

Graphical abstract
Keywords: 3CLpro, Severe acute respiratory syndrome, SARS, MERS, Coronavirus
Abstract
Herein we report the discovery and SAR of a novel series of SARS-CoV 3CLpro inhibitors identified through the NIH Molecular Libraries Probe Production Centers Network (MLPCN). In addition to ML188, ML300 represents the second probe declared for 3CLpro from this collaborative effort. The X-ray structure of SARS-CoV 3CLpro bound with a ML300 analog highlights a unique induced-fit reorganization of the S2–S4 binding pockets leading to the first sub-micromolar noncovalent 3CLpro inhibitors retaining a single amide bond.
Coronaviruses (CoV) are enveloped, large plus-strand RNA viruses associated with mild to severe respiratory symptoms, including the common cold and the Severe Acquired Respiratory Syndrome (SARS)-CoV.1, 2, 3 Identified as the etiological agent responsible for the global pandemic in 2003, SARS presents an atypical pneumonia that during the first major outbreak led to progressive respiratory failure in over 8000 individuals and about 800 deaths by July of that year.4 With the cooperation of leading nations, a rigorous public healthcare campaign was fortunately successful in controlling this outbreak. However, a reemergence of the SARS-CoV is considered a potential pandemic risk and new strains of human coronavirus continue to be identified. Since 2003, two additional human coronaviruses, NL63 and HKU1, have been identified in patients and the viruses have been characterized and found to be significantly less lethal than SARS-CoV.5, 6, 7 Most recently in 2012, a new SARS-like virus, designated the Middle East respiratory syndrome coronavirus (MERS-CoV), has been identified in 144 patients so far, 54 of whom died.8 There is now evidence for person-to-person transmission of MERS-CoV.9 Now, nearly a decade later, the possibility of another SARS-like pandemic appears even more palpable based upon the lethality and properties of the newly identified MEV-HCoV strain. Effective vaccines and small molecule antiviral agents to prevent or treat SARS-like infections still do not exist, thus tailored antiviral therapies are urgently needed in order to treat potential future outbreaks of SARs and related human coronaviruses.
The SARS and MERS coronaviruses encode two proteases, a papain-like protease (PLpro) and a 3-chymotrypsin-like protease (3CLpro), in their genome that are essential for viral replication. The viral polyprotein is cleaved at three unique sites by PLpro and 11 unique sites by 3CLpro. Initial reports of 3CLpro inhibitors in the literature focused on peptidomimetics, often four to five residues in length, bearing a reactive ‘warhead’ group, such as an aldehyde, halo-methyl ketone, or Michael acceptor at the terminus with several demonstrating a covalent interaction with the active site Cys-145 residue.10, 11, 12, 13, 14, 15, 16 Until recently, the majority of efforts to develop nonpeptidic 3CLpro inhibitors also relied on ‘warhead’ based design strategies (Fig. 1 , 1–5)17, 18, 19, 20, 21 and a number of these nonpeptidic inhibitors achieved sub-micromolar activity. In the case of pyridyl ester 4,20 this potent nanomolar mechanism-based enzyme inactivator led to cell based inhibition below 10 μM in SARS-CoV infected Vero E6 cells. Recently, we reported N-(tert-butyl)-2-(N-arylamido)-2-(pyridin-3-yl) acetamide 6 (Fig. 1, ML188) and its X-ray complex with 3CLpro (PDB: 3V3M) as a rare example of a noncovalent SARS-CoV 3CLpro inhibitor of moderate molecular weight with good enzyme and antiviral inhibitory activity.22 Herein, we describe the continuation of efforts to develop potent, noncovalent SARS-3CLpro inhibitors based upon a second chemical class of triazoles from our MLPCN screening campaign (7, Fig. 2 ) and progression of this lead series to a second generation probe ML300 (16e, Fig. 1) and beyond to arrive at sub-100 nM inhibitors. We propose from crystallography data that ML300 and related triazoles in this series inhibit 3CLpro via a novel mechanism of action and provide a new direction for additional noncovalent inhibitor design and refinement.
Figure 1.
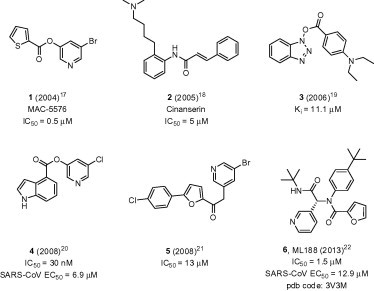
Representative nonpeptidic 3CLpro inhibitors utilizing a warhead and noncovalent mechanism of inhibition (1–6).
Figure 2.
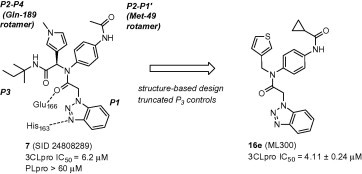
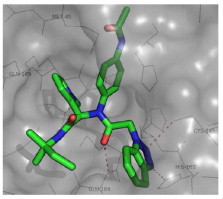
Binding orientation and properties of MLPCN 3CLpro HTS hit 7 and evolved probe molecule ML300(16e).
Using a designed expression construct which produces the post-proteolytic and authentic 3CLpro dimer, a screen against the NIH molecular libraries sample collection (∼293 K compounds) at the Scripps Research Institute Molecular Screening Center (SRIMSC) was undertaken. In addition to the diamide acetamide series represented by ML188 (6, Fig. 1),22 a related diamide series, represented by SID 24808289 (7, Fig. 2), was identified demonstrating a 3CLpro IC50 of 6.2 μM and good selectivity versus PLpro (IC50 > 60 μM) which is used as a control for cysteine-protease activity. Fortunately, quite early in the chemistry campaign an X-ray crystal structure of diamide 7 bound to 3CLpro was determined to 1.85 Å resolution. A solvent accessible surface depiction of 7 in the 3CLpro active site along with a wall-eye stereo view with key contact resides and hydrogen bonding contacts in depicted in Figure 3 . Interestingly, in contrast to the ML188-3CLpro crystal structure in which ML188 accommodates substrate sub-pockets in the enzyme active-site traditionally occupied by peptidomimetics, diamide 7 engenders an induced-fit complex resulting in a new surface dictated largely by a rearrangement of the Gln-189 and Met-49 residue side-chains.23 This induced fit accommodates the syn N-methyl pyrrole and anilido acetamide moieties of the inhibitor within subpockets that can be characterized as S2–S4 and S2–S1′ subpockets, respectively.
Figure 3.
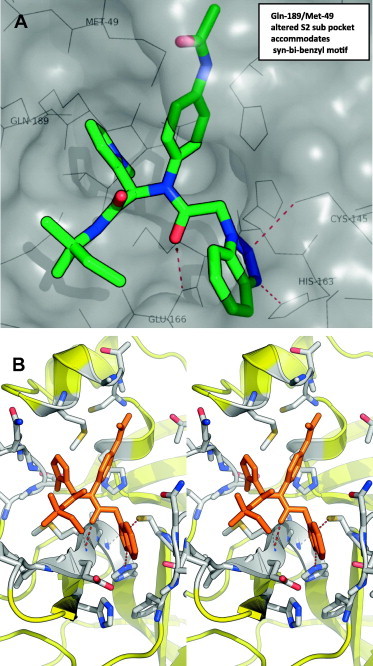
(A) Solvent accessible surface view of 7-3CLpro complex (PDB code: 4MDS, PubChem SID 24808289); (B) X-ray crystal structure of 7 (capped sticks in orange carbon) with SARS 3CLpro in wall-eye stereo view with key residues and hydrogen bonds.
Figure 2 schematically illustrates the inhibitor-active site interactions oriented in a manner similar as depicted in Figure 3. In addition to the P2–P4 and P2–P1′ groups the inhibitor partially occupies the S3 subpocket with a terminating 2-methylbutylamide. Key hydrogen bonding interactions can be found near the catalytic site with His-163 and the benzotriazole N-(3) engaged in a key interaction, with an interatomic distance of 2.9 Å. In addition a backbone Glu-166 NH interaction is evident with the central acetamide oxygen (N–O distance 2.8 Å).
Flexibility of the diamide scaffold (RotBon ∼7) coupled with the observed induced-fit within the active site of 3CLpro presents an added challenge with respect to in silico inhibitor approaches. Thus, our structure–activity-relationship (SAR) studies focused initially on three key areas within the diamide scaffold: (1) benzotriazole replacements with alternative hydrogen bond acceptor functionality to interact with His-163, (2) acetamide modifications within the P2–P1′ region, and (3) minimum pharmacophore deletion studies of the P3 2-methylbutylmide. The P2–P4 group was held constant for this investigation and based upon HTS and reconfirmation results (data not shown) the N-methyl pyrrole was replaced with an equipotent 3-thienyl moiety.
In parallel with efforts to obtain the 3CLpro-7 crystal structure, synthesis of first generation analogs to survey diversity of the benzotriazole unit were initiated using a modified version of our 4CC-Ugi strategy (Scheme 1 ) to allow for late stage azole introduction. Thus, Ugi reaction using t-butyl isocyanide, chloroacetic acid, thiophene-3-carbaldehyde, and N-(4-aminophenyl)acetamide proceeded smoothly to give chloride 8, which could be isolated in good yield after chromatography. Displacement of chloride 8 with azole NH heterocycles provided 9a–c. Alternatively, displacement of 8 with sodium azide and subsequent Huisgen cycloaddition reaction with an appropriate acetylene furnished 1,2,3-triazoles 10a–c in good overall yield.
Scheme 1.
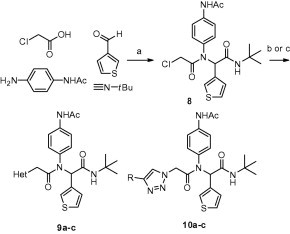
Synthesis of P1 analogs 9a–c and 10a–c. Reagents and conditions: (a) MeOH, 50 °C, 4 h, 95%, (b) (i) NaH, HetNH, DMF, (ii) 9, DMF, 65–80%, (c) (i) NaN3, DMF, 100 °C μwave 30 min, 95%, (ii) acetylene (R = Ph, TMS), DCE, 120 °C 16 h, 85–98%, (iii) R = TMS, TBAF, HOAc, 0 °C–rt, 45%. Final library compounds were purified by UV prep or mass-directed prep HPLC.
Synthesis of P2–P1′ amide analogs within the elaborated diamide were similarly prepared in an Ugi reaction using Boc-protected 4-(amino) aniline (Scheme 2 ) as the amine component. Deprotection of 11 using trifluoroacetic gave aniline 12 which was reacted with a variety of carboxylic acid derivatives under HATU coupling conditions or reacted with an acid chloride or sufonyl chloride in the presence of TEA to give final examples 13a–l.
Scheme 2.
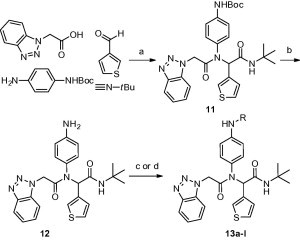
Synthesis of P2–P1 analogs 12 and 13a–l. Reagents and conditions: (a) MeOH, 50 °C, 4 h, 94%, (b) TFA, 95% (c) (i) HATU, DIPEA, DMF, RCO2H, 55–73% (d) RCO2Cl or RSO2Cl, TEA, DCM, 51–64%. Final library compounds were purified by UV prep or mass-directed prep HPLC.
Synthesis of P3 truncated analogs began with reductive amination using thiophene-3-carbaldehyde with either 4-bromoaniline or Boc-protected 4-(amino) aniline to give intermediates 14a–b in good yield. Amide coupling with HATU using benzotriazol-1-yl-acetic acid installed the requisite P1 groups to afford 15a–b. Initial efforts focused on preparing the identical amide library prepared in the elaborated series 13 (Scheme 3 ). This was readily accomplished as before; Boc-deprotection of 15a followed by amide coupling or acylation/sulfonylation, gave 16a–k. Subsequent synthesis of a series of biaryls as amide replacements commenced using a Suzuki cross-coupling with 15b and a variety of boronic acids to afford target molecules 17a–e.
Scheme 3.
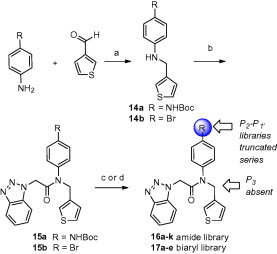
Synthesis of P2–P1′ analogs 16a–k and 17a–e within truncated series. Reagents and conditions: (a) NaHB(OAc)3, DCE, rt, 80% (b) benzotriazol-1-yl-acetic acid, HATU, TEA, DMF, rt, 74% (c) (i) 15a, TFA, DCM, 95%, (ii) HATU, DIPEA, DMF, RCO2H, 65–80%; RCO2Cl or RSO2Cl, TEA, DCM, 90–95%; NaHB(OAc)3, RCHO, DCE, 45–95% (d) 15b, Ar/HetB(OH)2, 1 M aq Na2CO3, 5 mol % Pd(PPh3)4, THF, 30–65%. Final library compounds were purified by UV prep or mass-directed prep HPLC.
SAR for 1,3-azole P1 replacements (9a–c, Fig. 4 ) indicated a strict requirement for the 1,2,3-triazole unit; benzimidazoles 9a–b and 2-methyl-1-imidozyl derivative 9c were uniformly inactive. Since the N-(3) nitrogen of 7 appeared to be involved in a hydrogen bond with His-163, it was somewhat surprising that 9a was not tolerated since the imidazole has the potential to maintain a N-(3)-His-163 hydrogen bond interaction. However, within the 3CLpro-7 structure the catalytic Cys-145 residue is located within 3.3 Å of the N-(2) nitrogen, indicating potential for a weak hydrogen bond and/or dipole–dipole stabilization interaction. This potential interaction may perhaps be responsible for the 1,2,3-triazole preference. Interestingly, 4-phenyl 1,2,3-triazole 10c was tolerated with an IC50 of 11 μM, suggesting additional avenues for optimization. Accommodation of the phenyl moiety of 10c within the active site S1 subpocket is not entirely clear at this time. Based on the 3CLpro-7 structure, Glu-166, Phe-140, and Glu-166 are predicted to be within close proximity. Unsubstituted triazole 10a and trimethyl silyl triazole 10b were inactive, demonstrating the importance of maintaining a proper aromatic ring in this subpocket.
Figure 4.
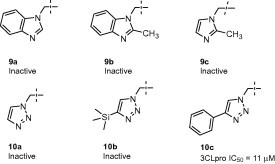
Representative azole replacements (9a–c and 10a–c).
Amide library 13a–l (Fig. 5 ) within the elaborated diamide series displayed a range of potency from moderate micromolar activity (13a, 13b–d, 13f–g) comparable to the HTS hit 7, to weakly active or inactive. Cyclic and acyclic acetamide congeners related to HTS lead 7 showed consistent activities below 10 μM with branched i-propyl derivative 13d and cyclobutyl amide 13g having the greatest activity below 5 μM. Modification to sulfonamide 13b resulted in a three-fold loss in inhibition relative to acetamide 13a. The smaller cyclopropyl (13f) or larger cyclohexyl (13h) cyclic amide generally resulted in loss of inhibition. Incorporating a sterically hindered t-butyl amide 13e also led to a modest three-fold loss in activity. Lastly, aromatic and heteroaromatic amides (13i–k) in addition to iso-butyl carbamate 13l were weak or inactive as 3CLpro inhibitors. Collectively, these data appear to be consistent with the 3CLpro-7 structure whereby a short helix-loop-helix motif (Val-42-Ile-43-Cys-44-Thr-45-Ala-46) and a proximal β-turn element (Thr-24-Thr-25) define the edge of this pocket with minimal volume for larger groups beyond acetamide 7.
Figure 5.
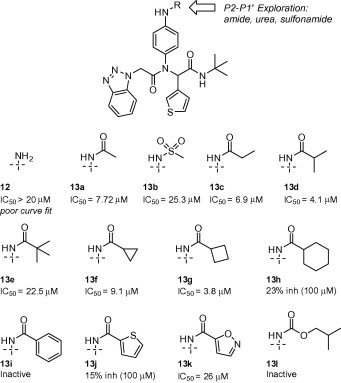
3CLpro activity from library 13.
With limited success from the above S1 and S2–S1′ studies we turned to P3 truncation to potentially identify a minimum pharmacophore to reduce overall MW and improve ligand efficiency (LE).24 Examination of the P3 group within the 7-3CLpro structure suggested this group was unfavorably solvent exposed relative to the t-butylamide–S3 interaction found within the ML188-3CLpro structure.22 Initial efforts led to 16a–k (Table 1 ). Gratifyingly truncated amides proved to have comparable activity versus the elaborated diamide counterparts (Fig. 5 see 13c–d, 13f–g, vs Table 1 see 16a–c, 16e–f). Interestingly, truncated series 16 appeared to better tolerate larger substituents, perhaps suggesting additional changes in the shape of the active site within this subpocket. For example cyclohexyl amide 16g was found to be a weak inhibitor and similarly carbamate 16i had moderate inhibitory activity of 10.3 μM while its related counterpart 13l (Fig. 5) was inactive.
Table 1.
| Cmpd | R | IC50a | Cmpd | R | IC50a |
|---|---|---|---|---|---|
| 16a | 2.9 | 16i | 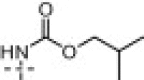 |
10.3 | |
| 16b | 3.6 | 16j | 2.1 | ||
| 16c | 13.3 | 16k | 1.5 | ||
| 16d | 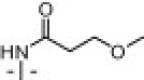 |
3.4 | 17a | 0.051 | |
| 16e | 4.1 | 17b | 0.97 | ||
| 16f | 8.1 | 17c | 0.70 | ||
| 16g | 22.1 | 17d | 2.0 | ||
| 16h | 18% (100 μM) | 17e | 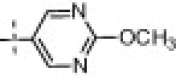 |
12.5 |
IC50 are the average of three independent determinations and represent a co-efficient of variation (CV) < 0.10.
At this stage in the project with efforts focused on P3 truncated analogs bearing a putative S2–S1′ interaction, we elected 16e for further characterization and probe declaration (ML300, Fig. 6). Relative to probe ML188 (6) and the equipotent diamide 13d, ML300 proved to offer improvements in several areas (Fig. 6 ). SARS 3CLpro inhibitor ML300 is ∼100 amu lower MW (MW = 431) relative to 13d with moderate ligand efficiency (LE). Deletion of the lipophilic P3 group reduces c Log P an order of magnitude (c Log P = 3.2) and thus greatly improves ligand-efficiency-dependent lipophilicity (LELP) versus ML188 and 13d. Probe molecules ML188 and ML300 were evaluated in an in-house25 in vitro DMPK panel including plasma protein binding, P450 enzyme inhibition, and intrinsic clearance using liver microsomes (Fig. 6). Both ML188 and ML300 possess good free fraction with ML300 being superior (1.5 and 4.0-fold improved human and rat fraction unbound, respectively); however, intrinsic clearance (CLHEP normalized to liver blood flow, Q h = 21 mL/min/kg, Q r = 70 mL/min/kg) indicates ML188 and both ML300 are both predicted to be highly cleared. ML188 and ML300 possess modest P450 enzyme inhibition, with ML300 maintaining 5–10 μM activity across four major CYP enzymes (Fig. 6). Probe ML300 was found to be highly selective in a Eurofins lead-profiling screen26 with only modest activity for melatonin MT1 receptor in a radioligand binding assay.
Figure 6.

Profiles of 3CLpro inhibitors 6 (ML188), 13d, and 163 (ML300).
Based on the absence of key hydrogen bonding interactions of the P2–P1′ amide of 7 with the 3CLpro active site, in addition to the poor metabolic instability and CYP profile of ML300, we opted to explore more diverse amide replacements as a means to improve metabolic stability, P450 activity, and 3CLpro inhibitory potency. Initial efforts identified representative N-methyl (16j) and N-benzyl (16k) anilines with potency comparable to probe ML300 (16e). The lack of activity for benzamide 16h versus the reduced benzylamine 16k is striking and indicates the enhanced flexibility of the N-benzyl group is permitting a productive interaction to occur where previously aromatic amides were not tolerated (see Fig. 5, 13i–j). A subsequent survey of biaryls was explored (Table 1, 17a–e) and on the basis of the 3CLpro-7 X-ray, we targeted 3-pyridyl (17b–c) and 4-pyrimidyl (17e) heterocycles as means to potentially engage a side-chain interaction from the hydroxyl groups of Thr-24 or Thr-25. These modifications afforded inhibitors with micromolar activity and in the case of 2-methoxypyridyl 17c submicromolar activity (IC50 = 700 nM). Unexpectedly the parent simple phenyl biaryl 17a proved to have a major impact on activity with a ∼7–10-fold increase relative to 17b–c. 3CLpro inhibitor 17a represented the first sub-100 nM inhibitor for the series and to our knowledge one of the most potent nonwarhead based SARS 3CLpro inhibitors to date. At this time inhibitor 17a is relatively unoptimized and thus current efforts are focused on targeted biaryl congeners to understand DMPK, cellular activity, as well as potential broad spectrum activity against other coronavirus strains including MERS-CoV.
In summary, we have described the identification and binding orientation and interactions for a second class of diamide SARS 3CLpro inhibitors, culminating in probe ML300 and subsequently improved inhibitors such as 17a, which possess LE > 0.3 and an LELP approaching 10. The X-ray crystal structure of HTS hit 7 bound to 3CLpro27 was instrumental in guiding optimization and the induced-fit of this inhibitor 3CLpro complex illustrates the challenges of divergent SAR and the limitations of virtual based screens. The four component Ugi reaction was utilized once more to rapidly generate SAR for the putative P2–P1′ and P1 subgroups. Importantly, P3 truncation was possible for this triazole series of 3CLpro inhibitors, allowing for significant MW reduction without diminishing potency. Collaborative efforts in these laboratories continue towards the identification active inhibitors within the truncated biaryl class. Integrated efforts with DMPK assessment continue in order to improve intrinsic clearance and diminish P450 activity, which are issues to be addressed within the series prior to in vivo proof-of-mechanism studies. ML300 is an MLPCN probe and is freely available upon request.
Acknowledgments
This work was supported in part by MLPCN (1U54 MH084659 and MH084512) and NIAID to ADM (AI060915, AI026603 and AI085089). The authors thank the synchrotron beamline (LS-CAT) personnel at the Advanced Photon Source at Argonne National Lab. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
References and notes
- 1.McIntosh K., Dees J.H., Becker W.B., Kapikian A.Z., Chanock R.M. Proc. Natl. Acad. Sci. U.S.A. 1967;57:933. doi: 10.1073/pnas.57.4.933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Myint S.H. In: Siddell S.G., editor. Plenum Press; New York: 1995. p. 389. (Human coronovirus infections). [Google Scholar]
- 3.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Eng. J. Med. 2003;348:1953. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 4.Ziebuhr J. Curr. Opin. Microbiol. 2004;7:412. doi: 10.1016/j.mib.2004.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pyrc K., Berkhout B., van der Hoek L. J. Virol. 2007;81:3051. doi: 10.1128/JVI.01466-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fielding B.C. Future Microbiol. 2011;6:153. doi: 10.2217/fmb.10.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cui L.-J., Zhang C., Zhang T., Lu R.-J., Xie Z.-D., Zhang L.-L., Liu C.-Y., Zhou W.-M., Ruan L., Ma X.-J., Tan W.-J. Adv. Virol. 2011:6. doi: 10.1155/2011/129134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention: http://www.cdc.gov/coronavirus/mers/index.html.
- 9.(a) Zaki A.M., van Boheemen S., Bestebroer T.M., Osterhaus A.D., Fouchier R.A. N. Eng. J. Med. 2012;367:1814. doi: 10.1056/NEJMoa1211721. [DOI] [PubMed] [Google Scholar]; (b) Debing Y., Jochmans D., Neyts J. Curr. Opin. Virol. 2013;3:217. doi: 10.1016/j.coviro.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ghosh A.K., Xi D., Johnson M.E., Baker S.C., Mesecar A.D. Ann. Rep. Med. Chem. 2006;41:183. doi: 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jain R.P., Pettersson H.I., Zhang J., Aull K.D., Fortin P.D., Huitema C., Eltis L.D., Parrish J.C., James M.N., Wishart D.S., Vederas J.C. J. Med. Chem. 2004;47:6113. doi: 10.1021/jm0494873. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh A.K., Xi K., Ratia K., Santarsiero B.D., Fu W., Harcourt B.H., Rota P.A., Baker S.C., Johnson M.E., Mesecar A.D. J. Med. Chem. 2005;48:6767. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]
- 13.Yang S., Chen S.J., Hsu M.F., Wu J.D., Tseng C.T., Liu Y.F., Chen H.C., Kuo C.W., Wu C.S., Chang L.W., Chen W.C., Liao S.Y., Chang T.Y., Hung H.H., Shr H.L., Liu C.Y., Huang Y.A., Chang L.Y., Hsu J.C., Peters C.J., Wang A.H., Hsu M.C. J. Med. Chem. 2006;49:4971. doi: 10.1021/jm0603926. [DOI] [PubMed] [Google Scholar]
- 14.Zhang J., Pettersson H.I., Huitema C., Niu C., Yin J., James M.N.G., Eltis L.D., Vederas J.C. J. Med. Chem. 2007;50:1850. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]
- 15.Xue X., Yu H., Yang H., Xue F., Wu Z., Shen W., Li J., Zhou Z., Ding Y., Zhao Q., Zhang X.C., Liao M., Bartlam M., Rao Z. J. Virol. 2008;82:2515. doi: 10.1128/JVI.02114-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akaji K., Konno H., Mitsui H., Teruya K., Shimamoto Y., Hattori Y., Ozaki T., Kusunoki M., Sanjoh A. J. Med. Chem. 2011;54:7962. doi: 10.1021/jm200870n. [DOI] [PubMed] [Google Scholar]
- 17.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen W.Q., Lu C.Y., Wong T.W., Ling W.H., Lin Z.N., Hao Y.T., Liu Q., Fang J.Q., He Y., Luo F.T., Jing J., Ling L., Ma X., Liu Y.M., Chen G.H., Huang J., Jiang Y.S., Jiang W.Q., Zou H.Q., Yan G.M. Emerg. Infect. Dis. 2005;11:89. doi: 10.3201/eid1101.040138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu C.Y., King K.Y., Kuo C.J., Fang J.M., Wu Y.T., Ho M.Y., Liao C.L., Shie J.J., Liang P.H., Wong C.H. Chem. Biol. 2006;13:261. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ghosh A.K., Gong G., Grum-Tokars V., Mulhearn D.C., Baker S.C., Coughlin M., Prabhakar B.S., Sleeman K., Johnson M.E., Mesecar A.D. Bioorg. Med. Chem. Lett. 2008;18:5684. doi: 10.1016/j.bmcl.2008.08.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J., Huitema C., Niu C., Yin J., James M.N.G., Eltis L.D., Vederas J.C. Bioorg. Chem. 2008;36:229. doi: 10.1016/j.bioorg.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jacobs J., Grum-Tokars V., Zhou Y., Turlington M., Saldanha S.A., Chase P., Eggler A., Dawson E.S., Baez-Santos Y.M., Tomar S., Mielech A.M., Baker S.C., Lindsley C.W., Hodder P., Mesecar A., Stauffer S.R. J. Med. Chem. 2013;56:534. doi: 10.1021/jm301580n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee T.W., Cherney M.M., Liu J., James K.E., Powers J.C., Eltis L.D., James M.N. J. Mol. Biol. 2007;366:916. doi: 10.1016/j.jmb.2006.11.078. For an example of ‘induced-fit’ and discussion on active-site flexibility in the context of inhibitor bound versus unbound SARS 3CLpro using an aza-peptide epoxide see: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hopkins A.L., Groom C.R., Alex A. Drug Discovery Today. 2004;9:430. doi: 10.1016/S1359-6446(04)03069-7. [DOI] [PubMed] [Google Scholar]
- 25.Morrison R.D., Blobaum A.L., Byers F.W., Santomango T.S., Bridges T.M., Stec D., Brewer K.A., Sanchez-Ponce R., Corlew M.M., Rush R., Felts A.S., Manka J., Bates B.S., Venable D.F., Rodriguez A.L., Jones C.K., Niswender C.M., Conn P.J., Lindsley C.W., Emmitte K.A., Daniels J.S. Drug Metab. Dispos. 2012;40:1834. doi: 10.1124/dmd.112.046136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.For information on MLPCN’s probe compound ancillary screen see Eurofins LeadProfilingScreen®: www.eurofinspanlabs.com.
- 27.The protein–ligand X-ray structure of 7-bound SARS-3CLpro has been deposited in PDB. RCSB ID code rcsb081783 and PDB ID code 4MDS.