gene expression and phenotypes remain unknown. To address this, DNA sequence data will have to be matched with well-defined phenotypes to make meaningful connections between structure, function and mechanism.
A potential hurdle to this approach is how to encourage “phenotypers” to report their observations. After the initial excitement (during the 1980s and 1990s) of identifying disease-causing molecular defects and the mechanisms by which they arise, the enthusiasm has declined such that it has become increasingly difficult to report small numbers of human variants in scientific journals. Consequently, many new variants associated with well-defined phenotypes and (equally important) variants which cause no change in phenotype remain unreported. Consequently a large amount of potentially valuable information remains inaccessible.
To overcome this problem we implemented a process for capturing such information with the incentive of microattribution, whereby the contribution of those collecting new detailed genotype/phenotype data is positively encouraged and appropriately acknowledged 1. We have applied the microattribution approach to inherited disorders affecting either the structure of hemoglobin (hemoglobinopathies such as sickle cell disease (SCD)), or the levels and balance of globin chain production, (the thalassemias). We also included variants that cause hereditary persistence of fetal hemoglobin (HPFH), a condition associated with increased production of gamma globin which ameliorates the clinical endpoints of SCD and β-thalassemia. The hemoglobinopathies and thalassemias are among the commonest inherited disorders in humans. Variants of the globin-encoding genes, residing in the α-like and β-like globin gene clusters, have provided key insights into the principles underlying human molecular genetics since the discipline was established in the 1950s 2.
Although most hemoglobinopathies are classic monogenic disorders affecting the structural gene, globin gene expression is the end product of a complex regulatory network (transcriptional and epigenetic) that emerges during terminal erythroid differentiation. Consequently, globin gene expression may also be affected by trans-acting mutations. Examples of such mutations were initially found in rare families with syndromal disorders of which α-thalassemia was one component, e.g. the ATR-X (MIM 301040) and ATMDS (MIM 300448) syndromes 3,4. Similarly, trichothiodystrophy (MIM 126340) was shown to be associated with β-thalassemia due to mutations in the XPD component of the general transcription factor complex TFIIH 5. The association of X-linked thrombocytopenia with β-thalassemia identified a mutation of the erythroid specific transcription factor GATA-1 6 and recently, systematic analysis of subjects with unexplained HPFH has identified mutations in the KLF1 erythroid transcription factor 7. Finally, the implementation of genome-wide association studies searching for quantitative trait loci that influence the level of fetal hemoglobin (HbF) has revealed several important regulators of HBG1/HBG2 gene expression, including the HBS1L-MYB 8 and BCL11A loci 9,10 on chromosomes 6 and 2, respectively. As genetic variation in the genes within the erythroid network are investigated in further detail we anticipate many more discoveries of trans-acting mutations which may provide target pathways for manipulating globin gene expression to ameliorate the symptoms of thalassemia and SCD. Therefore it is important that an effective database is created to accommodate all of the mutations affecting the globin genes and the network regulating their expression.
Here, we report the first example of implementing microattribution to systematically document genetic variation leading to human genetic disorders using the hemoglobinopathies and thalassemias as an example. Furthermore we demonstrate that microattribution can incentivise data contribution and, importantly show how an integrated human variant database (including the recently acquired microattribution data) has provided key insights into human genetic diseases. Microattribution provides an important mechanism and incentive for researchers to report all variants within a specific gene or disease network. Following the principles established for the globin gene disorders, these databases should provide a key resource for understanding the molecular pathology of human genetic diseases.
Developing the microattribution process
To ensure that all natural mutations and their associated phenotypes are accurately and efficiently recorded we have comprehensively documented genotype/phenotype information in patients with globin gene disorders in a series of interrelated LSDBs. Traditionally, credit has been given to discoverers of genetic variants by citations of their publications describing the variants. However, the increased rate of discovery through resequencing efforts far exceeds the capacity of citations of individual publications to give adequate credit. In order to be used effectively, published variants are deposited into databases such as those described here, nevertheless many variants may not be published. Alternatively variants may be discovered in large-scale collaborative project. . Credit can be given to the discoverers of variants deposited in databases through the novel process of microattribution 1. Each variant used in a paper is listed in Supplementary Tables 1–4 with its accession number and unique IDs for the discoverers, or “authors”, of the variant. In this paper, we have implemented “microcitations” to hemoglobinopathy-associated variants to provide incentives to data producers to deposit all of their data in these public resources 1. Depositing our Supplementary Tables 1–4 in a central repository (e.g. NCBI) provides a venue for quantitative microcitations for every unique author. Using this approach (first implemented in 2010) we have demonstrated a significant increase in the number of reported variants in the globin gene network (Supplementary Figure 1).
Database structure
The HbVar database of hemoglobin variants and thalassemia mutations (http://globin.bx.psu.edu/hbvar) is a publicly available LSDB which provides timely information to interested users including the globin research community, providers of genetic services and counselling, patients and their families, and pharmaceutical industries 11. The database is designed to allow regular entry updates and corrections, and has a user-friendly query interface that provides easy access to this information as an aid-in-diagnosis. Variant pathogenicity is established either directly from clinical findings or indirectly from linkage analysis, association, conservation and/or quantitative functional assays.
According to the literature available, variations in genes residing outside the human globin gene clusters are also implicated in producing or modifying the phenotypes associated with the hemoglobinopathies and thalassaemias. We have therefore developed separate LSDBs for such genes. We have based these LSDBs on the Leiden Open-Access Variation database (LOVD) management system 12. (See Supplementary Note.) We have developed LOVD-based LSDBs for a total of 37 genes, documenting some 1,889 unique genetic variants. Thirty-one of these LSDBs (ALOX5AP, AQP9, ARG2, ASS1, ATRX, BCL11A, CNTNAP2, CSNK2A1, EPAS1, ERCC2, FLT1, GATA1, GPM6B, HAO2, HBS1L, KDR, KL, KLF1, MAP2K1, MAP3K5, MAP3K7, MYB, NOS1, NOS2, NOS3, NOX3, NUP133, PDE7B, SMAD3, SMAD6, and TOX why is TFIIH/ XPD not in this list ? {Note that XPD is officially called ERCC2, it encodes on of the subunits of TFIIH}) correspond to genes encoding erythroid-expressed proteins, including transcription factors (e.g. GATA1, KLF1), chromatin associated co-factors (e.g. ATRX) and genes implicated in modulating HbF levels (e.g. AQP9, ARG2, BCL11A, HBS1L, MAP3K5, MAP3K7, MYB, PDE7B).
In order to bridge the wealth of information stored in HbVar with information deposited in the newly developed LOVD-based LSDBs that document genetic variation in other genes (referred to hereafter as HbVar-related LSDBs; http://lovd.bx.psu.edu), we needed to overcome the different architecture and design of these two databases. Therefore, we developed a LOVD copy of HbVar that serves as an intermediate between the HbVar-related LSDBs and HbVar. The LOVD copy of HbVar consists of 6 separate LSDBs for the functional globin genes (HBA2, HBA1, HBG2, HBG1, HBD, HBB), that intercommunicate with HbVar. This is necessary since the current capacity of the LOVD system cannot accommodate the wealth of information (particularly phenotypic data) presently deposited in HbVar.
Implementing microattribution
All genetic variation data have been collected and documented in these LSDBs with appropriate attribution of data contributors. These variants are reported in publicly available Microattribution Tables (also provided in Supplementary Table 1) that have been centrally deposited to NCBI (Supplemenatry Fig. 2). More specifically:
The first table contains the information required for submission of SNPs to the central depository, namely dbSNP (www.ncbi.nlm.nih.gov/projects/SNP). A blank form can be obtained by downloading from dbSNP along with the information specifying a SNP. When processing variant submissions in their official nomenclature (http://www.hgvs.org), NCBI 'stabilizes' the corresponding rs number for the variant and maintains the strand and orientation of the allele state.
The second table contains information needed for microattribution. It lists the variant nucleotide in the official nomenclature and the conventional, or common, variant name. For each variant, it includes both a local ID (e.g. from a LSDB) and an ID for the central repository (rs ID for dbSNP), as well as an ID for other general databases (OMIM variant ID and Swiss-Prot variant ID), when available. Each person involved in generating the biological information in the table (authors of papers, data submitters) can be listed as a unique ResearcherID, from the ResearcherID system of Thomson ISI (www.researcherid.com). This unique identifier is assigned to each variant and corresponds to a unique data contributing entity, the latter being from individual researchers to international research consortia. This approach allows instant views of an author’s citation metrics. Other systems that can be employed for this purpose include OpenID (http://openid.net) or Research Identification Primer (RIP; www.gen2phen.org).
The third table provides phenotype information. It depicts the phenotypic outcome of the genetic variant (hemoglobin variant, thalassemic condition, HbF modulation), hematological indices (Hb, MCH, MCV, HbA2, HbF), and clinical features (e.g. morphological alterations of erythrocytes, associated organ pathology). Depending on different diseases, these columns will differ accordingly.
The fourth table gives frequency information about each variant. Each record gives an ethnic group and the frequency at which the variant is found (either as a count or a percentage). This, as well as the former, table has more than one row for a variant.
In this protocol, data submitters can directly contribute data to HbVar to receive microattribution credit. These variants leading to hemoglobinopathies have received direct microattribution credit and have been recorded with ResearcherIDs, while in the case of previously published variants, the corresponding PubMedID was also used (Fig. 1). To date, 232 variants have been directly submitted to HbVar without being published in a peer-reviewed journal, some of which have been deposited with more than one ResearcherID. Seventy-six variants were “orphan”, namely variants for which there is neither PubMedID nor ResearcherID, due to the lack of valid contact details of the variant contributors or lack of response to our invitation. These variants have been deposited with an HbVar ResearcherID.
Figure 1. Graphical display of the HBB promoter variants recorded in HbVar, partitioned into unpublished variants contributed by investigators (blue) and published variants (purple).

The genomic position, sequence change and associated phenotype (β+ or β0 thalassemia) are given for each variant. Known protein-binding sites in the DNA sequence are boxed, with the name of the site and the binding protein above it. The transcription start site (+1) is in reverse type. The reverse complement of the genomic sequence is shown so that the gene is in the conventional left-to-right transcriptional orientation. The image was generated by displaying the results of a query on HbVar in the Pennsylvania State University genome browser followed by editing for clarity. Variants are given using the conventional nomenclature.
For all unpublished variants directly contributed to HbVar by the microattribution process, a very stringent evaluation of the information submitted takes place. Contributed variant data are evaluated by curators, all of whom are senior scientists with extensive editorial experience, especially in the field of hemoglobinopathies. The curators directly contact the data contributors, if needed, for clarifications related to issues pertaining to phenotypic description, method of variant identification, ethnicity of the individual with the variant, allele frequency and so on. Upon acceptance, contributed data become part of the main HbVar data collection recorded with the contributor(s) researcher ID.
Although microattribution can operate locally, i.e. within journals and databases each reporting quantitative citation of accessions, depositing the Microattribution Tables in a central repository of cited accessions, e.g. NCBI or EBI, allows the central registry to be mined for citations associated with unique author identities and with each author’s publications and database entries.
Mining the databases
In the case of globin gene disorders many variants were conventionally reported in genetics journals and these identified and/or elucidated many mechanisms underlying key aspects of gene regulation in-cis (e.g. promoters, enhancers, silencers, mRNA processing signals, translational signals) and in-trans (e.g. transcription factors, chromatin remodeling factors, protein chaperones. Furthermore, these variants helped to establish the mechanisms underlying human genetic disease. Implementation of the microattribution approach has significantly added to the repository of variants and use of this expanded database will continue to provide an important resource for generating and testing new hypotheses in the globin field (below, we provide some recent examples illustrating the value of comprehensive datasets in this system). The value of the comprehensive globin variant database (pre- and post-microattribution) clearly emphasizes the importance of developing similar databases for other genes and disease systems for which microattribution will become the main route to publication.
The first example of the value of the microattribution approach is the finding that the distribution of promoter mutations differs among globin genes. Although a great deal has been learnt about mammalian promoters from previous analysis of the globin genes, additional variants continue to develop our knowledge of how they are normally activated and how they are altered in human genetic disease. Globin gene promoter mutations contributing to β-like thalassemias and HPFH comprise approximately 10% of the total variants and result in various phenotypes, from the asymptomatic non-deletional HPFH conditions to the mild forms of β- and δ-thalassemia. The HBB promoter region harbours several genetic variants associated with β+ (expressing lower than normal levels of β-globin) and β0 (expressing no β-globin) thalassemia; these cluster in cis-regulatory elements known to bind transcription factors (Fig. 1). Many of these have been published, but an increasing number of unpublished variants have been contributed to HbVar from investigators around the world. The unpublished variants provide a more complete view of the contribution of genetic variants to phenotype. In this particular case, they reveal phenotypic consequences of variants in more positions of well known transcription factor binding sites (the “CACC” box and the “TATA” box), and show that additional substitutions in other binding sites contribute to phenotype (e.g. positions c.-80, c.-81, and c.-138). The HBB:c.-121C>T transition is adjacent to the CCAAT box. This motif was recognized 30 years ago as a component of some promoters, but the newly reported mutation is the first indication that genetic variation close to the motif affects HBB gene expression in humans.
In contrast to the promoters for HBB and HBD, variants are not found in the first 100 bp of the HBG1 and HBG2 promoters, but instead variants occur in the upstream region from approximately -100 to -200 bp (Fig. 2a). The HBG1/HBG2 gene promoters have several cis-regulatory elements in common with HBB and HBD promoters, such as a “TATA” box and a proximal “CCAAT” box, but no variants have been found in them. However, the “CCAAT” box is duplicated in the promoters of HBG1/HBG2 genes, and the upstream CCAAT box (and nucleotides very close to it) does carry variants associated with HPFH. A newly discovered, unpublished variant, c.-250C>T, calls attention to a tight cluster of mutations all associated with HPFH. An HPFH-associated variant has now been reported at each nucleotide from c.-251 to c.-248 (-198 to -195 related to the gene transcription start site), and a variant at c.-255 (-202) is associated with a similar phenotype (Fig. 2B). Given these phenotypes, this cluster of variants within the motif CCCTTCCC delineates a response element important for the silencing of the HBG1 and presumably HBG2 genes in adult erythroid cells (the same c.-250C>T mutation has been found in the promoter of the HBG2 gene; data not shown).
Figure 2. Functional role of HBG1 and HBG2 promoter variants.

HBG1 promoter variants are confined to the upstream region and associated with HPFH. The top line gives a schematic view of previously described binding sites for transcription factors, including the TATA box, the stage-selector element (SSE), the CCAAT boxes, GATA motifs bound by GATA1, and an octamer motif (OCT), plus the response element (RE) defined by a cluster of HPFH mutations. Motifs in which variants have been found are colored gray. The transcription start site (+1) is in reverse type. The image was generated by displaying the results of a query on HbVar in the Pennsylvania State University genome browser followed by editing for clarity. Variants are given using the conventional nomenclature. (b) Flow cytometry analysis of γ-globin+ erythrocytes from adult HBG1 c.-248C>G HPFH β-YAC transgenic lines. A mouse monoclonal γ-globin antibody was used to determine the percentage of F cells. Line and individual numbers are indicated at the top of the panels. Percent γ-globin–positive cells are indicated within each plot (see also Online Methods). Wild-type (wt) β-YAC mice served as negative controls, and HBG1 c.-170G>A HPFH β-YAC mice13 were used as positive controls. In parallel experiments, human β-globin was expressed in 92–97% of the cells analyzed for all lines (data not shown). (c) Human γ-globin gene expression in HBG1 c.-248C>G HPFH β-YAC transgenic lines. Percent γ-globin gene expression, copy number-corrected and normalized to per-copy mouse α-globin gene expression, is shown on the y axis. β-YAC construct and line numbers, where appropriate, are indicated at the bottom of the plot. Error bars represent standard deviation of triplicate experiments.
To test the hypothesis, derived from the documented variants, that this motif delineates a response element important for silencing of the HBG1 and HBG2 genes, we recently produced human β-globin locus (β-YAC) transgenic mice containing the -248 C>G Brazilian HPFH mutation in the HBG1 gene, which directly alters the CCCTTCCC sequence at the 3' C. Adult mice display a HPFH phenotype with an increased number of HbF-containing cells (Fig. 2b), and real-time quantitative RT-PCR analyses demonstrated that one line shows an 8–34 fold increase of HBG1 gene expression relative to wild-type β-YAC mice (Fig. 2c). By comparison, -117 Greek HPFH β-YAC transgenic mice display a 56-fold increase of γ-globin gene expression relative to wild-type β-YAC mice. Future experiments will examine the mechanism of repression at this region. Recent studies have shown that the transcription factor BCL11A acts to repress HBG1 and HBG2 expression in adult erythroid cells, acting with the protein SOX6 13. Although BCL11A showed no binding in HBG1 and HBG2 proximal promoters, SOX6 showed strong binding which overlapped with GATA1 binding in these regions. In this way the database has posed a new testable hypothesis. The CCCTTCCC element, which is adjacent to a GATA binding site, may bind a currently unknown protein that acts in concert with BCL11A to repress production of γ-globins.
Overall, comparative analysis of the globin gene promoter mutations revealed a distinct distribution pattern for each gene. In the HBD gene, promoter mutations are widely spread within the proximal promoter region and do not form mutational clusters around cis-regulatory elements (Fig. 2C). Interestingly, mutations at positions c.-81A>G and c.-80T>C have been found in the TATA boxes of the HBB and HBD genes, suggesting that they could be the result of genetic recombination events 14.
A second example of the value of the microattribution approach was the discovery of a-thalassemia resulting from inherited or acquired mutations in the ATRX gene. The comprehensive database originally identified and defined some of the key trans-acting factors in the globin gene system. The expanded database continues to refine our understanding of such trans acting factors. Unlike the common forms of α-thalassemia, resulting from cis-acting genetic defects, two rare forms of α-thalassemia are caused by trans-acting mutations in the X-linked ATRX gene. These mutations cause ATR-X syndrome, which is characterized by a severe form of syndromal mental retardation with characteristic dysmorphic faces, genital abnormalities, and a mild but variable form of hemoglobin H disease 3. In addition, acquired mutations in the ATRX gene are seen in patients who develop the ATMDS syndrome, a condition in which α-thalassemia (AT) is associated with myelodysplastic syndrome (MDS) 4. In both conditions, the levels of α-globin mRNA are reduced, suggesting that the ATRX gene is involved in the normal regulation of α-globin gene expression. To date, 107 unique inherited and/or acquired disease-causing missense mutations have been found, which are located predominantly in two highly conserved domains of the ATRX protein (Supplementary Fig. 4). These variants cluster within a globular domain that contains a plant homeodomain (PHD) which binds the N-terminal tails of histone H3, and the 7 helicase sub-domains which identify ATRX as a member of the SNF2 family of chromatin-associated proteins. Structure/function studies based on natural mutations in the comprehensive database (Figure 4) have elucidated precisely how ATRX is recruited to some of its targets via an interaction with the N-terminal tails of histone H3.
Notably, the degree of α-thalassemia seen in ATMDS patients (acquired ATRX gene mutations) is much greater than in patients with the ATR-X syndrome (inherited ATRX gene mutations), even when (by comparing mutations on the comprehensive database) we can see the same ATRX mutation occurs in either condition 15. Again analysis of the comprehensive variant database poses a new testable hypothesis. These findings suggest that another component of the ATRX pathway may frequently be mutated in patients with the common forms of MDS.
A third example of the value of microattribution is the discovery of variants in KLF leading to elevated HbF levels. KLF1 encodes a key erythroid transcriptional regulator that has many target genes with essential functions in erythroid cells including the globins, membrane proteins and heme synthesis enzymes17. The first report on KLF1 mutations in humans linked them to the rare blood group In(Lu) phenotype18, in which the expression of the Lutheran blood group antigens is diminished. The reported individuals carried eight different loss-of-function mutations and one mutation abolishing a GATA1 binding site in the KLF1 promoter. In all cases, the mutant KLF1 allele occurred in the presence of a normal KLF1 allele. A subsequent study on a large Maltese pedigree demonstrated that haploinsufficiency for KLF1 causes HPFH7. A mutation in KLF1, resulting in p.Lys288X, was present exclusively in all individuals in this family with HPFH. This mutation ablates the complete zinc finger domain and therefore abrogates DNA binding of the mutant KLF1 protein (Fig. 3 and Supplementary Table 2). The occurrence of HPFH in the individuals with In(Lu) has not been investigated. An analysis of archived blood samples from a number of these individuals with In(Lu) showed that their HbF levels were raised compared to those observed in control samples. Also, 30 out of 31 Sardinian individuals bearing four different KLF1 mutations showed raised HbF levels compared to control samples. In addition, two individuals suffering from dyserythropoietic anemia carried a KLF1 p.Glu325Lys alteration and had an HbF level of 40% (Fig. 3 and Supplementary Table 2)19, 20. Mutations at this position alter the DNA binding specificity of KLF1. We note that the mouse neonatal anemia mutant (Nan) has an alteration in the orthologous amino acid of Klf1, p.Glu339Asp21, 22. Adult heterozygous Nan animals show increased expression of embryonic globins, a condition akin to HPFH. Collectively, these data support the link between KLF1 and HPFH and highlight the importance of the second DNA-binding zinc finger for normal KLF1 function. This raises the possibility that some of the KLF1 mutations which result in altered DNA binding specificity may have increased impact on HbF levels. This hypothesis can now be experimentally tested in vitro by DNA binding assays and in vivo in animal models.
Figure 3. Correlation of the different KLF1 gene variants deposited into HbVar (shown as blue and red squares, depicting unpublished and published information, respectively) and their corresponding HbF levels (median value in cases of three or more individuals) compared to wild-type individuals (shown as green squares).
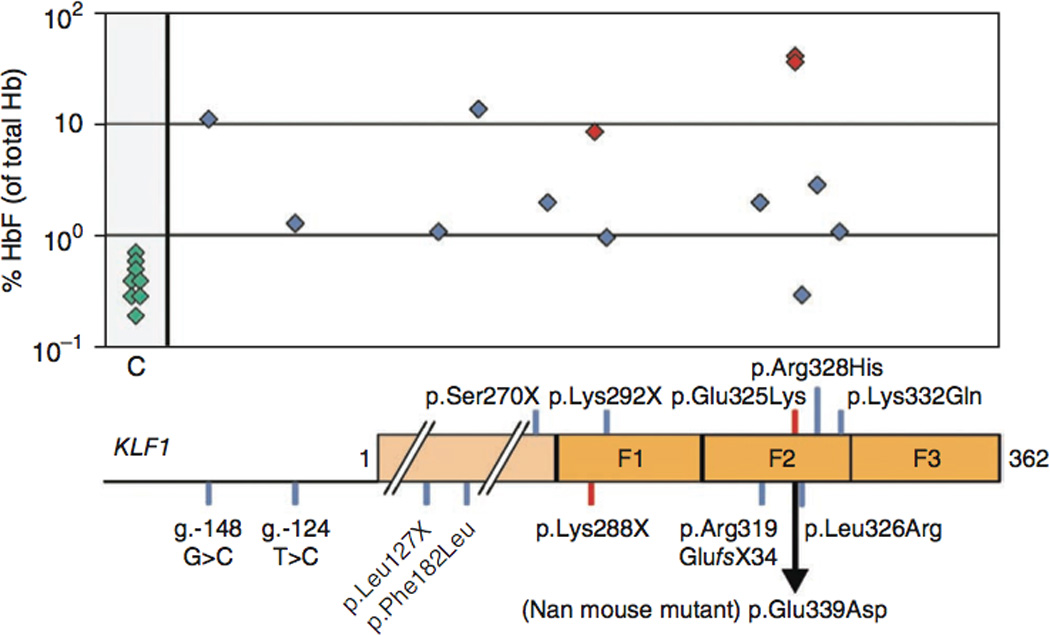
KLF1 is not shown to scale. A simplified diagram depicting the KLF1 promoter and protein is shown underneath. The positions of the zinc fingers are indicated (F1, F2 and F3). For the exact HbF levels corresponding to each KLF1 gene variant, see Supplementary Table 2.
A final example of the value of microattribution is the discovery of hemoglobin variants. A large proportion of genetic variation in the human globin genes leads to hemoglobin variants. Most hemoglobin variants are rare, result from single amino acid substitutions of a globin chain and have a negligible or even no effect on hemoglobin function.
The documented hemoglobin variants reside solely within exons and include: (a) Structural variants with a pleiotropic effect [e.g. HbS (HBB:c.20A>T), HbE (HBB:c.79G>A) and HbC (HBB:c.19G>A], (b) Variants (138 different variants) leading to unstable hemoglobin, where mutations affect the heme pocket of the globin chain, (c) Variants leading to methemoglobinemia, where the ferrous ion (Fe2+) of the heme group is oxidized to the ferric state (Fe3+). Most of these variants involve replacement by tyrosine of the histidine residues that anchor heme. (d) Variants (92 different variants) with altered oxygen affinity. Most of these result in increased oxygen affinity.
Although all of these correlations between structure and function have depended on the comprehensive database, new insights and questions continue to arise as new mutants are added to the repository, an initiative that sparked the implementation of the microattribution process for hemoglobinopathies. Notably, 14 hemoglobin variants result from the same mutation but on a different α-globin gene paralogue 16, i.e., involving related genes that have evolved from recent gene duplication and as such are subject to frequent gene conversion events. HbF-Sardinia and HbF-Lesvos provide another such example, involving the same mutation (c.227T>C) but on the paralogous HBG1 and HBG2 genes, respectively 17.
Discussion
The development of an integrated set of comprehensive LSDBs for a particular spectrum of human genetic diseases with microattribution, as described here for the hemoglobinopathies, provides an example of how such systems might be set up for a wide range of human genetic disorders in the future. In the past, the description of natural variants has been accommodated by the conventional literature and has made an enormous contribution to the field of human genetics. In addition it has demonstrated how some of these mutations have reached polymorphic frequencies via natural selection, while detailed analysis of natural mutants has also been invaluable in establishing many of the general principles underlying mammalian gene regulation and human molecular genetics.
The strength of such observations will continue to increase as new mutations enter the databases, even though these might not merit a full publication on their own. Furthermore, new patterns of mutation may emerge; the accumulation of coding mutations in particular regions of a protein often identify a functionally important domain, as illustrated by ATRX and KLF1 gene variants (Supplementary Fig. 4 and Fig. 3, respectively), and conversely, the identification of common neutral variants may rule out a major functional role for other regions. Similarly, DNA variants of key regulatory regions (promoters, enhancers, silencers, boundary elements, locus control regions) are often critical in identifying important cis-elements and yet other neutral variants may help map regions of little functional importance (Fig. 1 and Supplementary Fig. 3). At the nucleotide level, such variants can even help map transcription factor binding sites 18. The emergence of patterns of mutation may also point to mechanisms of mutation, exemplified by gene conversion events identified at the HBA1/HBA2 and HBG1/HBG2 genes. Additionally, subtle phenotypic differences, e.g. between δβ-thalassemia and deletional HPFH 2, can be attributed to the different junction points and the sequences that are removed or juxtaposed as a result of these deletions. Systematic documentation of these deletions in HbVar is currently under way and may allow the identification of novel regulatory elements that lie within the deleted or juxtaposed regions.
Perhaps the most important aspect of such comprehensive interacting databases is that they will pose and answer questions that could otherwise not be addressed at all, potentially leading to entirely new insights. These databases will not only be of value in establishing the phenotypes of natural variants but may also be used in the development of personalized medicine. In the globin field a great deal of effort is directed towards the development of drugs to increase the level of HbF and thereby ameliorate the clinical severity of β-thalassemia and SCD. Potential therapeutic agents identified to date include hydroxyurea and butyrate. The response to HbF-augmenting therapies is variable in patients with β-thalassemia and SCD with approximately 25% of the patients being poor or non-responders 19. Therefore, the ability to predict a patient’s response to HU and/or other HbF-augmenting drugs would help in optimising therapy. Polymorphisms in genes regulating HbF expression, HU metabolism and erythroid progenitor proliferation might modulate the patient’s response to HbF-inducing pharmacological agents 20. Data to support the use of pharmacogenetic testing for HU treatment for hemoglobinopathies are currently very limited. Several SNPs in the HAO2, ARG2, FLT1, and NOS1 genes have been associated with variable HbF response to HU treatment 20, while whole-genome transcription profiling efforts are expected to shed light on new pathways involved in this process (Ref. 21 and Phylactides et al., in preparation).
Since its establishment in 2001, we have witnessed a significant annual growth in HbVar content, while a fraction of data submitters were subsequently encouraged to submit a full or short report to the scientific journal Hemoglobin 22. We anticipate that microattribution will further encourage new data submitters to contribute their observations to HbVar to receive not only credit in the form of microcitations but also co-authorship in a future microattribution update. The microattribution process established here provides a template for similar ventures for other human genes, their associated systems and the variants that cause their associated genetic diseases.
In essence, this project represents a well-coordinated multicenter effort to systematically document genetic variation in globin and associated genes relevant to hemoglobinopathies and thalassaemias, and the first example of implementing microattribution to provide incentives for submitting data describing genetic variation. As such, it should serve as a model for the comprehensive documentation and analysis of genetic variations in other common or genetically complex disorders, the conduct of a thorough synopsis of other fields, or both.
URLs
HbVar Database of Hemoglobin Variants and Thalassemia Mutations, http://globin.bx.psu.edu/hbvar/; Golden Helix Server, http://www.goldenhelix.org/; Leiden Open-Access Variation Database, http://www.lovd.nl/; Frequencies of Inherited Disorders database, http://www.findbase.org/; dbSNP database, http://www.ncbi.nlm.nih.gov/projects/SNP/; Human Genome Variation Society, http://www.hgvs.org/; ResearcherID System of Thomson ISI, http://www.researcherid.com/; Open ID system, http://openid.net/; Genotype-to-Phenotype database project's Researcher Identification Primer (RIP), http://www.gen2phen.org/.
Methods
Quantitation of hemoglobin fractions
Twenty microliters of total blood was analyzed using cation-exchange high performance liquid chromatography (VARIANT, Bio-Rad Laboratories).
Construction of the HBG1 c.-248C>G HPFH β-YAC
A 213-kb yeast artificial chromosome (YAC) carrying the human β-globin locus with the HBG1 c.-248C>G point mutation (Aγ -195C>G), leading to the Brazilian type of non-deletional HPFH, which directly alters the CCCTTCCC sequence at the 3′ C, was synthesized as follows, using previously described methods30. Briefly, a marked HBG1 gene (Aγm) contained as a 5.4-kb SspI fragment (GenBank file U01317, coordinates 38,683-44,077) in the yeast-integrating plasmid (YIP) pRS406 was mutagenized using the QuikChange Site-Specific Mutagenesis Kit (Stratagene). The presence of the HBG1 c.-248C>G point mutation was confirmed by DNA sequencing, and the mutation was introduced into the β-YAC by 'pop-in', 'pop-out' homologous recombination in yeast. The mark in the Aγm-globin gene is a 6-bp deletion at +21 to +26 relative to the Aγ-globin translation start site, allowing preliminary discrimination of the modified β-YAC from the wild-type β-YAC by restriction enzyme digestion following homologous recombination. The presence of the mutation in clones passing this test was confirmed by DNA sequence analysis of a PCR-amplified fragment encompassing the mutated region. Transformation of yeast, screening of positive clones, purification of the β-YAC and mouse transgenesis were performed as described previously31.
Copy number determination
The relative β-YAC transgene copy number was calculated using the HBG1 and HBG2 genes and a standard curve generated from genomic DNA samples from our wild-type β-YAC transgenic mice. Samples of transgenic mouse genomic DNA were serially diluted from 100–0.01 ng and subjected to SYBR PCR with HBG1 or HBG2 primers. The copy number for each reaction was estimated by comparing the threshold cycle of each sample to the threshold cycle of the standards and normalizing to the wild-type β-YAC transgenic mouse samples.
Real-time quantitative RT-PCR
Total RNA, isolated from adult peripheral blood, was reverse-transcribed and the resultant complementary DNA was subjected to real-time quantitative RT-PCR analysis with SYBR green using a CFX96 system (Bio-Rad). Human γ-globin expression was normalized to mouse α-globin expression and corrected for transgene and endogenous gene copy number. PCR primer sequences were as previously described32. Results are averages of triplicates, with the standard error indicated.
F-cell detection by flow cytometry
We used a protocol adapted from references32 and 33. Essentially, mouse blood was collected from the tail vein in heparinized capillary tubes. Ten microliters of whole blood was washed in 1 ml PBS, centrifuged at 200g at 4 °C for five minutes, and the pellet was resuspended and fixed in 1 ml of 4% fresh paraformaldehyde and PBS at pH 7.5 (Sigma-Aldrich) for 40 min at 37 °C. The cells were centrifuged, and the pellets were resuspended in 1 ml of ice cold acetone and methanol (4:1) and incubated on ice for one minute. Following centrifugation, cells were washed twice in 1 ml ice-cold PBS and 0.1% BSA and resuspended in 800 µl of PBS, 0.1% BSA and 0.1% Triton X-100 (PBT). One microgram of γ-globin antibody (catalog number sc-21756 unconjugated, Santa Cruz Biotechnology) was added to 100 µl of the cell suspension and incubated for 20 min in the dark at room temperature (37 °C). One milliliter of ice-cold PBS and 0.1% BSA was added, the sample was centrifuged and the pellet was resuspended in 100 µl ice-cold PBT. One hundred microliters of Alexa 488 (catalog number 11001, Invitrogen Molecular Probes) secondary antibody, diluted 1:200 in ice-cold PBT, was added to the cell suspension and the sample was incubated at room temperature for 20 min in the dark. Cells were washed with 1 ml of ice-cold PBS and 0.1% BSA and the pellets were resuspended in 200 µl of PBS. Samples were analyzed using an Accuri C6 Flow Cytometer (Accuri Cytometers, Inc.) with a 530/30 nm (FITC/GFP) emission filter. Data from 30,000 cells were acquired for analysis using CFlow Software (Accuri Cytometers, Inc.); cells were gated to exclude dead cells. For, FL1-A, a 530/30 nm (FITC, GFP) filter was used to identify the Alexa 488–positive F cell population; For FL2-A a 585/40 nm (PE, PI) filter was used as a compensation to identify the Alexa 488–negative cell population. For M3, the mean fluorescent intensity, an increase in F cells is reflected by a peak shift and increase in the peak of fluorescence intensity. P4, distinct positive F cells.
Supplementary Material
Acknowledgements
This work was supported by US National Institutes of Health (NIH) grants R01 DK065806, RC HG005573. U01 HG004695 to RCH and European Commission grants [FP6-026539 (ITHANET), FP7-200754 (GEN2PHEN)] to GPP and the NIHR Biomedical Research Centre (Oxford).
References
- 1.Anonymous. Human variome microattribution reviews. Nat. Genet. 2008;40:1. [Google Scholar]
- 2.Patrinos GP, Antonarakis SE. Human hemoglobin. In: Speicher M, Antonarakis SE, Motulsky A, editors. Human Genetics: Problems and Approaches. Heidelberg, Germany: Springer-Verlag; 2010. pp. 366–401. [Google Scholar]
- 3.Gibbons RJ, Picketts DJ, Villard L, Higgs DR. Mutations in a putative global transcriptional regulator cause X-linked mental retardation with alpha-thalassemia (ATR-X syndrome) Cell. 1995;80:837–845. doi: 10.1016/0092-8674(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 4.Gibbons RJ, et al. Identification of acquired somatic mutations in the gene encoding chromatin-remodeling factor ATRX in the α-thalassemia myelodysplasia syndrome (ATMDS) Nat. Genet. 2003;34:446–449. doi: 10.1038/ng1213. [DOI] [PubMed] [Google Scholar]
- 5.Viprakasit V, et al. Mutations in the general transcription factor TFIIH result in β-thalassaemia in individuals with trichothiodystrophy. Hum. Mol. Genet. 2001;10:2797–2802. doi: 10.1093/hmg/10.24.2797. [DOI] [PubMed] [Google Scholar]
- 6.Yu C, et al. X-linked thrombocytopenia with thalassemia from a mutation in the amino finger of GATA-1 affecting DNA binding rather than FOG-1 interaction. Blood. 2002;100:2040–2045. doi: 10.1182/blood-2002-02-0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borg J, et al. Haploinsufficiency for the erythroid transcription factor KLF1 causes hereditary persistence of fetal hemoglobin. Nat. Genet. 2010;42:801–805. doi: 10.1038/ng.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thein SL, et al. Intergenic variants of HBS1L-MYB are responsible for a major quantitative trait locus on chromosome 6q23 influencing fetal hemoglobin levels in adults. Proc. Natl. Acad. Sci. USA. 2007;104:11346–11351. doi: 10.1073/pnas.0611393104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Menzel S, et al. A QTL influencing F cell production maps to a gene encoding a zinc-finger protein on chromosome 2p15. Nat. Genet. 2007;39:1197–1199. doi: 10.1038/ng2108. [DOI] [PubMed] [Google Scholar]
- 10.Sankaran VG, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2007;322:1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 11.Hardison RC, et al. HbVar: a relational database of human hemoglobin variants and thalassemia mutations at the globin gene server. Hum. Mutat. 2002;19:225–233. doi: 10.1002/humu.10044. [DOI] [PubMed] [Google Scholar]
- 12.Fokkema IF, den Dunnen JT, Taschner PE. LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum. Mutat. 2005;26:63–68. doi: 10.1002/humu.20201. [DOI] [PubMed] [Google Scholar]
- 13.Peterson KR, et al. Use of yeast artificial chromosomes (YACs) in studies of mammalian development: production of β-globin locus YAC mice carrying human globin developmental mutants. Proc. Natl. Acad. Sci. USA. 1995;92:5655–5659. doi: 10.1073/pnas.92.12.5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu J, et al. Transcriptional silencing of γ-globin by BCL11A involves long-range interactions and cooperation with SOX6. Genes Dev. 2010;24:783–798. doi: 10.1101/gad.1897310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borg J, Georgitsi M, Aleporou-Marinou V, Kollia P, Patrinos GP. Genetic recombination as a major cause of mutagenesis in the human globin gene clusters. Clin. Biochem. 2009;42:1839–1850. doi: 10.1016/j.clinbiochem.2009.07.014. [DOI] [PubMed] [Google Scholar]
- 16.Steensma DP, Gibbons RJ, Higgs DR. Acquired α-thalassemia in association with myelodysplastic syndrome and other hematologic malignancies. Blood. 2005;105:443–452. doi: 10.1182/blood-2004-07-2792. [DOI] [PubMed] [Google Scholar]
- 17.Drissen R, et al. The erythroid phenotype of EKLF-null mice: defects in hemoglobin metabolism and membrane stability. Mol. Cell. Biol. 2005;25:5205–5214. doi: 10.1128/MCB.25.12.5205-5214.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singleton BK, Burton NM, Green C, Brady RL, Anstee DJ. Mutations in EKLF/KLF1 form the molecular basis of the rare blood group In(Lu) phenotype. Blood. 2008;112:2081–2088. doi: 10.1182/blood-2008-03-145672. [DOI] [PubMed] [Google Scholar]
- 19.Arnaud L, et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am. J. Hum. Genet. 2010;87:721–727. doi: 10.1016/j.ajhg.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singleton BK, et al. A novel EKLF mutation in a patient with dyserythropoietic anemia: the first association of EKLF with disease in man. Blood. 2009;114:72. [Google Scholar]
- 21.Siatecka M, et al. Severe anemia in the Nan mutant mouse caused by sequence-selective disruption of erythroid Kruppel-like factor. Proc. Natl. Acad. Sci. USA. 2010;107:15151–15156. doi: 10.1073/pnas.1004996107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Heruth DP, et al. Mutation in erythroid specific transcription factor KLF1 causes hereditary spherocytosis in the Nan hemolytic anemia mouse model. Genomics. 2010;96:303–307. doi: 10.1016/j.ygeno.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moradkhani K, et al. Mutations in the paralogous human α-globin genes yielding identical hemoglobin variants. Ann. Hematol. 2009;88:535–543. doi: 10.1007/s00277-008-0624-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Papadakis MN, Patrinos GP, Drakoulakou O, Loutradi-Anagnostou A. HbF-Lesvos: an HbF variant due to a novel G gamma mutation (:G gamma 75 ATAACA) detected in a Greek family. Hum. Genet. 1996;97:260–262. doi: 10.1007/BF02265278. [DOI] [PubMed] [Google Scholar]
- 25.Patrinos GP, et al. Improvements in the HbVar database of human hemoglobin variants and thalassemia mutations for population and sequence variation studies. Nucleic Acids Res. 2004;32:D537–D541. doi: 10.1093/nar/gkh006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Steinberg MH, et al. Fetal hemoglobin in sickle cell anemia: determinants of response to hydroxyurea. Multicenter study of hydroxyurea. Blood. 1997;89:1078–1088. [PubMed] [Google Scholar]
- 27.Ma Q, et al. Fetal hemoglobin in sickle cell anemia: genetic determinants of response to hydroxyurea. Pharmacogenomics J. 2007;7:386–394. doi: 10.1038/sj.tpj.6500433. [DOI] [PubMed] [Google Scholar]
- 28.Patrinos GP, Grosveld FG. Pharmacogenomics and therapeutics of hemoglobinopathies. Hemoglobin. 2008;32:229–236. doi: 10.1080/03630260701680367. [DOI] [PubMed] [Google Scholar]
- 29.Patrinos GP, Wajcman H. Recording human globin gene variation. Hemoglobin. 2004;28:v–vii. [PubMed] [Google Scholar]
- 30.Harju S, Navas PA, Stamatoyannopoulos G, Peterson KR. Genome architecture of the human β-globin locus affects developmental regulation of gene expression. Mol. Cell. Biol. 2005;25:8765–8778. doi: 10.1128/MCB.25.20.8765-8778.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harju-Baker S, Costa FC, Fedosyuk H, Neades R, Peterson KR. Silencing of Agamma-globin gene expression during adult definitive erythropoiesis mediated by GATA-1-FOG-1-Mi2 Complex binding at the-566 GATA site. Mol. Cell. Biol. 2008;28:3101–3113. doi: 10.1128/MCB.01858-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Böhmer RM. Flow cytometry of erythroid cells in culture: bivariate profiles of fetal and adult hemoglobins. Methods Cell Biol. 2001;64:139–152. doi: 10.1016/s0091-679x(01)64011-1. [DOI] [PubMed] [Google Scholar]
- 33.Amoyal I, Fibach E. Flow cytometric analysis of fetal hemoglobin in erythroid precursors of β-thalassemia. Clin. Lab. Haematol. 2004;26:187–193. doi: 10.1111/j.1365-2257.2004.00606.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.