

Abstract
Many Borrelia burgdorferi Erp outer surface proteins have been demonstrated to bind the host complement regulator factor H, which likely contributes to the ability of these organisms to evade the host innate immune system. B. burgdorferi controls Erp protein synthesis throughout the bacterial infectious cycle, producing the proteins during mammalian infections but repressing their synthesis during tick infections. Defining the mechanism by which B. burgdorferi regulates the expression of these virulence determinants will provide important insight into the biological and pathogenic properties of the Lyme disease spirochete. The present study demonstrates that two highly conserved DNA sequences located 5′ of erp operons specifically bind bacterial proteins. Analyses with B. burgdorferi of transcriptional fusions between erp promoter/operator DNAs and the gene for green fluorescent protein indicated that the expression of these operons is regulated at the level of transcriptional initiation. These analyses also indicated significant differences in the promoter strengths of various erp operons, which likely accounts for reported variations in expression levels of different Erp proteins. Mutagenesis of promoter-gfp fusions demonstrated that at least one of the proteins which bind erp operator DNA functions as a repressor of transcription.
The spirochete Borrelia burgdorferi is maintained in nature through an infectious cycle involving warm-blooded vertebrates and Ixodes sp. ticks. These bacteria regulate the expression of a number of different proteins during the infectious cycle, among which are the Erp outer surface lipoproteins. Individual bacteria encode multiple members of the Erp family, with the type strain, B31, being known to carry 17 erp genes at 10 separate loci (12, 13, 50). Each locus occupies an allelic position on a different plasmid of the cp32 family. Although cp32 plasmids are largely identical in their sequences, many are compatible with each other, with 12 apparent incompatibility groups identified to date (16, 47). Many Erp proteins have been demonstrated to bind host factor H, a regulator of complement activation, and may thereby help protect the bacteria from the alternative pathway of complement-mediated killing (4, 5, 25-29, 34, 46). Consistent with that function, B. burgdorferi expresses Erp proteins during transmission between the tick vector and the vertebrate host, times when the bacterium is exposed to host serum and the innate immune system (22-24, 36, 37). For unknown reasons, B. burgdorferi down-regulates Erp expression during infections of ticks (22, 36).
A number of in vitro studies have provided insights into the regulatory mechanisms by which B. burgdorferi controls Erp synthesis. The culture temperature significantly impacts Erp expression levels, with bacteria grown at 34°C producing higher levels of Erp proteins than do bacteria cultivated at 23°C (24, 44, 48). These temperatures mimic those experienced by feeding and unfed ticks, respectively (40). The bacteria also respond to quorum sensing and to other, unidentified chemical signals that potentially represent components of host blood or other tissues encountered during infection (1, 3, 8, 15, 23, 24, 41, 52, 53). The regulation of Erp expression appears to occur at the mRNA level (24, 44), although evidence has been presented suggesting that some erp operons are regulated by additional mechanisms (24).
Erp proteins have been given various other names by different researchers, including OspE, p21, Bbk2.10, pG, OspF, and Elp (50). The primary structures of Erp proteins may vary widely, with different proteins exhibiting anywhere between 18 and 100% identical amino acid sequences (2, 47, 50). These differences have led to the suggestion that the Erp family can be divided into three or more subfamilies (2). However, all erp genes hold a number of features in common, the most significant being that each erp operon is preceded by a highly conserved DNA sequence (Fig. 1) (2, 12, 13, 24, 32, 44, 49, 50). The transcriptional start site has been mapped to a point just 5′ of the translational initiation codon (24). Electrophoretic mobility shift assays (EMSAs) have demonstrated that an unidentified B. burgdorferi protein(s) specifically binds to DNA immediately 5′ of all tested erp operons (8). The expression of erp operons is not detectably affected by DNA supercoiling (6, 19), suggesting that the regulation of erp transcription is due to the binding of an activator and/or repressor protein(s) to operator sites.
FIG. 1.
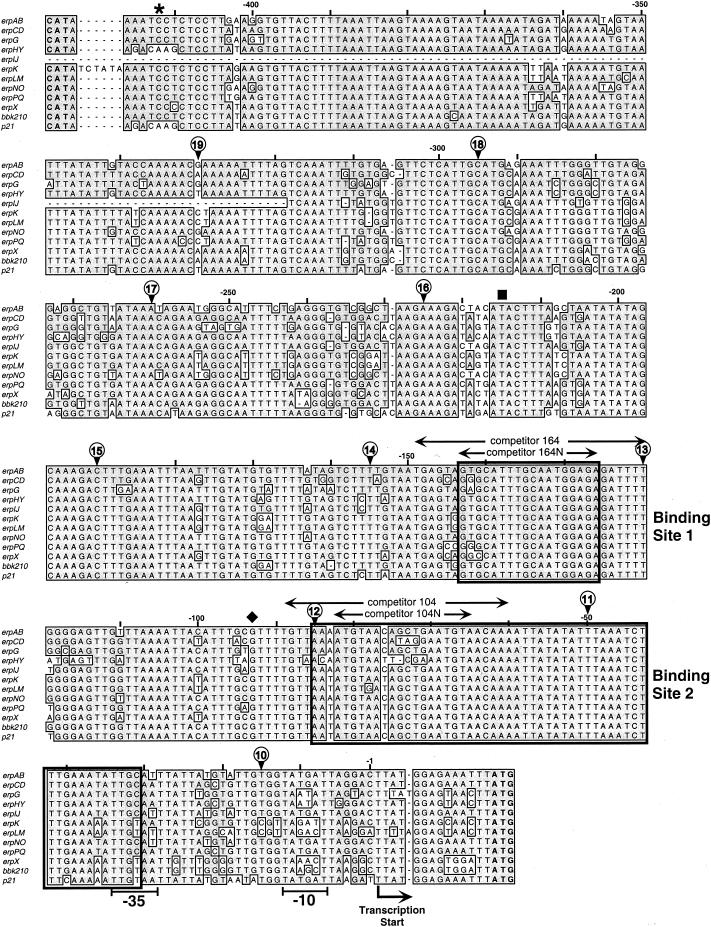
Alignment of the DNA sequences located 5′ of the 10 erp operons of strain B31, the bbk2.10 operon of strain 297, and the p21erp22 operon of strain N40. The complete sequence 5′ of the B31 erpIJ operon has not been determined (12, 13). Identical nucleotides found in 50% or more of the sequences are boxed and shaded. Start codons (ATG) of the first gene of each erp operon are indicated in bold at the lower right. The start codon of each divergently transcribed bppC gene (CAT) is indicated in bold at the upper left. The identified start of erp transcription (24) and probable −10 and −35 sequences are indicated. Nucleotides are numbered above each line relative to the start of transcription. The5′-most nucleotides of DNAs used in initial EMSA competition experiments are indicated by circled numbers as follows: the 5′ end of G10-AC is labeled “10,” the 5′ end of G11-AC is marked “11,” and so on. All of the competitors extended 3′ beyond the sequences illustrated. Competitor G9-AC was composed of DNA sequences entirely within the erpG gene and therefore is not indicated in this figure. Heavily lined boxes indicate the boundaries of protein-binding sites 1 and 2. The asterisk above bp −412 indicates the 5′ ends of full-length promoter/operator fragments used for the construction of most Perp::gfp fusions, the black square above bp −215 indicates the 5′ ends of fragments used to produce pEAP1 and pGPB, and the diamond above bp −93 indicates the 5′ ends of fragments used for the construction of pEAP2 and pGPL.
To better understand the mechanisms by which B. burgdorferi regulates Erp expression, we have performed a detailed characterization of erp promoter/operator DNAs. EMSAs were utilized to precisely map protein-binding sites within these regions. Recent advances have enabled the use of transcriptional fusions with reporter genes for the study of the nature of promoter/operator elements in B. burgdorferi (11, 16). This technique was utilized to define the roles of 5′ DNA in the transcriptional regulation of erp operons and to explore reasons for differences in promoter activity. In addition, promoter-reporter fusions were used to examine erp operons that were previously believed to possess regulatory mechanisms not found in other members of the erp family.
MATERIALS AND METHODS
Bacteria.
The B. burgdorferi strains used for this work are listed in Table 1. All strains were grown in modified Barbour-Stoenner-Kelly medium (BSK-II) (9). Unless otherwise noted, B. burgdorferi strains were cultured at 34°C. By the use of PCR with previously described oligonucleotide primer pairs (35, 38, 42), strain B31-e2 was determined to contain only plasmids cp26, cp32-1, cp32-3, cp32-4, lp17, lp38, and lp54.
TABLE 1.
Bacterial strains used for this work
| Strain | Description |
|---|---|
| B31 | Wild type, B. burgdorferi type strain |
| 297 | Wild type |
| N40 | Wild type |
| B31-RML | Subculture of strain B31 |
| B31-e2 | Clone of strain B31 |
| KS10 | B31-e2 containing pBLS590 (pJAH2 containing promoterless gfp) |
| KS11 | B31-e2 containing pBLS591 (pBLS590 containing PerpAB from −442 to start) |
| KS12 | B31-e2 containing pBLS592 (pBLS590 containing PerpG from −442 to start) |
| KS13 | B31-e2 containing pBLS593 (pBLS590 containing PerpAB from −442 to −10 and PerpG from 3′ of −10 to start) |
| KS15 | B31-e2 containing pBLS594 (pBLS590 containing PerpG from −442 to 5′ of −10 and PerpAB from −10 to start) |
| KS16 | B31-e2 containing pEAP1 (pBLS590 containing PerpAB from −215 to start) |
| KS17 | B31-e2 containing pEAP2 (pBLS590 containing PerpAB from −93 to start) |
| KS18 | B31-e2 containing pBLS595 (pBLS590 containing Pbbk2.10 from −442 to start) |
| KS19 | B31-e2 containing pBLS598 (pBLS590 containing PerpAB from −442 to start, with PerpG −10 site) |
| KS20 | B31-e2 containing pBLS599 (pBLS590 containing PerpAB from −34 to start) |
| KS21 | B31-e2 containing pBLS600 (pBLS590 containing Pp21erp22 from −442 to start) |
| KS23 | B31-e2 containing pBLS603 (pBLS590 containing erp ribosome-binding site only) |
| GPB | B31-e2 containing pCJB1 (pBLS590 containing PerpG from −215 to start) |
| GPL | B31-e2 containing pCJB2 (pBLS590 containing PerpG from −93 to start) |
Temperature shift studies were performed as previously described (48). Briefly, cultures of B. burgdorferi were grown at 23°C until they reached mid-exponential phase (approximately 107 bacteria per ml). Aliquots of each culture were then diluted 1:100 into fresh medium and grown to mid-exponential phase at 34°C.
Erp protein expression by strain B31-e2 was examined by immunoblotting using monospecific rabbit polyclonal antisera (19). Aliquots of bacterial lysates containing equal amounts of total protein were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were then electrotransferred to nylon membranes and incubated with antisera, and bound antibodies were detected by enhanced chemiluminescence (Amersham Pharmacia, Piscataway, N.J.) (48).
EMSAs.
Whole-cell lysates were prepared from B. burgdorferi B31-RML cultured at either 34 or 23°C as previously described (8). EMSAs were performed essentially as described previously (8), with the major change being the use of biotinylated DNA probes rather than 32P-labeled DNA. Probes were produced by PCR using a biotinylated 5′ primer and an unlabeled 3′ primer (Integrated DNA Technologies, Coralville, Iowa), with previously cloned erp promoter DNAs as templates (49). Two different probes were utilized in these studies: one probe, created by a PCR using oligonucleotides Bio-E43 and E-8, extended from the start codon of erpG upstream to bp −330, and a shorter probe, created with oligonucleotides Bio-104 and R-8, spanned nucleotides −31 through −89 of erpG (Table 2). PCR products were purified through Centricon-100 microconcentrators (Amicon, Beverly, Mass.) (18).
TABLE 2.
Oligonucleotides used for this work Sequences of specific restriction endonuclease recognition sites are underlined
| Name | Sequence (5′ to 3′)a |
|---|---|
| G9-A | TAAGAAAATGAAAAATTTAAT |
| G9-C | AATTCTTGAAATTTCTTTCTA |
| G10-A | TGGTAATATTAGTACTTTATG |
| G10-C | TTTTTTTCTTTTATCTATTTT |
| G11-A | TTAAATCTTTGAAATATTGCA |
| G11-C | GAATTTCTTTTTCTTTCTCTT |
| G12-A | AAAATGTAACAGCTGAATGTA |
| G12-C | TGACCACCATCTTTTGCTTGC |
| G13-A | TGGCGAGTTGGTTAAAATTAC |
| G13-C | GGGTGTGTTATCGTGGGAACT |
| G14-A | TTGTAATGAGTAGAGCATTTG |
| G14-C | GTGGTGGATTAAACAGACTGT |
| G15-A | CTTGAAAATTTAATTTGTATG |
| G15-C | CCTTGCATTAACTCTTTATCT |
| G16-A | AAAGATAGTATACTTTGTGTA |
| G16-C | TTTTTCTTTTACATTTTGTTT |
| G17-A | ACAGAAGTAGTGATTTTTGAG |
| G17-C | TCGCATCAATCTTGCAAGAAA |
| G18-A | CATGCAAAATCTGGGCTGTAG |
| G18-C | ACTGCACAAATAATTAAATTT |
| Bio-E43 | biotin-AAAATTTTAGTCAAATTTGGAGTG |
| Bio-104 | biotin-TGTTAAAATGTAACAGCTGAATGTAACAAA |
| R-8 | GCAATATTTCAAAGATTTAAA |
| E-8 | CATAAGTTACTCC |
| E-43 | AAAATTTTAGTCAAATTTGGAGTG |
| 104-F | TGTTAAAATGTAACAGCTGAATGTAACAAA |
| 104-R | TTTGTTACATTCAGCTGTTACATTTTAACA |
| 104N-F | ATGTAACAGCTGAATGTA |
| 104N-R | TACATTCAGCTGTTACAT |
| 164-F | TGAGTAGAGCATTTGCAATGGAGAGATTTT |
| 164-R | AAAATCTCTCCATTGCAAATGCTCTACTCA |
| 164N-F | GAGCATTTGCAATGGAGA |
| 164N-R | TCTCCATTGCAAATGCTC |
| BSV-G | AAAAAACTCGAGTGGCCGAGGAGCAGGACTGACACG |
| BSV-H | AAAAAACTCGAGTCTGATTAGAAAAACTCATCGAG |
| GFP-1 | ATAATAGGATCCATACATATGAGTAAAGGAGAAG |
| GFP-2 | ATAATAAAGCTTATTATTTGTATAGTTCATCCATGCCATG |
| G-1 | AAAAAAGGTACCATATATAGCAAAG |
| G-2 | TTTTTTGGATCCTCCATAAAGTACTAATATTAC |
| G-2A | TTTTTTGGATCCTCCATAAAGTACTAAT |
| G-3 | AAAAAAGGTACCGTTTTGTTAAAATGTAACAGCTG |
| A-1 | AAAAAAGGTACCCCTCTCCTTGAAGGTG |
| A-2 | TTTTTTGGATCCTCCATAAGTCCTAATCATAC |
| A-4 | ATAATGGATCCTCCATAAGTCCTAATATTACCAC |
| A-10 | ATAATAGGTACCTTGCATTTATTATGTATTGTGG |
| E-116 | ATATAATTTTGTTACATTCAG |
| BBK-2 | CATAATGGATCCTCCATAAGCCTTAAGCTTAC |
| 21-1 | ATAATAGGTACCACAAGCTCCTTATAAGTG |
| 21-2 | TTTTTTGGATCCTCCATAAATCTTAATCATAC |
| PRO-1 | CGCGCTTATGGAG |
| PRO-2 | GATCCTCCATAAGCGCGGTAC |
Sequences of specific restriction endonuclease recognition sites are underlined.
Competitor DNAs G-9 through G-18 were produced by PCRs using an erpG clone as a template, with oligonucleotide pairs G-9A plus G-9C, G-10A plus G-10C, etc. Three other competitor DNAs were also produced by PCRs using the same template, and they were named according to the distance of their 5′ and 3′ ends from the start of erp transcription. Competitor −149/−31 was produced with oligonucleotide primers 164-F and R-8, competitor −149/−79 was produced with oligonucleotides 164-F and R-350, and competitor −89/−31 was produced with oligonucleotides 104-F and R-8. An additional competitor, E-43/A-2, encompassed the entire bppC-erpAB intergenic region and was produced by a PCR using oligonucleotides E-43 and A-2. All PCR products were purified as described above. Other, shorter competitor DNAs were produced by annealing two complementary oligonucleotides to produce double-stranded DNAs. Competitor 104 was produced from oligonucleotides 104-F and 104-R, competitor 104N was produced from oligonucleotides 104N-F and 104N-R, competitor 164 was produced from oligonucleotides 164-F and 164-R, and competitor 164N was produced from oligonucleotides 164N-F and 164N-R.
A LightShift Chemiluminescent EMSA kit (Pierce Biotechnology) was utilized according to the manufacturer's recommendations. Briefly, 10-μg aliquots of bacterial extract were incubated either with or without 4 pmol of unlabeled specific competitor DNA in LightShift buffer containing 2 μg of poly(dI-dC) (nonspecific competitor). Samples were incubated for 5 min at room temperature to allow protein binding to poly(dI-dC) and/or the competitors. Next, 20-fmol aliquots of labeled DNA probe were added to each sample, such that each contained a 200-fold excess of competition. Samples were then subjected to electrophoresis through a 7% polyacrylamide gel (37.5:1 acrylamide-bisacrylamide). After electrophoresis, DNAs were transferred to a nylon membrane, cross-linked by UV irradiation, and then blocked for 15 min at room temperature with LightShift blocking buffer. The membrane was next incubated with streptavidin-horseradish peroxidase conjugate for 15 min, followed by a 5-min incubation in Luminol enhancer substrate solution. Labeled DNAs were visualized by exposure to X-ray film. The relative abundance of erp operator binding proteins was determined as described above, except the amounts of bacterial extract examined in each assay ranged between 0.7 and 5.1 μg.
erp promoter-gfp fusion plasmids.
With the recent development of independently replicating plasmid vectors suitable for transformation into B. burgdorferi, new avenues have been opened for dissecting the DNA elements involved in the regulation of erp gene expression. To that end, we constructed a series of plasmids that contain transcriptional fusions with gfp (Fig. 2). The inserts of all recombinant plasmids were sequenced on both strands to confirm that no mutations were introduced during the cloning process.
FIG. 2.

Construction of Perp::gfp fusion plasmids. A nonessential region of the B. burgdorferi-E. coli shuttle vector pBSV2 was removed to produce pJAH2. The mutant 3 allele of gfp was then cloned into pJAH2 to produce pBLS590. To the 5′ side of gfp are KpnI and BamHI sites that facilitate the easy cloning of any promoter element, with the GG bases of the BamHI site oriented to comprise the 3′ end of any inserted ribosome binding site (consensus sequence, 5′-GGAGG-3′).
In the initial step of construction, the B. burgdorferi-Escherichia coli shuttle vector pBSV2 (51) was made smaller by the removal of 600 bp of nonessential DNA. To do so, we removed the gene encoding zeocin resistance by PCR with oligonucleotides BSV-G and BSV-H (Table 2), digestion with XhoI, and ligation with T4 DNA ligase. The resulting plasmid was designated pJAH2. The mutant 3 allele of gfp (14) was then amplified by PCR using oligonucleotides GFP-1 and GFP-2. The resultant gfp amplicon and pJAH2 were each digested with BamHI and HindIII and then were ligated.
With the oligonucleotides listed in Table 2, fragments of DNA were then amplified by PCRs from recombinant plasmid clones of the erpAB and erpG operons (49) or from uncloned PCR amplicons of the bbk2.10 locus of strain 297 and the p21erp22 locus of strain N40 (47). Each oligonucleotide pair contained a KpnI and a BamHI site in the 5′ and 3′ oligonucleotide, respectively. In addition, all 3′ oligonucleotides contained an erp consensus ribosome binding site. Oligonucleotide pairs for the production of each promoter-reporter fusion plasmid were as follows: for pBLS591, A-1 plus A-2; for pBLS592, A-1 plus G-2; for pBLS593, A-1 plus G-2A; for pBLS594, A-1 plus A-2; for pBLS595, A-1 plus BBK-2; for pBLS598, A-1 plus A-4; for pBLS600, 21-1 plus 21-2; for pEAP1, G-1 plus A-2; for pEAP2, G-3 plus A-2; for pCJB1, G-1 plus G-2; and for pCJB2, G-3 plus G-2. Amplicons were digested with KpnI and BamHI and ligated between the KpnI and BamHI sites of pBLS590, such that the ribosome binding sites were located the same distance from the translational start codon as in most erp operons.
An additional plasmid was produced in which only the DNA 3′ of the putative −35 sequence of erpAB was included in the fusion. To do so, we first subjected pBLS591 to PCR with oligonucleotides A-10 and E-116, which amplified in opposite directions out from the erpAB promoter. This amplicon was then digested with KpnI and self-ligated.
Control plasmid pBLS603 contained only a ribosome binding site located 5′ of gfp. This plasmid was constructed by annealing oligonucleotides PRO-1 and PRO-2 and then ligating the resulting double-stranded DNA between the KpnI and BamHI sites of pBLS590.
Transformation of B. burgdorferi.
B. burgdorferi B31-e2 was transformed with recombinant plasmids under standard electroporation conditions (39). After an overnight incubation in BSK-II lacking antibiotics, bacterial cultures were plated in semisolid BSK-II containing 1.2% agarose (w/vol) and kanamycin at a concentration of 200 μg/ml (10, 30). Plates were incubated at 34°C in a 5% CO2 atmosphere. Antibiotic-resistant colonies were screened for the presence of recombinant plasmids by PCRs using oligonucleotides GFP-1 and GFP-2. B. burgdorferi strains carrying pBLS590 and derivative plasmids are listed in Table 1.
To ascertain whether sequences of heterologous plasmids remained stable after passaging through B. burgdorferi, plasmids of several transformed strains were reisolated and sequenced. B. burgdorferi cultures (100 ml) were grown to mid-exponential phase, and total plasmids were purified by use of Qiagen Midiprep kits (Qiagen, Chatsworth, Calif.). Aliquots (approximately 1 μg) of each purified plasmid preparation were used to transform E. coli, and bacteria were plated on Luria-Bertani agar containing kanamycin at a concentration of 50 μg/ml. Plasmids of transformed E. coli were then purified and sequenced.
Analysis of GFP expression.
The effects of the culture temperature on green fluorescent protein (GFP) expression from each promoter-reporter construct were assessed by both flow cytometry and immunoblot analysis. Cultures of each transformed B. burgdorferi strain were grown to mid-exponential phase in BSK-II broth either at a constant 23°C or with a shift from 23 to 34°C. Bacteria were collected by centrifugation, washed twice with phosphate-buffered saline (PBS), and then resuspended in PBS at concentrations of approximately 106 bacteria per ml. Aliquots of each bacterial suspension were analyzed in a FACSCalibur flow cytometer (Becton Dickinson, San Jose, Calif.), with excitation and detection wavelengths of 488 and 530 nm, respectively. At least 70,000 events were measured for each flow cytometry analysis. GFP expression levels of transformed bacteria were further analyzed by immunoblotting. Additional aliquots of bacteria were lysed in a boiling water bath, and 1 μg of total protein from each was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis. Proteins were transferred to nylon membranes and incubated with rabbit anti-GFP polyclonal antisera (Oncogene, San Diego, Calif.), and bound antibodies were detected by enhanced chemiluminescence (Amersham Pharmacia).
RESULTS
Identification of protein-binding sites.
Members of our laboratory previously demonstrated that cultured B. burgdorferi produces at least one protein that specifically binds to regions 5′ of all tested erp genes (8). In those studies, two DNA-protein complexes were observed when labeled DNA fragments were incubated with B. burgdorferi cell extracts, suggesting that proteins bound to two locations. To more precisely define the two protein-binding sites, we performed EMSAs with labeled erpG 5′ DNA, with a variety of unlabeled DNA fragments as competitors for protein binding.
Initial studies used overlapping fragments of the erpG gene and its 5′ noncoding DNA as competitors. Neither DNA fragment G9-AC, G10-AC, nor G11-AC competed for binding with protein-DNA complex 2, whereas fragments G12-AC through G18-AC all competed away protein binding to site 2 (Fig. 3A). Fragments G9-AC through G13-AC failed to compete for binding with site 1, while fragments G14-AC through G18-AC all competed for protein binding (Fig. 3B). These results indicate that the DNA component of complex 2 includes DNA located up through the 5′ end of fragment G12-AC and that of complex 1 includes DNA up through the 5′ end of fragment G14-AC (Fig. 1).
FIG. 3.

EMSAs identifying the two protein-binding sites of erp operator DNAs. All assays used protein extracts of B. burgdorferi cultured at 34°C. Experimental data shown in panels A, B, and C were obtained with a labeled probe extending from +18 to −330, while those for panel D were obtained with a probe spanning −31 to −89. For all panels, lanes contained either free DNA or DNA, poly(dI-dC), protein extract, and the DNA competitor noted above the lane. (A and B) Short and long exposures of the same EMSA indicating DNA competition of protein binding to site 2 (A) and site 1 (B). (C) Identification of maximal boundary of site 1. (D) Identification of maximal boundary of site 2. As previously observed (8), complex 1 was less abundant than complex 2, and longer exposure times were required to detect complex 1 on X-ray film. As a result, signals from complex 2 often saturated films that were used to detect complex 1.
DNA fragment G14-AC contained a short sequence of the erp 5′ noncoding region that was lacking from G13-AC (Fig. 1). EMSAs using a 30-bp DNA fragment with part of this sequence as a competitor, called competitor 164, indicated efficient competition for protein binding with DNA site 1 (Fig. 3C). It was noted that this DNA sequence included a short inverted repeat, TTGCAA (Fig. 1). Competitor 164N, an 18-bp DNA segment centered on this inverted repeat, also competed with DNA site 1 for protein binding (Fig. 3C). This DNA sequence is almost completely conserved in all erp operon 5′ regions (Fig. 1). Control studies showed that segments of DNA encompassing site 2 did not compete for protein binding to site 1, indicating that different proteins bind each site (Fig. 3C and data not shown).
The boundaries of binding site 2 proved to be much larger than those of site 1. Competition was observed only when the competitor DNA included the 59-bp segment found between nucleotides −31 and −89 (Fig. 3D). This region overlaps the putative −35 sequence of the promoter and is extremely well conserved in all erp operons (Fig. 1).
Relative abundance of erp operator-binding proteins.
We hypothesized that at least one of the proteins binding to the erp 5′ noncoding DNA influences erp transcriptional activity. If so, it was expected that the relative abundance of such a regulatory protein would be proportional to the abundance of Erp proteins. As noted above, Erp proteins are expressed at higher levels by bacteria cultured at 34°C than by those cultured at 23°C. EMSAs using various amounts of protein extract from B. burgdorferi indicated that bacteria grown at 23°C contained several times more of the protein that binds site 2 than did bacteria cultured at 34°C (Fig. 4). The inverse relationship between the levels of DNA-binding protein and of Erp proteins suggests that the protein that binds site 2 may function as a repressor. No detectable differences in the level of the site 1 binding protein were observed (data not shown).
FIG. 4.

Determination of relative abundance of the site 2 binding protein. Cell extracts from B. burgdorferi cultured at either 23 or 34°C were assayed by EMSA using labeled erpG promoter DNA. The total amounts of protein extract incubated with the DNA are indicated above each lane.
Promoter-reporter fusion analyses of B31 erp operons.
To characterize the functions of the protein-binding sites for erp expression, we created a series of transcriptional fusions between erp 5′ noncoding DNAs and gfp (Fig. 2; Table 2). Each plasmid was introduced into B. burgdorferi strain B31-e2, a subculture of type strain B31 that we were able to transform at a very high efficiency (data not shown). Importantly, B31-e2 bacteria demonstrate a temperature-dependent differential expression of Erp synthesis in patterns that are indistinguishable from those of low-passage-number, infectious bacteria (data not shown). Therefore, B31-e2 contains the mechanisms necessary to sense changes in environmental temperature and to control erp expression, and it is a suitable strain for the elucidation of those regulatory pathways.
Two concerns needed to be addressed prior to the use of pBVS2-derived plasmids for the analysis of B. burgdorferi promoter elements, namely the existence of other promoters in pBSV2 that might influence reporter gene expression and the genetic stability of heterologous DNA in B. burgdorferi. Due to the manner in which the parental pBSV2 plasmid was constructed, a putative B. burgdorferi promoter, that of bppA (paralog family 165), is located 5′ of gfp in pBLS590 (12, 19, 51). Derivatives of pBSV2 also contain an E. coli lac promoter that directs transcription towards the site in which the gfp gene of pBLS590 was inserted (51). However, B. burgdorferi strain KS10, which carries pBLS590 (Table 1), did not express GFP at levels that were detectable by either flow analysis or immunoblotting (Fig. 5 and data not shown). In addition, GFP expression was not detected for strain KS23, which carries control plasmid pBLS603, which has a consensus ribosome binding site located 5′ of gfp. We conclude that transcripts arising from the bppA promoter of pBLS590 and its derivatives terminate prior to reaching the gfp gene or that this promoter is inactive in these plasmids. These negative results also demonstrated that the E. coli lac promoter does not function in B. burgdorferi. During the course of this work, Carroll and coworkers reached the same conclusions from studies with similar pBSV2-derived promoter-gfp fusion plasmids (11). We addressed our second concern by reisolating and sequencing recombinant plasmids from several transformed B. burgdorferi strains. No differences were detected, indicating that heterologous DNA maintained by B. burgdorferi is genetically stable.
FIG. 5.

Representative GFP expression by untransformed B. burgdorferi B31-e2 or bacteria carrying a promoterless gfp (pBLS590), gfp with a 5′ ribosome-binding site only (pBLS603), PerpAB::gfp (pBLS591), or PerpG::gfp (pBLS592). Bacteria were either grown to mid-exponential phase at a constant 23°C (shaded) or were shifted from 23 to 34°C after reaching mid-exponential phase (unshaded).
All erp loci are flanked at the 5′ end by a divergently transcribed bppC gene, with the two genes being separated by approximately 430 bp of DNA (19, 49). Since the DNA sequences required for erp expression were unknown prior to this study, the entire sequences between the start codons of the B31 erpAB and erpG operons and their adjacent bppC genes were cloned to produce transcriptional fusions with gfp (Table 1; Fig. 1). The resultant plasmids, pBLS591 and pBLS592, were introduced into B31-e2 to produce strains KS11 and KS12, respectively (Table 1). Studies were then undertaken to determine whether the Perp::gfp fusion constructs exhibited temperature-dependent differential expression. KS11 and KS12 were cultured to mid-exponential phase at either 23 or 34°C, and GFP expression levels were examined by both flow cytometry and immunoblotting (Fig. 5 and data not shown). Expression of GFP was observed for both strains KS11 and KS12, with both strains producing higher levels of GFP when grown at 34°C than when cultivated at the lower temperature. These data indicate the regulation of Erp proteins at the level of transcriptional initiation and are consistent with earlier studies of erp genes and their encoded proteins (8, 24, 44, 48). Furthermore, these studies demonstrate that the temperature-dependent differential expression of erp genes is due to DNA elements located in the bppC-erp intergenic region.
To investigate the roles of 5′ noncoding DNA sequences, we next constructed promoter-gfp fusions with deletions distal to the start of transcription. Plasmids pEAP1 and pCJB1 contain DNAs from bp −215 through the start of transcription of PerpAB and PerpG, respectively, and include both protein-binding sites (Table 1; Fig. 1). Plasmids pEAP2 and pCJB2 contain DNAs from bp −93 to the start of transcription of PerpAB and PerpG, respectively, and contain only the second protein-binding site. pBLS599 contains only the RNA polymerase contact sites of PerpAB and thus lacks both protein-binding sites. The deletion of protein-binding site 1 from either 5′ DNA element had no detectable effect on GFP expression levels (Fig. 6), indicating that site 1 does not by itself greatly affect erp transcription in cultured bacteria. However, the deletion of both sites 1 and 2 resulted in elevated GFP expression, indicating that site 2 is involved in the repression of erp transcription.
FIG. 6.

Representative analyses of B. burgdorferi carrying deletions in the operators of PerpAB::gfp and PerpG::gfp. Bacteria were either grown to mid-exponential phase at a constant 23°C (shaded) or were shifted from 23 to 34°C after reaching mid-exponential phase (unshaded).
Intriguingly, strain KS11 produced significantly higher levels of GFP than did KS12, indicating that the erpAB promoter is stronger than that of erpG. Noting that these two promoters contain different −10 sequences, we produced three chimeric promoter-gfp fusions (Fig. 7). The promoter element of pBLS593 is identical to that of erpAB but contains the sequence of erpG 3′ of the −10 site. pBLS594 contains the 5′ element of erpG but has the −10 site and 3′ DNA of erpAB. pBLS598 contains the 5′ DNA of erpAB and the −10 site and 3′ DNA of erpG. Strain B31-e2 was transformed with pBLS593, pBLS594, or pBLS598 to produce strain KS13, KS15, or KS19, respectively. The three chimeras exhibited GFP expression levels intermediate to those of the wild-type promoters (Fig. 7).
FIG. 7.

Representative analyses of GFP expression by B. burgdorferi carrying chimeric PerpAB/PerpG::gfp transcriptional fusions. Bacteria were either grown to mid-exponential phase at a constant 23°C (shaded) or were shifted from 23 to 34°C after reaching mid-exponential phase (unshaded). Diagrams below each panel illustrate regions of the two erp promoter/operator regions contained in each construct.
Analyses of erp operons from other strains.
Characterizations of B. burgdorferi strains other than B31 have largely found identical temperature-dependent expression patterns of erp genes and proteins (1, 4, 23, 24, 28). However, some studies failed to detect expression of other strains' Erp proteins, leading to suggestions that a few erp operons are regulated through mechanisms that are different from that of most erp genes (1, 3, 15, 24, 52, 53). We therefore examined the promoter elements of those erp genes, using promoter-gfp fusion constructs.
Strain 297 encodes an Erp protein named Bbk2.10 (3). An initial characterization of cultured 297 bacteria did not detect Bbk2.10 by immunoblotting, and its mRNA was not detected by Northern blotting (1, 3). Subsequent reverse transcription-PCR (RT-PCR) analysis identified the bbk2.10 transcript in cultured bacteria (24). We constructed a Pbbk2.10::gfp transcriptional fusion plasmid, pBLS595, and introduced it into B31-e2 to create strain KS18. GFP expression analysis of KS18 confirmed that Pbbk2.10 is indeed functional in cultured bacteria, although it is a weak transcriptional promoter that yields very little protein product (Fig. 8).
FIG. 8.

Representative GFP analyses of B. burgdorferi carrying Pbbk2.10::gfp (pBLS595) or Pp21erp22::gfp (pBLS600). Bacteria were either grown to mid-exponential phase at a constant 23°C (shaded) or were shifted from 23 to 34°C after reaching mid-exponential phase (unshaded).
Two erp operons, ospEF and p21erp22, of strain N40 have been well characterized (7, 15, 31, 37, 45, 47, 48, 52). ospEF mRNA and both OspE and OspF proteins have been detected in cultured organisms, and they exhibit a temperature-dependent expression pattern identical to that of other erp operons (15, 31, 37, 48). However, two studies failed to detect the p21 transcript in cultured bacteria (15, 52), although erp22 mRNA has been detected by RT-PCR (7) (referred to as “erpD” in that report). Plasmid pBLS600 was constructed to contain Pp21erp22 fused with gfp and was introduced into B31-e2 to produce strain KS21. Flow cytometry indicated that Pp21erp22 is a very active promoter in cultured bacteria and that it exhibits temperature-dependent differential expression similar to that from other erp promoters (Fig. 8).
DISCUSSION
B. burgdorferi regulates Erp protein synthesis during the natural infectious cycle, producing these proteins during transmission between ticks and warm-blooded hosts but repressing their synthesis during persistent infection of the tick midgut (22, 23, 36, 37). Previous studies found that this regulation occurs at the mRNA level (24, 44). We have now demonstrated that B. burgdorferi proteins specifically bind to two DNA regions 5′ of the erp transcription start site. Since there are no obvious similarities between the two protein-binding sites and since neither DNA competed with the other for protein binding, we concluded that different proteins bind to each. The nature of the site 1 binding protein cannot yet be postulated, since the deletion of site 1 alone did not have appreciable effects on transcription in cultured bacteria. The deletion of site 2 resulted in elevated levels of transcription, indicating that the protein which binds that site is a transcriptional repressor. Also supporting that conclusion is the observation that bacteria cultured under conditions that repress Erp expression (i.e., 23°C) contained elevated levels of the site 2 binding protein. Since site 2 is immediately adjacent to and possibly overlaps the −35 site of the erp promoter, it is highly probable that a repressor binding to site 2 physically inhibits interactions between RNA polymerase and the promoter DNA. Studies are currently under way in our laboratory to identify the proteins that bind erp operator DNA, with the intention of elucidating their roles in the regulation of erp expression, and ultimately, how B. burgdorferi controls their functions.
The present study indicated that different erp promoter elements differ in transcriptional activity. Both the B31 erpAB and the N40 p21erp22 promoter/operator regions directed relatively high levels of transcription, while PerpG was a less active promoter and Pbbk2.10 was weaker still. The DNA sequences of both sites 1 and 2 are nearly identical for all four of these operons, and both PerpAB and PerpG compete with each other for protein binding (8). However, it is not impossible that the few nucleotide differences between them change their affinities for the DNA-binding proteins and thereby alter promoter strengths. The four examined operons also differ in the sequences of their −10 and −35 sites, which may affect the binding of RNA polymerase and the formation of open complexes. B. burgdorferi encodes only three sigma factors, but it is currently unknown which one is used for erp transcription. While the consensus promoter sequence has not yet been determined for any B. burgdorferi RNA polymerase holoenzyme, the differences in promoter strength observed in our studies suggest that PerpAB and Pp21erp22 are closer to the consensus than are PerpG or Pbbk2.10. An exchange of the −10 sites of PerpAB and PerpG did not result in the reversal of their relative strengths, so other DNA sequences must also be responsible for the differences in their activities. Clearly, additional studies will be required to conclusively determine the roles of changes in the promoter and operator sequences in the different transcriptional activities of various erp operons.
It was previously noted that B. burgdorferi cultured at either 23 or 34°C synthesizes proteins that bind to erp promoter DNAs (8). Because at least one of the DNA-binding proteins probably functions as a transcriptional repressor, it appears that bacteria cultured at 34°C do not fully derepress erp expression. Erp proteins are expressed at significantly higher levels during mammalian infections than during cultivation at any temperature (1, 15, 24), suggesting that B. burgdorferi produces far fewer of the repressor proteins during mammalian infections than in culture. B. burgdorferi cells grown in artificial culture medium are apparently exposed to a confusing variety of external signals, since they produce both proteins that are generally synthesized during mammalian infections and others that are specific for tick infections. We propose that the culture medium provides a cue(s) to B. burgdorferi that causes it to produce the repressor protein that binds site 2, resulting in the suboptimal expression of erp operons. In addition to being affected by temperature, the expression of Erp proteins is affected by unknown chemicals and by autoinducer 2 quorum sensing (1, 8, 24, 41), any of which may be involved in the inhibition of Erp synthesis during cultivation.
The observations that a repressor of erp transcription is produced during cultivation and that erp promoters vary significantly in strength may explain why certain Erp proteins have not been detected in cultured B. burgdorferi cells. The strain 297 Pbbk2.10 is relatively very weak, being such a poor promoter that mRNAs from cultured bacteria can only be detected by the sensitive technique of RT-PCR (3, 24). It is quite possible that this low level of transcription results in the production of very little Bbk2.10 protein, so little that the protein could not be detected by immunoblotting or an immunofluorescence assay with the previously used antibody (1, 3, 23, 24). An initial characterization of the pG Erp protein of strain ZS7 also failed to detect that protein by immunoblotting of cell lysates, and pG mRNA could not be detected by Northern blotting either (53). However, pG is absolutely identical to erpG of strain B31, with both operons containing identical 5′ and 3′ flanking sequences (49, 53). Furthermore, it has been demonstrated in this work and elsewhere that PerpG is active during cultivation (17, 19, 44) and that ZS7 exhibits temperature-dependent expression of other erp operons (27, 28). Since the use of low-titer antisera can cause one to miss the detection of proteins (19), it is very likely that previous inability to detect the 297 Bbk2.10 and ZS7 pG proteins was due to the use of antibody preparations that were not of sufficiently high titers to detect the products of these poorly expressed genes.
It is not clear why previous studies failed to detect the production of strain N40 p21 mRNA in cultured bacteria (7, 15, 52). The present study demonstrated that Pp21erp22 is comparatively strong, being similar to B31 PerpAB. Consistent with this observation, mRNA for the second gene in the operon, erp22 (previously referred to as N40 erpD), has been detected by RT-PCR of cultured organisms (7). Either bacteria must process p21erp22 mRNA to rapidly degrade the first gene but retain the second or the RT-PCR oligonucleotides and/or reaction conditions used in earlier studies were suboptimal for detection of the p21 message. Studies are currently on-going in our laboratory to identify which of these two possibilities is correct.
All examined Lyme disease spirochete isolates carry multiple, different erp loci (2, 13, 47, 50). Comparisons of erp promoter/operator regions have indicated extensive conservation, including protein-binding sites 1 and 2 (Fig. 1) (2, 3, 13, 32, 44, 47, 49). Furthermore, protein binding to one erp 5′ noncoding region can be effectively competed away with the homologous region of another erp operon (this work; also see reference 8). These observations strongly suggest that when a bacterium produces the proteins that bind erp operator sites 1 and 2, those proteins bind to each of the bacterium's many erp loci. Thus, a bacterium will simultaneously regulate the expression of its entire repertoire of erp genes. Studies using double-labeling immunofluorescence analysis indicated that cells of strain B31 behaved exactly as predicted: all Erp proteins were coexpressed (19). While similar studies of strain 297 failed to detect the simultaneous coexpression of every Erp protein (23, 24), those results were quite likely due to an inability to detect weakly expressed proteins, as discussed above. However, it has been proposed that B. burgdorferi sequentially expresses different erp operons during an infection, requiring that the cells repress the transcription of some erp loci while activating the expression of others (33). That hypothesis was based on a study that observed the production of antibodies directed against some Erp proteins early in infection, while antibodies against other proteins were not detected until a few weeks later (33). Other studies have demonstrated that all examined Erp proteins are produced by B. burgdorferi at the time of transmission from a tick to a mammal but that the immune system responses to those proteins sometimes differ in their intensities (23, 24, 36). Those data, together with the results of the present study, make it highly probable that antibodies directed against Erp proteins are sometimes detected at different times of infection because promoter variations cause differences in Erp expression levels, rather than because some erp operons are expressed while other loci are completely silenced.
Both this and another study (11) observed that the E. coli lac promoter carried by pBSV2 and its derivatives is not functional in B. burgdorferi. Several other promoters that function in E. coli are also nonfunctional in the Lyme disease spirochete, to the end that most antibiotic resistance genes used as selectable markers of transformation in B. burgdorferi had to be constructed by the placement of a strong spirochetal promoter 5′ of a resistance gene (10, 20, 21, 43). As noted above, none of the promoter consensus sequences for any B. burgdorferi RNA polymerase-sigma holoenzyme have been determined, but it certainly appears that the preferred sequences differ dramatically from those of E. coli. This highlights the importance of examining the expression of B. burgdorferi promoter elements in that bacterium only, rather than using unrelated organisms, such as E. coli, as proxies.
In conclusion, we have characterized the promoter/operator sequences of erp gene family members. At least two different proteins bind to DNA sites immediately 5′ of erp promoters, one of which functions as a transcriptional repressor. Various erp promoters exhibit different relative promoter activities, which may account for the reported inability to detect certain Erp proteins in cultured bacteria as well as for differences in Erp-directed antibody titers during mammalian infections. The variations in promoter strengths appeared to be due to differences in the sequences of erp promoters and operators. These data strengthen our hypotheses that all erp operons are regulated through a common mechanism and that individual bacteria express their entire repertoire of Erp proteins simultaneously. Many Erp proteins have been demonstrated to bind host factor H, which probably helps protect bacteria from killing by the host innate immune system. Therefore, elucidating the regulatory mechanisms controlling Erp synthesis will be important for understanding the pathogenic properties of B. burgdorferi as well as for directing the development of novel antibacterial agents.
Acknowledgments
This work was supported by grants AI-44254 and AI-49795 from the National Institutes of Health.
We thank Jay Carroll for sharing unpublished results; Melissa Hines for the construction of pJAH2; Philip Stewart for providing pBSV2; Greg Bowman and Jennifer Strange for flow cytometry; Natalie Mickelsen for technical assistance; and Jay Carroll, Robert Geraghty, Sean Riley, Tony Sinai, Philip Stewart, Kate von Lackum, Rachel Wattier, and Wolf Zückert for helpful comments on this work and on the manuscript.
REFERENCES
- 1.Akins, D. R., K. W. Bourell, M. J. Caimano, M. V. Norgard, and J. D. Radolf. 1998. A new animal model for studying Lyme disease spirochetes in a mammalian host-adapted state. J. Clin. Investig. 101:2240-2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Akins, D. R., M. J. Caimano, X. Yang, F. Cerna, M. V. Norgard, and J. D. Radolf. 1999. Molecular and evolutionary analysis of Borrelia burgdorferi 297 circular plasmid-encoded lipoproteins with OspE- and OspF-like leader peptides. Infect. Immun. 67:1526-1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Akins, D. R., S. F. Porcella, T. G. Popova, D. Shevchenko, S. I. Baker, M. Li, M. V. Norgard, and J. D. Radolf. 1995. Evidence for in vivo but not in vitro expression of a Borrelia burgdorferi outer surface protein F (OspF) homologue. Mol. Microbiol. 18:507-520. [DOI] [PubMed] [Google Scholar]
- 4.Alitalo, A., T. Meri, H. Lankinen, I. Seppälä, P. Lahdenne, P. S. Hefty, D. Akins, and S. Meri. 2002. Complement inhibitor factor H binding to Lyme disease spirochetes is mediated by inducible expression of multiple plasmid-encoded outer surface protein E paralogs. J. Immunol. 169:3847-3853. [DOI] [PubMed] [Google Scholar]
- 5.Alitalo, A., T. Meri, L. Rämö, T. S. Jokiranta, T. Heikkilä, I. J. T. Seppälä, J. Oksi, M. Viljanen, and S. Meri. 2001. Complement evasion by Borrelia burgdorferi: serum-resistant strains promote C3b inactivation. Infect. Immun. 69:3685-3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alverson, J., S. F. Bundle, C. D. Sohasky, M. C. Lybecker, and D. S. Samuels. 2003. Transcriptional regulation of the ospAB and ospC promoters of Borrelia burgdorferi. Mol. Microbiol. 48:1665-1677. [DOI] [PubMed] [Google Scholar]
- 7.Anguita, J., S. Samanta, B. Revilla, K. Suk, S. Das, S. W. Barthold, and E. Fikrig. 2000. Borrelia burgdorferi gene expression in vivo and spirochete pathogenicity. Infect. Immun. 68:1222-1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Babb, K., N. El-Hage, J. C. Miller, J. A. Carroll, and B. Stevenson. 2001. Distinct regulatory pathways control the synthesis of Borrelia burgdorferi infection-associated OspC and Erp surface proteins. Infect. Immun. 69:4146-4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbour, A. G. 1984. Isolation and cultivation of Lyme disease spirochetes. Yale J. Biol. Med. 57:521-525. [PMC free article] [PubMed] [Google Scholar]
- 10.Bono, J. L., A. F. Elias, J. J. Kupko, B. Stevenson, K. Tilly, and P. Rosa. 2000. Efficient targeted mutagenesis in Borrelia burgdorferi. J. Bacteriol. 182:2445-2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carroll, J. A., P. E. Stewart, P. Rosa, A. F. Elias, and C. F. Garon. 2003. An enhanced GFP reporter system to monitor gene expression in Borrelia burgdorferi. Microbiology 149:1819-1828. [DOI] [PubMed] [Google Scholar]
- 12.Casjens, S., N. Palmer, R. van Vugt, W. M. Huang, B. Stevenson, P. Rosa, R. Lathigra, G. Sutton, J. Peterson, R. J. Dodson, D. Haft, E. Hickey, M. Gwinn, O. White, and C. Fraser. 2000. A bacterial genome in flux: the twelve linear and nine circular extrachromosomal DNAs of an infectious isolate of the Lyme disease spirochete Borrelia burgdorferi. Mol. Microbiol. 35:490-516. [DOI] [PubMed] [Google Scholar]
- 13.Casjens, S., R. van Vugt, K. Tilly, P. A. Rosa, and B. Stevenson. 1997. Homology throughout the multiple 32-kilobase circular plasmids present in Lyme disease spirochetes. J. Bacteriol. 179:217-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cormack, B. P., R. H. Valdvia, and S. Falkow. 1996. FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173:33-38. [DOI] [PubMed] [Google Scholar]
- 15.Das, S., S. W. Barthold, S. Stocker Giles, R. R. Montgomery, S. R. Telford, and E. Fikrig. 1997. Temporal pattern of Borrelia burgdorferi p21 expression in ticks and the mammalian host. J. Clin. Investig. 99:987-995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eggers, C. H., M. J. Caimano, M. L. Clawson, W. G. Miller, D. S. Samuels, and J. D. Radolf. 2002. Identification of loci critical for replication and compatibility of a Borrelia burgdorferi cp32 plasmid and use of a cp32-based shuttle vector for the expression of fluorescent reporters in the Lyme disease spirochaete. Mol. Microbiol. 43:281-295. [DOI] [PubMed] [Google Scholar]
- 17.El-Hage, N., K. Babb, J. A. Carroll, N. Lindstrom, E. R. Fischer, J. C. Miller, R. D. Gilmore, Jr., M. L. Mbow, and B. Stevenson. 2001. Surface exposure and protease insensitivity of Borrelia burgdorferi Erp (OspEF-related) lipoproteins. Microbiology 147:821-830. [DOI] [PubMed] [Google Scholar]
- 18.El-Hage, N., L. D. Lieto, and B. Stevenson. 1999. Stability of erp loci during Borrelia burgdorferi infection: recombination is not required for chronic infection of immunocompetent mice. Infect. Immun. 67:3146-3150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.El-Hage, N., and B. Stevenson. 2002. Simultaneous coexpression of Borrelia burgdorferi Erp proteins occurs through a specific, erp locus-directed regulatory mechanism. J. Bacteriol. 184:4536-4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elias, A. F., P. E. Stewart, D. Grimm, M. J. Caimano, C. H. Eggers, K. Tilly, J. L. Bono, D. R. Akins, J. D. Radolf, T. G. Schwan, and P. Rosa. 2002. Clonal polymorphism of Borrelia burgdorferi strain B31 MI: implications for mutagenesis in an infectious strain background. Infect. Immun. 70:2139-2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frank, K. L., S. F. Bundle, M. E. Kresge, C. E. Eggers, and D. S. Samuels. 2003. aadA confers streptomycin resistance in Borrelia burgdorferi. J. Bacteriol. 185:6723-6727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gilmore, R. D., Jr., M. L. Mbow, and B. Stevenson. 2001. Analysis of Borrelia burgdorferi gene expression during life cycle phases of the tick vector Ixodes scapularis. Microbes Infect. 3:799-808. [DOI] [PubMed] [Google Scholar]
- 23.Hefty, P. S., S. E. Jolliff, M. J. Caimano, S. K. Wikel, and D. R. Akins. 2002. Changes in the temporal and spatial patterns of outer surface lipoprotein expression generate population heterogeneity and antigenic diversity in the Lyme disease spirochete, Borrelia burgdorferi. Infect. Immun. 70:3468-3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hefty, P. S., S. E. Jolliff, M. J. Caimano, S. K. Wikel, J. D. Radolf, and D. R. Akins. 2001. Regulation of OspE-related, OspF-related, and Elp lipoproteins of Borrelia burgdorferi strain 297 by mammalian host-specific signals. Infect. Immun. 69:3618-3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hellwage, J., T. Meri, T. Heikkilä, A. Alitalo, J. Panelius, P. Lahdenne, I. J. T. Seppälä, and S. Meri. 2001. The complement regulatory factor H binds to the surface protein OspE of Borrelia burgdorferi. J. Biol. Chem. 276:8427-8435. [DOI] [PubMed] [Google Scholar]
- 26.Kraiczy, P., K. Hartmann, J. Hellwage, C. Skerka, V. Brade, P. F. Zipfel, R. Wallich, and B. Stevenson. 2003. Immunological characterization of the complement regulator factor H-binding CRASP and Erp proteins of Borrelia burgdorferi. Int. J. Med. Microbiol. 293(Suppl. 37):152-157. [DOI] [PubMed] [Google Scholar]
- 27.Kraiczy, P., J. Hellwage, C. Skerka, M. Kirschfink, V. Brade, P. F. Zipfel, and R. Wallich. 2003. Immune evasion of Borrelia burgdorferi: mapping of a complement inhibitor factor H-binding site of BbCRASP-3, a novel member of the Erp protein family. Eur. J. Immunol. 33:697-707. [DOI] [PubMed] [Google Scholar]
- 28.Kraiczy, P., C. Skerka, V. Brade, and P. F. Zipfel. 2001. Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infect. Immun. 69:7800-7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kraiczy, P., C. Skerka, M. Kirschfink, V. Brade, and P. F. Zipfel. 2001. Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconectin and factor H. Eur. J. Immunol. 31:1674-1684. [DOI] [PubMed] [Google Scholar]
- 30.Kurtti, T. J., U. G. Munderloh, R. C. Johnson, and G. G. Ahlstrand. 1987. Colony formation and morphology in Borrelia burgdorferi. J. Clin. Microbiol. 25:2054-2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lam, T. T., T.-P. K. Nguyen, R. R. Montgomery, F. S. Kantor, E. Fikrig, and R. A. Flavell. 1994. Outer surface proteins E and F of Borrelia burgdorferi, the agent of Lyme disease. Infect. Immun. 62:290-298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marconi, R. T., S. Y. Sung, C. A. N. Hughes, and J. A. Carlyon. 1996. Molecular and evolutionary analyses of a variable series of genes in Borrelia burgdorferi that are related to ospE and ospF, constitute a gene family, and share a common upstream homology box. J. Bacteriol. 178:5615-5626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDowell, J. V., S. Y. Sung, G. Price, and R. T. Marconi. 2001. Demonstration of the genetic stability and temporal expression of select members of the Lyme disease spirochete OspF protein family during infection in mice. Infect. Immun. 69:4831-4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metts, M. S., J. V. McDowell, M. Theisen, P. R. Hansen, and R. T. Marconi. 2003. Analysis of the OspE determinants involved in binding of factor H and OspE-targeting antibodies elicited during Borrelia burgdorferi infection. Infect. Immun. 71:3587-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miller, J. C., J. L. Bono, K. Babb, N. El-Hage, S. Casjens, and B. Stevenson. 2000. A second allele of eppA in Borrelia burgdorferi strain B31 is located on the previously undetected circular plasmid cp9-2. J. Bacteriol. 182:6254-6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller, J. C., K. von Lackum, K. Babb, J. D. McAlister, and B. Stevenson. 2003. Temporal analysis of Borrelia burgdorferi Erp protein expression throughout the mammal-tick infectious cycle. Infect. Immun. 71:6943-6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nguyen, T.-P. K., T. T. Lam, S. W. Barthold, S. R. Telford, R. A. Flavell, and E. Fikrig. 1994. Partial destruction of Borrelia burgdorferi within ticks that engorged on OspE- or OspF-immunized mice. Infect. Immun. 62:2079-2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purser, J. E., and S. J. Norris. 2000. Correlation between plasmid content and infectivity in Borrelia burgdorferi. Proc. Natl. Acad. Sci. USA 97:13865-13870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Samuels, D. S., K. E. Mach, and C. F. Garon. 1994. Genetic transformation of the Lyme disease agent Borrelia burgdorferi with coumarin-resistant gyrB. J. Bacteriol. 176:6045-6049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwan, T. G., J. Piesman, W. T. Golde, M. C. Dolan, and P. A. Rosa. 1995. Induction of an outer surface protein on Borrelia burgdorferi during tick feeding. Proc. Natl. Acad. Sci. USA 92:2909-2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stevenson, B., and K. Babb. 2002. LuxS-mediated quorum sensing in Borrelia burgdorferi, the Lyme disease spirochete. Infect. Immun. 70:4099-4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stevenson, B., and S. W. Barthold. 1994. Expression and sequence of outer surface protein C among North American isolates of Borrelia burgdorferi. FEMS Microbiol. Lett. 124:367-372. [DOI] [PubMed] [Google Scholar]
- 43.Stevenson, B., J. L. Bono, A. Elias, K. Tilly, and P. Rosa. 1998. Transformation of the Lyme disease spirochete Borrelia burgdorferi with heterologous DNA. J. Bacteriol. 180:4850-4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stevenson, B., J. L. Bono, T. G. Schwan, and P. Rosa. 1998. Borrelia burgdorferi Erp proteins are immunogenic in mammals infected by tick bite, and their synthesis is inducible in cultured bacteria. Infect. Immun. 66:2648-2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stevenson, B., S. Casjens, R. van Vugt, S. F. Porcella, K. Tilly, J. L. Bono, and P. Rosa. 1997. Characterization of cp18, a naturally truncated member of the cp32 family of Borrelia burgdorferi plasmids. J. Bacteriol. 179:4285-4291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stevenson, B., N. El-Hage, M. A. Hines, J. C. Miller, and K. Babb. 2002. Differential binding of host complement inhibitor factor H by Borrelia burgdorferi Erp surface proteins: a possible mechanism underlying the expansive host range of Lyme disease spirochetes. Infect. Immun. 70:491-497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stevenson, B., and J. C. Miller. 2003. Intra- and interbacterial genetic exchange of Lyme disease spirochete erp genes generates sequence identity amidst diversity. J. Mol. Evol. 57:309-324. [DOI] [PubMed] [Google Scholar]
- 48.Stevenson, B., T. G. Schwan, and P. A. Rosa. 1995. Temperature-related differential expression of antigens in the Lyme disease spirochete, Borrelia burgdorferi. Infect. Immun. 63:4535-4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stevenson, B., K. Tilly, and P. A. Rosa. 1996. A family of genes located on four separate 32-kilobase circular plasmids in Borrelia burgdorferi B31. J. Bacteriol. 178:3508-3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stevenson, B., W. R. Zückert, and D. R. Akins. 2001. Repetition, conservation, and variation: the multiple cp32 plasmids of Borrelia species, p. 87-100. In M. H. Saier and J. García-Lara (ed.), The spirochetes: molecular and cellular biology. Horizon Press, Oxford, United Kingdom. [PubMed]
- 51.Stewart, P. E., R. Thalken, J. L. Bono, and P. Rosa. 2001. Isolation of a circular plasmid region sufficient for autonomous replication and transformation of infectious Borrelia burgdorferi. Mol. Microbiol. 39:714-721. [DOI] [PubMed] [Google Scholar]
- 52.Suk, K., S. Das, W. Sun, B. Jwang, S. W. Barthold, R. A. Flavell, and E. Fikrig. 1995. Borrelia burgdorferi genes selectively expressed in the infected host. Proc. Natl. Acad. Sci. USA 92:4269-4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wallich, R., C. Brenner, M. D. Kramer, and M. M. Simon. 1995. Molecular cloning and immunological characterization of a novel linear-plasmid-encoded gene, pG, of Borrelia burgdorferi expressed only in vivo. Infect. Immun. 63:3327-3335. [DOI] [PMC free article] [PubMed] [Google Scholar]