

Abstract
Among transplant recipients, those who produce antibodies against the donor's human leukocyte antigens (HLAs) are at higher risk for antibody-mediated rejection and transplant vasculopathy, which is a progressive, vasculo-occlusive disease that results in ischemic injury and deterioration of organ function. Antibodies against HLA class I (HLA-I) molecules are thought to contribute to transplant vasculopathy by triggering signals that elicit the activation and proliferation of endothelial cells. Here, we demonstrate a molecular association between HLA-I and the integrin β4 subunit after the stimulation of endothelial cells with HLA-I–specific antibodies. Knockdown of integrin β4 in these cells abrogated the ability of HLA-I to stimulate the phosphorylation of the kinases Akt, extracellular signal–regulated kinase (ERK), and Src, as well as cellular proliferation. Similarly, reducing the abundance of HLA-I suppressed integrin β4–mediated phosphorylation of ERK and the migration of endothelial cells on laminin-5, a component of the extracellular matrix. These results indicate a mutual dependency between HLA-I and the integrin β4 subunit to stimulate the proliferation and migration of endothelial cells, which may be important in promoting transplant vasculopathy and tumor angiogenesis.
Introduction
Antibody-mediated rejection is emerging as a leading cause of allograft rejection and graft loss (1, 2). Numerous studies have shown that patients who produce antibodies after transplant against donor major histocompatibility complex (MHC) antigens, also referred to in humans as human leukocyte antigens (HLAs), are at a higher risk of chronic rejection and transplant vasculopathy, which is characterized by concentric hyperplasia with intimal proliferation of the vessels of the allograft (3–6). The HLA class I (HLA-I) molecules include HLA-A, HLA-B, and HLA-C, and they are heterodimeric glycoproteins that consist of a heavy chain that is noncovalently associated with β2-microglobulin. The most distinct feature of HLA-I molecules is their high degree of polymorphism, which is a manifestation of their role in presenting a diverse range of antigenic peptides to responding T cells. In addition to their well-known role in antigen presentation, HLA-I molecules transduce signals in various cell types that elicit responses such as apoptosis and proliferation (7). Cross-linking of HLA-I molecules on the surface of endothelial cells with antibodies triggers phosphorylation of the kinases Src and focal adhesion kinase (FAK), which in turn causes activation of the phosphatidylinositol 3-kinase (PI3K)–Akt, S6 ribosomal protein, and extracellular signal–regulated kinase (ERK) signaling pathways, as well as the proliferation of endothelial cells (8–10). Signaling stimulated by antibodies against HLA-I also induces cytoskeleton reorganization and promotes the adherence of leukocytes to endothelial cells (8, 11). Several studies suggest that the signaling events that occur in endothelial cells during interactions with HLA-I–specific antibodies contribute to the process of transplant vasculopathy (3). Passive transfer of antibody against donor MHC class I (MHC-I) molecules in immunodeficient mice leads to the development of transplant vasculopathy (12, 13). Furthermore, the extent of phosphorylation of signaling molecules involved in MHC-I–dependent proliferation and survival pathways is increased in mice treated with MHC-I–specific antibody relative to that in isotype control immunoglobulin G (IgG)–treated mice (14).
The proximal molecular events at the plasma membrane that regulate the triggering of the HLA-I–dependent signaling cascade remain poorly understood. Given that HLA-I molecules do not have intrinsic kinase activities, it is conceivable that they physically associate with other molecules that have the capacity to transduce signals. In this respect, HLA-I interacts with the insulin receptor and the epidermal growth factor receptor (EGFR) to modify receptor function (15–17). MHC-I molecules also play a role in synaptic plasticity and neuronal development (18). Collectively, these data imply that HLA-I signaling has previously unsuspected physiological consequences beyond those related to immune recognition.
Integrins are cell adhesion molecules that mediate attachment between a cell and the extracellular matrix (ECM). Integrins also transduce intracellular signals that regulate cell proliferation, survival, and migration. Integrins are heterodimeric receptors consisting of an α and a β subunit, and they bind to components of the ECM (such as fibronectin, vitronectin, collagen, and laminin) and link the ECM with the cytoskeleton. Upon ligand binding, integrins activate various kinases, including FAK, Src, PI3K, and ERK (19). The integrin β4 subunit pairs with the α6 subunit to form a functional dimer to bind to laminin. The integrin β4 subunit differs from other integrin subunits by having a long cytoplasmic tail that interacts with FAK and Src to activate signaling pathways that elicit cell survival and proliferation (20, 21). Integrin β4 promotes angiogenesis and tumorigenesis through ERK, PI3K, or ErbB2 signaling (21).
Here, we investigated the structural requirements of HLA-I that were required for stimulating the proliferation and migration of human endothelial cells. Because antibody-mediated cross-linking of HLA-I molecules in endothelial cells elicits protein phosphorylation cascades that are similar to those mediated by the integrin β4 subunit (21, 22), we hypothesized that HLA-I associated with the β4 subunit to transduce signals in endothelial cells. We found that HLA-I and integrin β4 formed a molecular complex that was required to transduce signals that led to the proliferation of endothelial cells. Our results indicate a mutual dependency between HLA-I and integrin β4 to stimulate proliferation and migration of endothelial cells.
Results
Cross-linking of HLA-I stimulates formation of a complex between HLA-I and the integrin β4 subunit
To examine whether the integrin β4 subunit was a partner of HLA-I in signal transduction, we characterized their potential physical and functional interactions. First, we performed coimmunoprecipitation experiments to examine whether HLA-I and integrin β4 formed a complex. Stimulation of human endothelial cells with W6/32, a monoclonal antibody against HLA-I, increased the amount of the complex formed between HLA-I and integrin β4, but not integrin β1, β3, or β5 (Fig. 1A). Similar results were obtained when cells were stimulated with the F(ab′)2 fragment of W6/32 and samples were subjected to immunoprecipitation with an antibody against HLA-A2, an allele of the HLA-A locus (Fig. 1B).
Fig. 1.
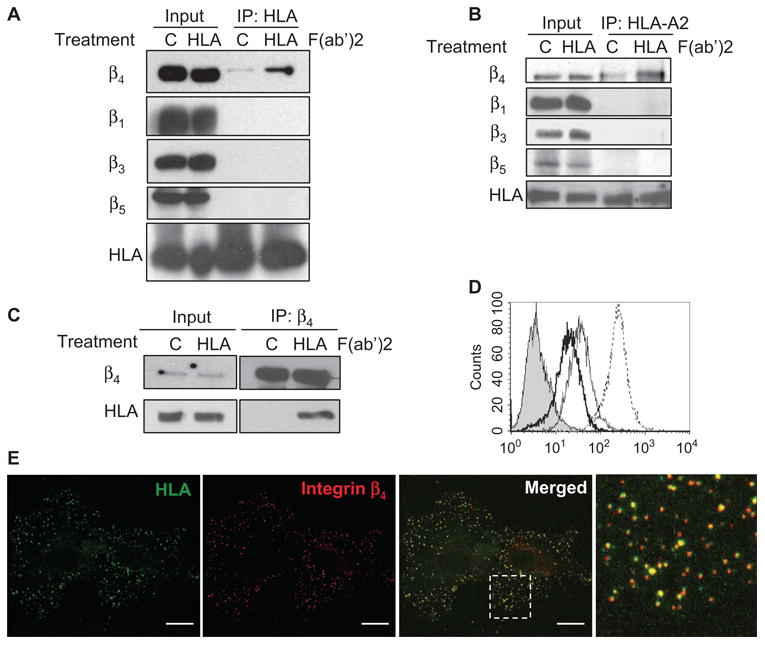
Ligation of HLA-I on endothelial cells triggers formation of a complex containing the integrin β4 subunit and HLA-I. (A) Endothelial cells (EC1) were stimulated with the F(ab′)2 fragments of W6/32 (an antibody against HLA-I) or with control IgG. Cell lysates were subjected to immunoprecipitation (IP) with W6/32 and analyzed by Western blotting with antibodies against the heavy chain of HLA-I (EMR8-5) and integrins β4, β1, β3, and β5, as indicated to the left of the blots. For each sample, 10% of the total cell lysate that was used in the immunoprecipitation was loaded as an input control. Data are representative of three independent experiments. (B) Endothelial cells (EC1) were stimulated as described in (A), lysates were subjected to immunoprecipitation with an antibody against HLA-A2, and samples were analyzed by Western blotting with antibodies against the HLA-I heavy chain (EMR8-5) and integrins β4 (205 kD), β1 (130 kD), β3 (105 kD), and β5 (88 kD). For each sample, 2.5% of the total cell lysate used in the immunoprecipitation reaction was loaded as an input control. Data are representative of two independent experiments. (C) Cells (EC3) were stimulated as described in (A), lysates were subjected to immunoprecipitation with an antibody against the integrin β4 subunit, and samples were analyzed by Western blotting with EMR8-5 or antibody against integrin β4. For each sample, 5% of the total cell lysate used in the immunoprecipitation reaction was loaded as an input control. Data are representative of four independent experiments. (D) Surface abundances of integrins were analyzed by flow cytometry. Unstained control cells are shown as a filled peak. The thick, thin, and dotted lines represent the surface expression of integrins β4, β3, and β1, respectively. Data are representative of four independent experiments. (E) Incubation of endothelial cells (EC1) with antibodies against the extracellular domains of HLA-I (green) and integrin β4 (red). The last panel is an enlarged field that is indicated by the white square in the merged image. Localization of both HLA-I and integrin β4 at the basal membrane was positively correlated as indicated by a correlation coefficient (r) of 0.63 and 0.72 for the merged image and boxed region, respectively. Scale bars, 20 μm. The data are representative of four independent experiments.
To further confirm the association between HLA-I and integrin β4, we performed reciprocal coimmunoprecipitation experiments with endothelial cells from a different donor (EC3). Specifically, immunoprecipitation with an antibody against the integrin β4 subunit demonstrated that the cross-linking of HLA-I increased the amount of the complex formed between integrin β4 and the heavy chain of HLA-I (Fig. 1C), whereas immunoprecipitation with an antibody against the integrin β1 subunit failed to pull down the HLA-I heavy chain and integrin β4 (fig. S1). The specific association between HLA-I and integrin β4 was not as a result of the higher abundance of β4 relative to that of other integrin subunits, because flow cytometric analysis showed that the amount of integrin β4 on the surface of endothelial cells was less than that of integrins β1 and β3 (Fig. 1D). That integrin β4 coimmunoprecipitated with MHC-I and W6/32, a monoclonal antibody that recognizes a conformational epitope formed by the assembly of the HLA-I heavy chain and β2-microglobulin, indicated that the integrin β4 subunit formed a complex with native HLA-I.
We next examined the localization of HLA-I and integrin β4 in endothelial cells by confocal microscopy. The integrin β4 subunit was distributed in a punctate pattern in confocal images that represented the optical plane corresponding to the basal cell surface, suggesting that integrin β4 was concentrated on the basal surface of the cell and therefore might function in cell adhesion (Fig. 1E). Indeed, the integrin β4 subunit mediates the adherence of cells to the ECM by forming type II hemidesmosomes in endothelial cells (23). Incubation of endothelial cells with W6/32 showed that HLA-I was also localized in a punctuate pattern on the basal surface of the cells. We quantitated the extent of colocalization of integrin β4 and HLA-I with the algorithm of Costes et al. (24). The merged image showed extensive colocalization of both proteins in a punctuate pattern (r = 0.72) (Fig. 1E). Their colocalization in concentrated regions on the basal surface suggests that HLA-I and the integrin β4 subunit may associate at sites of cell adhesion. In agreement with our findings, HLA-I and integrin β4 have a punctuate distribution in other cell types, including neurons and endothelial cells (23, 25).
To further characterize the molecular interaction between HLA-I and the integrin β4 subunit, we engineered recombinant mutant HLA-A2 molecules in which either the cytoplasmic (C-HLA-A2) or the extracellular (E-HLA-A2) domain was deleted and replaced with green fluorescent protein (GFP) (Fig. 2A). To avoid interference by endogenous HLA-I, we used adenoviral vectors to express the recombinant mutant proteins in primary endothelial cells that do not contain the HLA-A2 allele. Flow cytometric analysis showed that 90% of the cells that were infected with the adenovirus encoding E-HLA-A2 contained GFP and were detectable with an antibody against GFP (demonstrating that GFP was at the cell surface) (Fig. 2B), but not against HLA-A2 (Fig. 2B). Similarly, we found that 99% of the cells that were infected with adenovirus encoding C-HLA-A2 contained GFP and were detectable with an antibody against HLA-A2 (Fig. 2B), but not against GFP (Fig. 2B). Cross-linking of E-HLA-A2 with antibodies against GFP increased its association with integrin β4 relative to that in cells treated with antibodies against HLA-A2, which did not bind to E-HLA-A2. In contrast, the C-HLA-A2 protein did not associate with integrin β4 after cross-linking with antibodies against HLA-A2 (Fig. 2C). These results suggest that the cytoplasmic tail of the heavy chain of HLA-I was required to form a complex with the integrin β4 subunit.
Fig. 2.
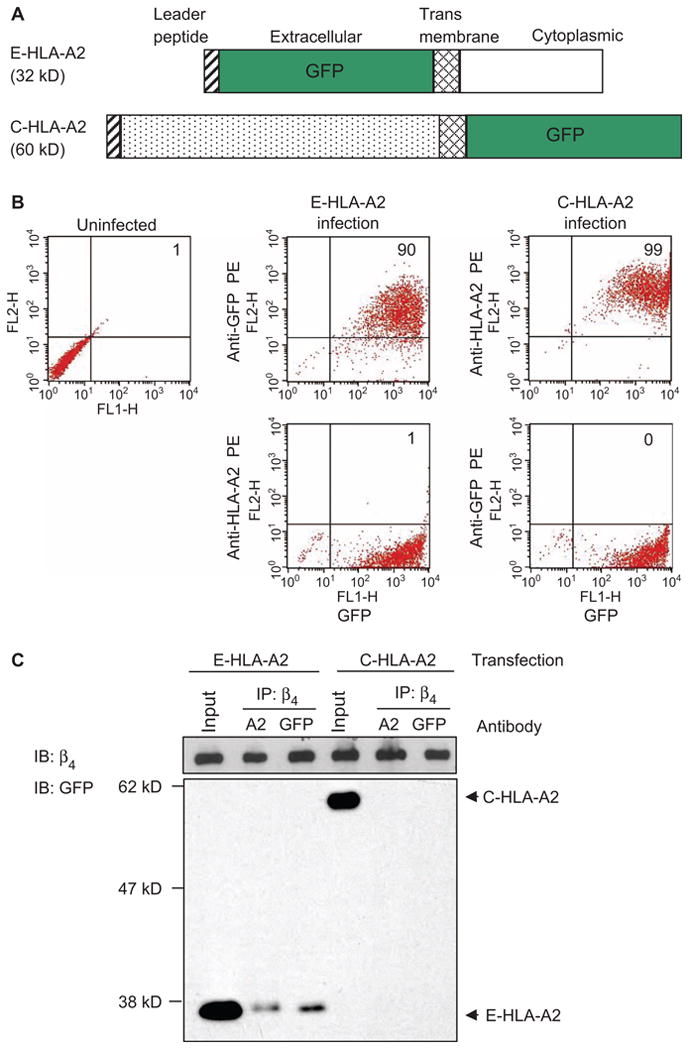
The cytoplasmic domain of the HLA-I heavy chain mediates the interaction between HLA-I and integrin β4. (A) Schematic representation of the mutant HLA-A2 molecules that have either the extracellular (E-HLA-A2) or the cytoplasmic (C-HLA-A2) domains deleted and replaced with GFP. (B) Endothelial cells were infected with adenoviruses encoding either E-HLA-A2 or C-HLA-A2. More than 90% of the infected cells contained the mutant proteins, as determined by flow cytometric analysis of GFP. Data are representative of 10 independent experiments. (C) Endothelial cells containing E-HLA-A2 or C-HLA-A2 were stimulated with antibodies against either HLA-A2 or GFP, cells were lysed and subjected to immunoprecipitation with Sepharose beads conjugated to antibody against integrin β4, and samples were analyzed by Western blotting with an antibody against GFP. Endothelial cells from donor EC2 were used in these experiments. For each sample, 10% of the total cell lysate used in the immunoprecipitation was loaded as an input control. Data are representative of three independent experiments.
Knockdown of the integrin β4 subunit impairs HLA-I–mediated phosphorylation of target proteins
We reasoned that if HLA-I associated with integrin β4 to transduce signals, depletion of integrin β4 should inhibit HLA-I–mediated phosphorylation of target proteins. Thus, we used small interfering RNA (siRNA) to knock down integrin β4. Transfection of endothelial cells with a single integrin β4–specific siRNA resulted in knockdown of integrin β4 but did not affect the amounts of other proteins involved in the HLA-I signaling pathway, including the mammalian target of rapamycin (mTOR), FAK, Src, Akt, and ERK (Fig. 3A). Furthermore, transfection of cells with the integrin β4–specific siRNA did not alter the abundance of β2-microglobulin, which associates with the heavy chain of HLA-I to form heterodimers (Fig. 3A). In addition, knockdown of the integrin β4 subunit did not decrease the amount of HLA-I at the cell surface, as demonstrated by flow cytometry (Fig. 3B).
Fig. 3.
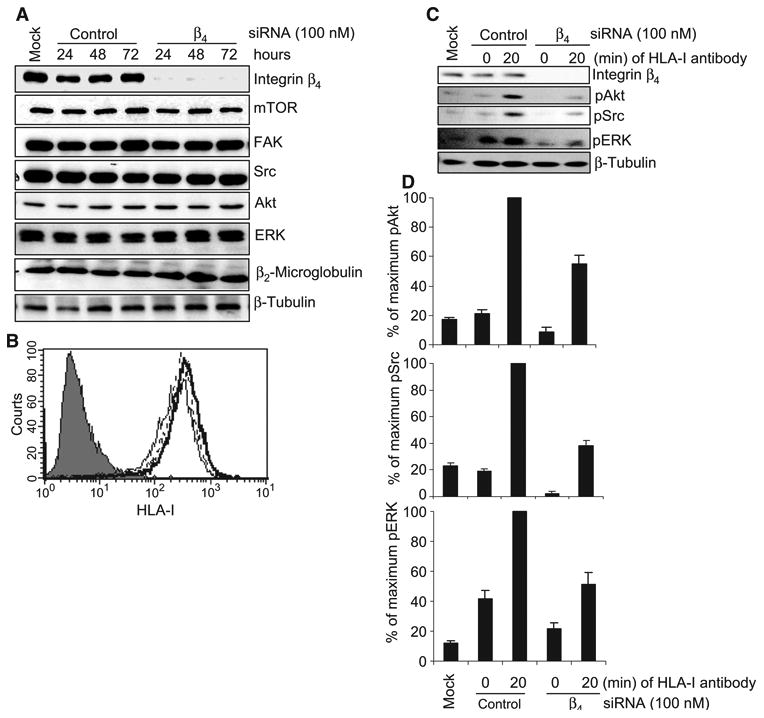
Knockdown of integrin β4 inhibits HLA-I–mediated phosphorylation of target proteins in endothelial cells. (A) Endothelial cells were transfected with 100 nM single duplex siRNA against the integrin β4 subunit (sequence: 5′-GAGAGCAGCUUCCAAAUCA-3′) or with control siRNA and were incubated for 24, 48, or 72 hours. Cells were lysed and subjected to Western blotting analysis with antibodies against integrin β4, mTOR, FAK, Src, Akt, ERK, and β2-microglobulin, with β-tubulin serving as a loading control. (B) Expression of HLA-I on the surface of endothelial cells transfected with siRNAs was analyzed by flow cytometry with the HLA-I–specific antibody W6/32. Negative control cells are shown as a filled peak, whereas the thin, thick, and dotted lines represent mock-transfected cells, cells transfected with integrin β4–specific siRNA, and cells transfected with control siRNA, respectively. (C) Endothelial cells were transfected with integrin β4–specific siRNA or control siRNA (100 nM each). After 48 hours, cells were stimulated with W6/32 (1 μg/ml) for 20 min, lysed, and analyzed by Western blotting with antibodies against pAkt (Ser473), pSrc (Tyr416), and pERK (Thr202/Tyr204). β-Tubulin was detected to determine equal loading. Data shown are representative of three independent experiments. (D) Protein bands shown in (C) were quantified by densitometry and the results are expressed as the mean ± SEM percentage of the maximal extent of protein phosphorylation stimulated by W6/32. Endothelial cells (EC1) were used in these experiments. The data presented in (A) to (C) are representative of at least three independent experiments. (D) Pooled data from three independent experiments.
We next determined the effect of knockdown of integrin β4 on HLA-I–mediated signaling. Cross-linking HLA-I on control endothelial cells induced the phosphorylation of Src, Akt, and ERK (Fig. 3, C and D), whereas HLA-I–mediated phosphorylation of these kinases was reduced in cells transfected with integrin β4–specific siRNA, which suggested that HLA-I depended on integrin β4 for signaling (Fig. 3, C and D). To rule out potential off-target effects of this siRNA, we used two additional siRNAs that targeted different regions within integrin β4 messenger RNA (mRNA). We found that knockdown of integrin β4 with either of these siRNAs also decreased HLA-I–mediated phosphorylation of Src and Akt relative to that in control cells (fig. S2).
To exclude the possibility that inhibition of HLA-I–mediated phosphorylation of the target kinases by integrin β4–specific siRNA was caused by disruption of the cell-adhesive properties of integrin β4, we performed parallel experiments in which cultured endothelial cells were held in suspension, stimulated with antibodies against integrin β4 or HLA-I, lysed, and subjected to Western blotting analysis. Antibody-dependent cross-linking of the integrin β4 subunit increased the extent of phosphorylation of FAK, Akt, Src, and ERK in suspended endothelial cells relative to that in untreated cells (Fig. 4A). A similar pattern of protein phosphorylation was observed when suspended endothelial cells were stimulated with antibodies against HLA-I (Fig. 4B). Knockdown of the integrin β4 subunit impaired HLA-I–mediated phosphorylation of FAK, Akt, Src, and ERK in endothelial cells cultured in suspension (Fig. 4B).
Fig. 4.
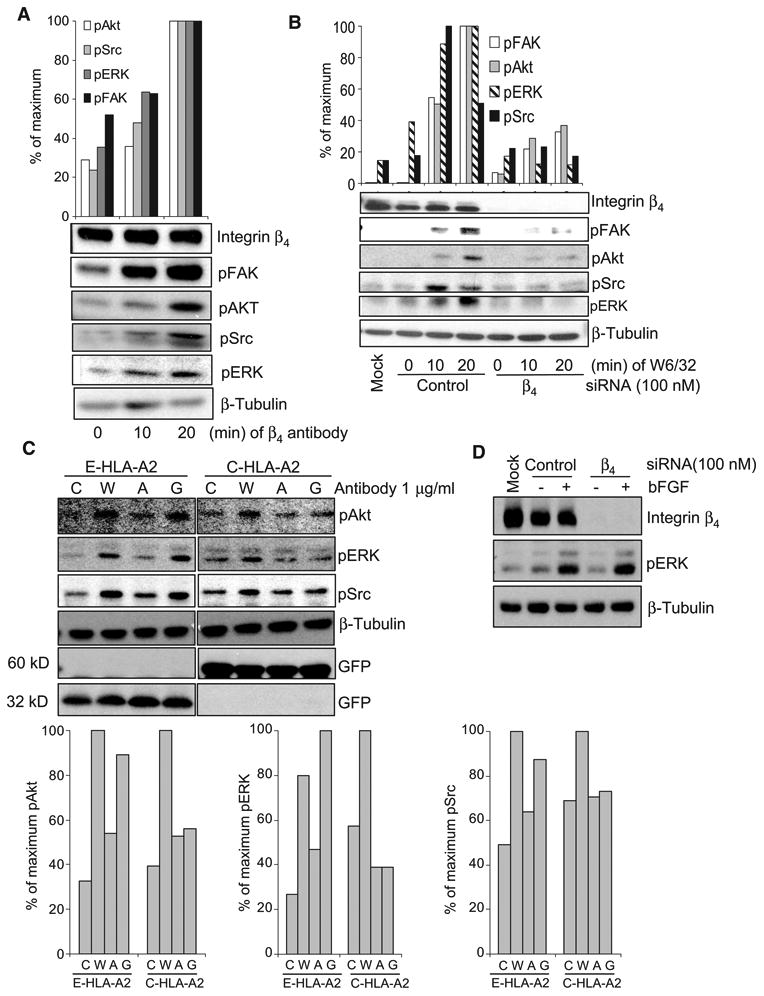
Deletion of the cytoplasmic domain of the heavy chain of HLA-I blocks HLA-I signaling. (A) Endothelial cells (EC1) cultured in suspension were stimulated with antibodies against the integrin β4 subunit. Cells were lysed and analyzed by Western blotting with antibodies against pFAK, pSrc, pAkt, and pERK. Densitometric analysis was performed and the results are expressed as the percentage of the maximal extent of phosphorylation stimulated by the integrin β4–specific antibody. The data are representative of two independent experiments. (B) Endothelial cells (EC1) transfected with control or integrin β4–specific siRNA were cultured in suspension, stimulated with W6/32, and analyzed by Western blotting to determine the extent of phosphorylation of target proteins. Results from densitometric analysis are expressed as the percentage of maximal extent of phosphorylation stimulated by W6/32. The data are representative of three independent experiments. (C) Endothelial cells (EC2) were infected with adenoviruses encoding E-HLA-A2 or C-HLA-A2 and were stimulated with control mouse IgG (C), W6/32 (W), antibody against HLA-A2 (A), or antibody against GFP (G). Cells were lysed and analyzed by Western blotting with antibodies against pAkt, pERK, and pSrc. Membranes were also incubated with antibody against GFP to determine the abundances of E-HLA-A2 and C-HLA-A2, and β-tubulin was used as a loading control. Protein bands were quantified by densitometry, and the results are expressed as the percentage of the maximal extent of phosphorylation of target proteins stimulated by W6/32 and are shown in the bar graphs. Data are representative of three independent experiments. (D) Endothelial cells (EC1) were transfected with control or integrin β4–specific siRNA, stimulated with bFGF, and analyzed by Western blotting to determine the extent of phosphorylation of ERK. The data are representative of two independent experiments.
Because the cytoplasmic domain of the heavy chain of HLA-I is required for its association with integrin β4 (Fig. 2C), we reasoned that deletion of this region would abolish the capacity of HLA-I to transduce signals. To test this, we infected endothelial cells lacking endogenous HLA-A2 with adenovirus encoding either C-HLA-A2 or E-HLA-A2 and stimulated the cells with antibodies against GFP or HLA-A2. The HLA-I–specific monoclonal antibody W6/32 was used as a positive control to stimulate signals through endogenous HLA-I molecules. Cross-linking of E-HLA-A2 molecules with antibody against GFP increased the extent of phosphorylation of Akt, Src, and ERK relative to that in cells treated with control IgG or antibody against HLA-A2 (Fig. 4C). In contrast, cross-linking of C-HLA-A2 molecules with antibody against HLA-A2 failed to stimulate phosphorylation of the same kinases (Fig. 4C). An increased extent of phosphorylation was observed, however, when endothelial cells containing C-HLA-A2 were treated with W6/32, which confirmed signaling through endogenous HLA-I. We did not observe an increase in the extent of protein phosphorylation in the absence of the binding of antibodies against GFP or HLA-A2 to E-HLA-A2 and C-HLA-A2, respectively (Fig. 4C). These results indicate that deletion of the cytoplasmic, but not the extracellular, domain of the HLA-I heavy chain impaired HLA-I signaling. Given that the cytoplasmic tail of HLA-I is required for its association with integrin β4 (Fig. 2C), these data suggest that activation of the HLA-I signaling pathway requires an association between HLA-I and the integrin β4 subunit.
To assess the specificity of the association between HLA-I and integrin β4, we determined whether this integrin subunit was required for growth factor–mediated signaling. We transfected endothelial cells with siRNA targeting integrin β4 and stimulated them with basic fibroblast growth factor (bFGF). Knockdown of integrin β4 did not impair bFGF-induced phosphorylation of ERK (Fig. 4D). These results suggest that integrin β4 is required for HLA-I signaling but not for FGF receptor signaling.
Knockdown of the integrin β4 subunit inhibits HLA-I–mediated cell proliferation
Ligation of HLA-I stimulates cell proliferation through the activation of FAK, PI3K-Akt, and mTOR signaling pathways (8, 9). We therefore tested whether ligation of HLA-I stimulated the proliferation of endothelial cells through integrin β4. We transfected endothelial cells with integrin β4–specific siRNA or negative control siRNA, stimulated the cells with W6/32, and measured cell proliferation by monitoring the dilution of the intravital dye carboxyfluorescein succinimidyl ester (CFSE) (26). Antibody-mediated cross-linking of HLA-I stimulated the proliferation of endothelial cells transfected with control siRNA relative to that of cells treated with isotype control IgG (Fig. 5A). Knockdown of integrin β4 prevented the HLA-I–stimulated increase in cell proliferation, which demonstrated that integrin β4 was required for HLA-I–mediated proliferation of endothelial cells.
Fig. 5.

Integrin β4 is required for HLA-I–mediated cell proliferation. (A) Endothelial cells (EC1) were transfected with control or integrin β4–specific siRNA. Twenty-four hours later, cells were labeled with CFSE and stimulated with W6/32 for 72 hours. Cell proliferation was analyzed with ModFit LT software. The proliferation index represents the number of proliferating cells in test cultures as a ratio of the number of proliferating cells in the control cultures. Data are presented as mean proliferation index ± SEM from three independent experiments. *P < 0.05 by one-way ANOVA, Fisher's LSD. (B) Endothelial cells (EC2) were infected with adenoviruses expressing E-HLA-A2 or C-HLA-A2 and were stimulated with antibodies against GFP or HLA-A2 in the presence of BrdU overnight. Cells stimulated with mouse IgG or W6/32 served as negative and positive controls, respectively. Data are presented as the mean percentage of BrdU-positive cells ± SEM of three independent experiments. The flow cytometry plot of one representative experiment is presented in fig. S3.
To further elucidate the molecular basis of HLA-I–dependent cell proliferation, we infected endothelial cells with adenovirus encoding C-HLA-A2 or E-HLA-A2, stimulated the cells with antibodies against HLA-A2 or GFP, and determined cell proliferation by measuring the incorporation of BrdU (5-bromo-2′-deoxyuridine) into the cells. Treatment of cells containing C-HLA-A2 with antibody against HLA-A2 failed to induce proliferation (Fig. 5B and fig. S3), whereas the ligation of E-HLA-A2, which has an intact cytoplasmic tail, stimulated proliferation of the cells (Fig. 5B and fig. S3). These results further document the importance of the cytoplasmic domain of the heavy chain of HLA-I for cell proliferation induced by antibody against HLA-I.
Knockdown of HLA-I impairs laminin-5–stimulated phosphorylation of ERK
Our findings prompted us to determine whether HLA-I played a role in the function of the integrin β4 subunit. Integrin β4 is a cell adhesion molecule that regulates cell migration and survival (21), so we reasoned that HLA-I might be required for integrin β4–induced cell migration. We transfected HLA-A2– and HLA-B56–positive endothelial cells with siRNA against the heavy chain of HLA-I or the integrin β4 subunit or with control siRNA and subsequently plated the cells on collagen I or laminin-5 to activate integrins β1 and β4, respectively. Endothelial cells plated on poly-l-lysine served as a negative control. Transfection of endothelial cells with siRNA against the HLA-I heavy chain substantially reduced the abundance of HLA-I on the surface of the cells, as demonstrated by the decreased extent of binding of specific antibodies against HLA-A2 and HLA-B56 and of the pan-reactive antibody W6/32 (Fig. 6, A and B). The siRNA against the HLA-I heavy chain had no effect on the abundance of integrin β4 or ERK (Fig. 6C). ERK is a well-established downstream effector in the integrin β4–dependent signaling pathway that leads to migration (27). Plating on collagen I stimulated substantial phosphorylation of ERK in endothelial cells transfected with control siRNA, integrin β4–specific siRNA, or siRNA against the HLA-I heavy chain (Fig. 6D). In contrast, transfection with siRNAs against either the integrin β4 subunit or the HLA-I heavy chain decreased the extent of phosphorylation of ERK in cells plated on laminin-5 relative to that in endothelial cells transfected with control siRNA (Fig. 6D). Consistent with this finding, transfection with another siRNA that targeted a different region of the mRNA encoding the HLA-I heavy chain also inhibited laminin-5–induced phosphorylation of ERK (fig. S4).
Fig. 6.
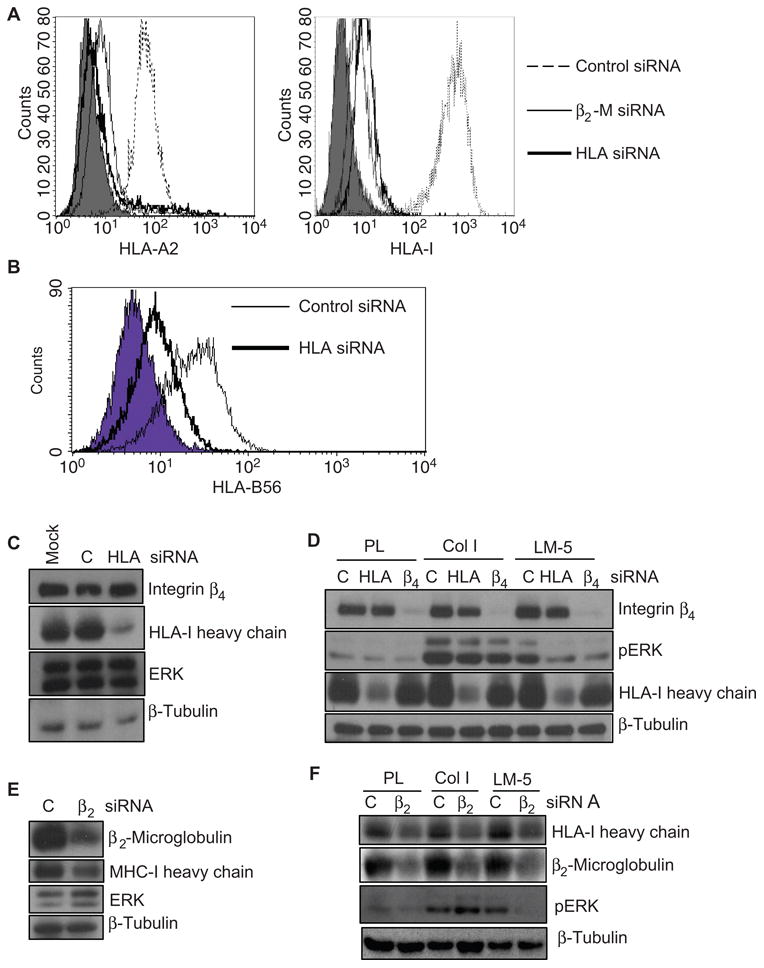
Knockdown of the heavy chain of HLA-I or of β2-microglobulin inhibits the phosphorylation of ERK in endothelial cells. (A) The abundance of HLA-I was analyzed by flow cytometry with the pan-reactive HLA-I–specific antibody, W6/32, or the allele-specific antibody against HLA-A2 in cells that had been transfected with control siRNA or with siRNAs specific for the HLA-I heavy chain or β2-microglobulin. Negative control cells are shown as a filled peak. (B) The abundance of HLA-I was analyzed by flow cytometry with an antibody against the allele-specific HLA-B56 in cells transfected with control siRNA or siRNA against the HLA-I heavy chain. Negative control cells are shown as a filled peak. The data are representative of two independent experiments. (C) Endothelial cells were transfected with siRNA against the HLA-I heavy chain and analyzed by Western blotting with antibodies against integrin β4, the HLA-I heavy chain, ERK, and β-tubulin. (D) Cells transfected with siRNAs against HLA-I heavy chain or the integrin β4 subunit or with control siRNA were plated on dishes coated with poly-l-lysine (PL), collagen I (Col I), or laminin-5 (LM-5), after which they were analyzed by Western blotting with antibodies against integrin β4, pERK, and the HLA-I heavy chain (n = 3 experiments). (E) Cells were transfected with β2-microglobulin–specific siRNA and analyzed by Western blotting with antibodies against β2-microglobulin, ERK, and the HLA-I heavy chain. (F) Cells transfected with β2-microglobulin–specific siRNA were plated onto laminin-5 to simulate the phosphorylation of ERK. Cells were analyzed as described in (D). The data presented in (A) and (C) to (F) are representative of three independent experiments.
To further exclude potential off-target effects of the HLA-I–specific siRNAs, we indirectly reduced the abundance of HLA-I with β2-microglobulin–specific siRNA, as previously described (28). We transfected endothelial cells with siRNA against β2-microglobulin and then stimulated the cells by plating them on poly-l-lysine, collagen I, or laminin-5. Transfection of endothelial cells with β2-microglobulin–specific siRNA decreased the amounts of β2-microglobulin and HLA-I at the cell surface (Fig. 6, A and E). Similar to knockdown of the HLA-I heavy chain, knockdown of β2-microglobulin was accompanied by a loss in the capacity of laminin-5 to induce the phosphorylation of ERK (Fig. 6F). Together, these results demonstrate a role for HLA-I in integrin β4–mediated signaling.
Knockdown of HLA-I reduces the extent of endothelial cell migration
Integrin β4 signaling promotes the migration of endothelial cells through the activation of ERK (27). To examine the role of HLA-I in integrin β4–induced cell migration, we transfected endothelial cells with control siRNA, siRNA against the HLA-I heavy chain, or siRNA against the integrin β4 subunit, plated the cells on laminin-5–coated dishes, and examined their migration in an in vitro wound-healing assay. We found that endothelial cells transfected with control siRNA and stimulated with FGF migrated across the wound and completely closed the gap within 12 hours of incubation (Fig. 7A). In contrast, the extent of migration of endothelial cells transfected with siRNAs against either the integrin β4 subunit or the heavy chain of HLA-I was substantially lower.
Fig. 7.
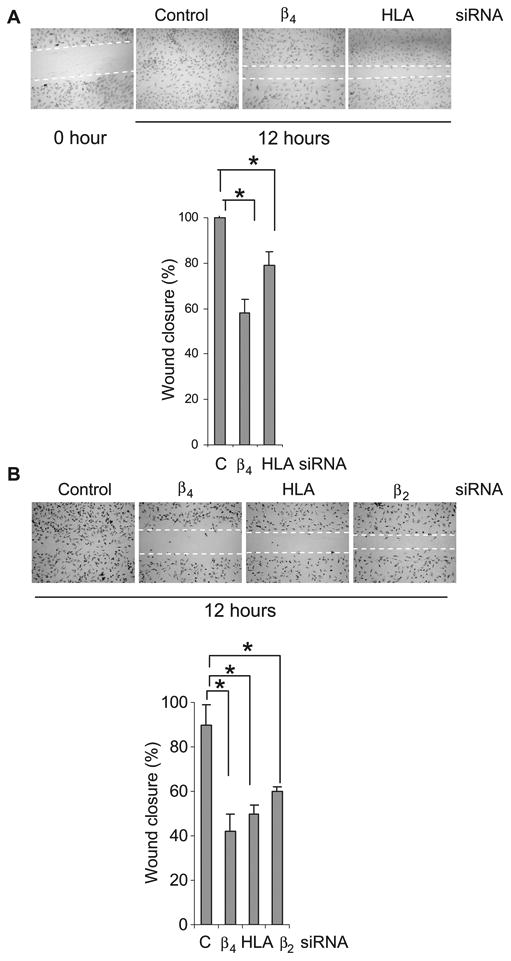
Knockdown of HLA-I inhibits migration of endothelial cells. (A) Cells transfected with control siRNA or with siRNA against the integrin β4 subunit or the HLA-I heavy chain were plated on laminin-5 and stimulated with bFGF, and their migration across an artificial wound was measured. (B) Cells were transfected with control siRNA or with siRNA against the integrin β4 subunit, the HLA-I heavy chain, or β2-microglobulin and were pretreated with mitomycin C to inhibit cell proliferation before being assayed for their ability to migrate. The graphs show the mean percentage ± SEM of wound closure. *P < 0.05 by one-way ANOVA, Fisher's LSD (n = 9 experiments). Endothelial cells (EC1) were used in these experiments.
It was possible that knockdown of integrin β4 could have reduced the number of cells in the denuded area of the wound by inhibiting cell proliferation, rather than migration. Thus, we also examined the migration of endothelial cells plated on laminin-5 that had been pretreated with mitomycin C to prevent cell proliferation. Transfection of cells with siRNA targeting integrin β4, the HLA-I heavy chain, or β2-microglobulin decreased the extent of cell migration relative to that of cells transfected with control siRNA and treated with mitomycin C (Fig. 7B). These results are consistent with previous findings showing that knockdown of β2-microglobulin inhibits cell migration and invasion (29). These data demonstrate that integrin β4–mediated cell migration is dependent on its interactions with HLA-I molecules.
Discussion
Here, we demonstrated a previously uncharacterized physical and functional association between the integrin β4 subunit and HLA-I. Our results suggest that heterotypic, cis interactions between HLA-I and the integrin β4 subunit are essential for transducing intracellular signals that elicit the migration and proliferation of endothelial cells, because the knockdown of HLA-I impaired laminin-5–induced integrin β4 signaling and inhibited the migration of endothelial cells. Likewise, in the absence of integrin β4, antibody-induced cross-linking of HLA-I molecules failed to elicit proliferative signaling.
Deletion of the cytoplasmic tail, but not the extracellular domain, of the heavy chain of HLA-I abolished its interaction with the integrin β4 subunit. The importance of the cytoplasmic domain of the HLA-I heavy chain was also supported by our finding that it was required for HLA-I–mediated phosphorylation of target proteins and cell proliferation. The cytoplasmic tail of HLA-I mediates several important functions, including endocytosis, degradation, and trafficking (30–33). Previous studies by Gur et al. indicate that the cross-linking of HLA-I molecules that lack cytoplasmic domains can still stimulate activation signals in T lymphocytes (34). Integrin β4 is not found on mature T or B cells (35), which suggests that the proximal signaling events in lymphocytes and endothelial cells are different and are likely related to the co-receptors that partner with MHC-I molecules to elicit signaling events. In this respect, immune and nonimmune receptors physically associate with cell surface MHC-I molecules, and these include the insulin receptor, EGFR, CD25, CD1a, CD71, CD82, and CD8 (7).
Our demonstration of a mutual dependency between integrin β4 and HLA-I to stimulate migration and proliferation raises the key question of how signal transduction is orchestrated through these molecular interactions. Our data are consistent with a model whereby cross-linking of HLA-I molecules with antibodies leads to the recruitment of integrin β4 and the subsequent activation of intracellular signals involving Src, FAK, PI3K-Akt, and ERK. Merdek et al. showed that dimerization of the cytoplasmic domain of the integrin β4 subunit leads to the activation of Src signaling (36), which is an early event in the HLA-I signaling pathway (8, 37). Alternatively, the cytoplasmic domain of integrin β4 interacts with FAK directly, which can lead to the phosphorylation of FAK (20).
HLA-I–mediated transduction of signals is most likely of importance in the setting of antibody-mediated rejection of allografts, in which antibody-mediated modifications of endothelial cell function are thought to promote the development of chronic rejection and transplant vasculopathy (3, 8–12, 38, 39). Associations between the integrin β4 subunit and HLA-I might also play an important function in tumor angiogenesis and cancer progression. Given the role of the integrin β4 subunit in promoting the migration, proliferation, invasion, and metastasis of tumor cells (21, 22, 27, 40), an increased abundance of HLA may promote angiogenesis by augmenting integrin β4–dependent signaling. Indeed, an increase in the amounts of the HLA-I heavy chain and β2-microglobulin was found in gastric, squamous, and renal cell carcinomas (29, 41, 42), which correlates with the extent of activation of the PI3K and ERK pathways and with tumor progression (43). On the other hand, the reduced abundance of HLA-I in non–small-cell lung cancer and breast cancer is associated with improved survival (44, 45); a reduction in the abundance of HLA may inhibit tumor angiogenesis by impairing integrin β4–dependent signaling.
In addition to tumor angiogenesis and transplant vasculopathy, the interaction between HLA-I and integrin β4 might be important in neuronal development. Studies by Van der Zee et al. demonstrated that conditional deletion of the integrin β4 subunit in Schwann cells leads to delayed regeneration of axons (46). Similarly, Oliveira et al. found that the lack of surface MHC-I in mice deficient in TAP1 (transporter associated with antigen processing–1) or β2-microglobulin impeded the ability of neurons to regenerate axons (47). In addition, Sabha et al. showed that the mouse strain A/J, which has a stronger axonal growth potential, shows a clear increase in the abundance of MHC-I, whereas C57BL/6J mice, which have a comparatively poor regenerative potential, display a lower extent of increase in the amount of MHC-I in the spinal cord after injury (48). Our study provides a potential mechanistic explanation for these studies. Specifically, it is plausible that the integrin β4 subunit and HLA-I form a signaling complex that mediates axonal growth in neurons. Our elucidation of the interaction between the integrin β4 subunit and HLA-I may lead to the development of therapeutic strategies to prevent antibody-mediated transplant rejection and tumor progression.
Materials and Methods
Antibodies
The murine hybridoma (HB-95) cell line, which produces the monoclonal antibody W6/32, was purchased from the American Type Culture Collection, and W6/32 was purified by protein A/G agarose affinity chromatography. W6/32 recognizes a conformational epitope on all HLA-A, B, and C heavy chains when they are in association with β2-microglobulin (49). The antibody EMR8-5, which recognizes the denatured heavy chain of HLA-I and was used for Western blotting analysis, was obtained from Medical and Biological Laboratories. The allele-specific monoclonal antibody against HLA-A2 (H37) and the monoclonal antibody against HLA-B56 were gifts from J. Lee (One Lambda Inc.). The mouse IgG isotype control and antibodies against vinculin and β2-microglobulin were purchased from Sigma-Aldrich. Rabbit antibodies against phosphorylated Akt (pAkt, Ser473), pSrc (Tyr416), pERK (Thr202/Tyr236), mTOR, Akt, ERK, and integrin β3 were purchased from Cell Signaling Technology. Antibodies against integrins β1, β3, and β4 were purchased from BD Biosciences and were used in flow cytometry studies. Polyclonal antibody against pFAK (Tyr397) was obtained from BioSource International. Rabbit polyclonal antibodies against FAK, c-Src, β-tubulin, integrin β1, and integrin β4, as well as protein A/G plus agarose, were obtained from Santa Cruz Biotechnology. Fluorescein isothiocyanate (FITC)–conjugated goat antibody against mouse IgG was purchased from Jackson ImmunoResearch Laboratories.
Cell culture
Primary human aortic endothelial cells were purchased from Cambrex Corporation or were isolated from human aortas, as previously described (50). Cells were cultured in M199 complete medium containing 1 mM sodium pyruvate (Irvine Scientific), penicillin (100 U/ml, Invitrogen), streptomycin (100 μg/ml, Invitrogen), 20% (v/v) fetal bovine serum (FBS, HyClone), heparin (90 μg/ml, Sigma-Aldrich), and endothelial cell growth supplement (20 μg/ml, BD Biosciences). The HLA-A and B genotypes of the endothelial cells used in this study were EC1 (HLA-A*02, A*11, B*44, and B*56), EC2 (HLA-A*01, A*33, B*44, and B*57), and EC3 (HLA-A*01, A*02, B*08, and B*60). Endothelial cells were used for experiments during passages 3 to 8.
Coimmunoprecipitations
Endothelial cells (∼1.5 × 106 to 3 × 106) were treated with the F(ab′)2 fragments of W6/32 or control mouse IgG for 20 min. The F(ab′)2 fragments of W6/32 were used instead of the whole IgG to prevent the Fc fragment from interfering with immunoprecipitation with protein A/G agarose beads. Our previous studies showed that stimulation of endothelial cells with the F(ab′)2 fragment of W6/32 gives results comparable to those observed in cells stimulated with the intact IgG (14, 51, 52). Cells were lysed in buffer containing 1% CHAPS, 150 mM NaCl, 50 mM tris-HCl (pH 7.4), 50 mM NaF, 1.5 mM Na3VO4, 0.5 mM phenylmethylsulfonyl fluoride (PMSF), aprotinin (0.2 μg/ml), and leupeptin (0.5 μg/ml), and cell debris was pelleted by centrifugation at 10,000g for 2 min at 4°C. Antibodies against the integrin β4 subunit or HLA-I (W6/32) were added to the cleared supernatant, and samples were placed on the rotator overnight, after which they were incubated with protein A/G beads for 3 hours. Cells infected with adenoviruses were treated with antibodies and immunoprecipitated with Sepharose 4B beads (GE Healthcare) coupled to antibody against the integrin β4 subunit (BD Biosciences) according to the manufacturer's directions. Immunoprecipitates were washed three times in buffer containing 40 mM Hepes (pH 7.5), 120 mM NaCl, 1 mM EDTA, 10 mM β-glycerophosphate, 50 mM NaF, 1.5 mM Na3VO4, 0.3% CHAPS, aprotinin (0.2 μg/ml), and leupeptin (0.5 μg/ml), and samples were resolved on an SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel. Of the total amount of protein used in the immunoprecipitation, 5% was loaded onto the gel as a lysate control.
Western blotting analysis
Cells were grown in conditions of low serum concentration (2% FBS) for 12 hours, stimulated with W6/32 for 10, 20, or 30 min, and lysed in RIPA-like buffer [250 mM NaCl, 0.1% SDS, 2 mM dithiothreitol (DTT), 0.5% NP-40, 50 mM tris-HCl (pH 7.4), 50 mM NaF, 1.5 mM Na3VO4, 0.5 mM PMSF, aprotinin (0.2 μg/ml), and leupeptin (0.5 μg/ml)]. Cell lysates were resolved by SDS-PAGE and proteins were transferred overnight onto Immobilon-P membranes (Millipore). Membranes were blocked with 5% milk in tris-buffered saline containing 1% Tween-20 (TBST) for 1 hour at room temperature and then incubated overnight with the appropriate primary antibodies. Immunoreactive bands were visualized with horseradish peroxidase (HRP)–conjugated secondary antibodies. Protein bands were scanned and quantitated with the ImageJ analysis program (http://rsb.info.nih.gov/ij/).
Confocal microscopy
Endothelial cells were plated onto coverslips coated with laminin-5 or collagen I and were fixed with freshly prepared 4% paraformaldehyde for 20 min without permeabilization. This fixation method is reported to primarily label cell surface proteins (53). The fixed cells were then incubated with W6/32 and rabbit antibody against integrin β4 followed by FITC-conjugated goat antibody against mouse IgG (Santa Cruz Biotechnology) and Rhodamine Red-X–conjugated goat antibody against rabbit IgG (Invitrogen). Basal and apical membrane locations were determined visually in the Z plane by light-field microscopy. The colocalization of integrin β4 and HLA-I at the basal membrane was examined with a Zeiss LSM 5 Pascal laser scanning microscope with a Plan-Apochromat 63×/1.4 oil DIC (differential interference contrast) objective lens. Images were acquired by the sequential scanning mode to avoid bleed-through. To perform quantitative statistical colocalization, we analyzed merged images with ImageJ software with the JACoP plug-in, according to the algorithm of Costes et al. (24).
siRNA design and transfection of cells
The three integrin β4–specific siRNAs (5′-GAGAGCAGCUUCCAAAUCA-3′, 5′-CAGAAGAUGUGGAUGAGUU-3′, and 5′-GAGCUGCACGGAGUGUGUC-3′) (54), the siRNAs against the HLA-I heavy chain targeting the HLA-A and B alleles (5′-GCAGAGAUACACCUGCCAU-3′ and 5′-GAGCUCAGAUAGAAAAGGA-3′) (28), the β2-microglobulin–specific siRNA (5′-UUGCUAUGUGUCUGGGUUU-3′) (43), and the negative control siRNA against firefly GL2 luciferase (5′-CGUACGCGGAAUACUUCGA-3′) (55) were synthesized by Dharmacon Inc. Endothelial cells were cultured from 60 to 80% confluence and transfected with siRNA (100 nM) with the TransIT-TKO transfection reagent (Mirus Bio); control cells were treated with transfection reagent alone according to the manufacturer's instructions. Experiments were conducted 48 hours (or as noted) after transfection.
Construction and expression of HLA mutant proteins
To generate the HLA mutant proteins C-HLA-A2 and E-HLA-A2, we extracted mRNA from primary endothelial cells carrying the HLA-A*0201 allele and reverse-transcribed the mRNA to generate complementary DNA (cDNA). To generate C-HLA-A2, we amplified the cDNA encoding the extracellular and transmembrane regions (corresponding to amino acid residues 1 to 338) of HLA-A2 by polymerase chain reaction (PCR) assay with the following primers: primer 1 (forward), 5′-gggctagccATGGCCGTCATGGCG-3′, and primer 2 (reverse), 5′-ggagATCTGAGCTCTTCCTCCTCCACAT-3′. Purified PCR products were digested with Nhe I and Bgl II and subcloned into Nhe I and Bgl II sites of pEGFP-N1 to generate constructs that encoded HLA-mutant-GFP fusion proteins. To generate E-HLA-2, we amplified the cDNA encoding the leader peptide (corresponding to amino acid residues 1 to 23) with the following primers: primer 1 (forward, as described earlier) and primer 3 (reverse), 5′-ggggatCCCAGGTCTGGGTCAGG-3′, digested with Nhe I and Bam HI and subcloned into pEGFP, which had been digested with Nhe I and Bam HI. Next, the cDNA encoding the transmembrane and intracellular domains of HLA-A2 (corresponding to amino acid residues 304 to 365) was amplified with primer 4 (forward, 5′-ggtgtaca-GCCCACCATCCCCATC-3′) and primer 5 (reverse, 5′-gactagTCACACTTTACAAGCTGTGAGA-3′), digested with Brs GI and Spe I, and inserted into pEGFP-N1 that had been digested with Nhe I and Xba I, which had the leader peptide inserted. All of the plasmids were sequenced to confirm that the inserts were correctly inserted and contained no mutations. The cDNAs encoding the HLA-mutant-GFP fusion proteins were then subcloned into the adenovirus-based vector pAd/PL-DEST. Recombinant adenoviruses encoding the HLA mutant proteins were generated and purified as described by the manufacturer (Invitrogen), and titered by analyzing GFP fluorescence by flow cytometry. To express the HLA-A2 mutant proteins, we incubated primary human aortic endothelial cells (EC2) that do not carry an endogenous HLA-A2 allele with the adenoviruses at a multiplicity of infection (MOI) of 2:1.
Cell proliferation assays
Endothelial cells were transfected with the appropriate siRNA and then starved overnight in M199 medium containing 2% FBS. The transfected cells were labeled with CFSE (2 μM, Invitrogen) at 37°C for 15 min, washed twice in warm phosphate-buffered saline (PBS), and stimulated with W6/32 or control mouse IgG for 72 hours. The cells were detached with 0.25% trypsin and 0.05% EDTA, washed, and analyzed by flow cytometry. A decrease in the amount of intracellular fluorescence of CFSE was an indicator of cell proliferation. Data were analyzed with CellQuest Pro (BD Biosciences) and ModFit LT software (Verity Software House). Cell proliferation was calculated with the Proliferation Wizard Model (Verity Software House). In other experiments, endothelial cells were infected with adenoviruses encoding C-HLA-A2 or E-HLA-A2 and then stimulated overnight with control mouse IgG, W6/32, antibody against GFP, or antibody against HLA-A2 (1 μg/ml) in M199 medium containing 2% FBS and 15 μM BrdU. Cells were fixed and then incubated with an antibody against BrdU, and a minimum of 10,000 gated events (cells) were acquired on a FACSCalibur flow cytometer. Cell proliferation was determined by counting the number of BrdU-containing cells.
In vitro wound-healing assay
The in vitro wound-healing assay was performed as described previously (27) with slight modifications. Briefly, cells transfected with siRNAs against integrin β4 or HLA-I or with control siRNA were plated on dishes coated with laminin-5. For some experiments, the cells were pretreated with mitomycin C (10 μg/ml) for 2 hours to inhibit cell proliferation (56). The cells were washed with PBS and incubated in medium without mitomycin C. Cell monolayers were scratched with a pipette tip and stimulated with FGF for 12 hours. The cells were stained with Wright-Giemsa (Sigma-Aldrich), and wound closure was monitored by microscopy. A total of nine wounds were measured for each test condition. The degree of wound closure was determined by calculating the percentage of the area covered by migrating cells in the initial wound.
Statistical analysis
The one-way analysis of variance (ANOVA) with Fisher's least significant difference (LSD) test was used for comparisons, with P < 0.05 considered significant. Data are presented as mean ± SEM.
Supplementary Material
Fig. S1. Immunoprecipitation with an integrin β1–specific antibody fails to pull down integrin β4 and the HLA-I heavy chain in endothelial cells.
Fig. S2. Knockdown of integrin β4 blocks HLA-I–mediated phosphorylation of target proteins in endothelial cells.
Fig. S3. The cytoplasmic domain of the HLA-I heavy chain is required for HLA-I–mediated cell proliferation.
Fig. S4. Knockdown of the HLA-I heavy chain inhibits the phosphorylation of ERK.
Acknowledgments
We thank M. Edidin for his valuable comments.
Funding: This work was supported by the National Institute of Allergy and Infectious Diseases grant RO1 AI 042819 (E.F.R.); NIH grant U01AI077821 (E.F.R.); the National Heart, Lung and Blood Institute grant RO1 HL 090995 (E.F.R.); and NIH grants R0-1 DK 55003 (E.R.), R0-1 DK56930 (E.R.), R21CA137292 (E.R.), and P30 DK41301 (E.R.).
Footnotes
Author contributions: X.Z. and E.F.R. carried out project planning, experimental design, experimental work, data analysis, and manuscript preparation; E.R. carried out experimental design, data analysis, and manuscript preparation; and E.F.R. supervised the project.
Competing interests: The authors declare that they have no competing interests.
References
- 1.Reed EF, Demetris AJ, Hammond E, Itescu S, Kobashigawa JA, Reinsmoen NL, Rodriguez ER, Rose M, Stewart S, Suciu-Foca N, Zeevi A, Fishbein MC. International Society for Heart and Lung Transplantation, Acute antibody-mediated rejection of cardiac transplants. J Heart Lung Transplant. 2006;25:153–159. doi: 10.1016/j.healun.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 2.Singh N, Pirsch J, Samaniego M. Antibody-mediated rejection: Treatment alternatives and outcomes. Transplant Rev. 2009;23:34–46. doi: 10.1016/j.trre.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Reed EF. Effect of antibodies on endothelium. Am J Transplant. 2009;9:2459–2465. doi: 10.1111/j.1600-6143.2009.02819.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leffell MS, Zachary AA. Antiallograft antibodies: Relevance, detection, and monitoring. Curr Opin Organ Transplant. 2010;15:2–7. doi: 10.1097/MOT.0b013e3283342798. [DOI] [PubMed] [Google Scholar]
- 5.Colvin RB. Pathology of chronic humoral rejection. Contrib Nephrol. 2009;162:75–86. doi: 10.1159/000170814. [DOI] [PubMed] [Google Scholar]
- 6.Stegall MD, Gloor JM. Deciphering antibody-mediated rejection: New insights into mechanisms and treatment. Curr Opin Organ Transplant. 2010;15:8–10. doi: 10.1097/MOT.0b013e3283342712. [DOI] [PubMed] [Google Scholar]
- 7.Arosa FA, Santos SG, Powis SJ. Open conformers: The hidden face of MHC-I molecules. Trends Immunol. 2007;28:115–123. doi: 10.1016/j.it.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 8.Jin YP, Korin Y, Zhang X, Jindra PT, Rozengurt E, Reed EF. RNA interference elucidates the role of focal adhesion kinase in HLA class I-mediated focal adhesion complex formation and proliferation in human endothelial cells. J Immunol. 2007;178:7911–7922. doi: 10.4049/jimmunol.178.12.7911. [DOI] [PubMed] [Google Scholar]
- 9.Jindra PT, Jin YP, Rozengurt E, Reed EF. HLA class I antibody-mediated endothelial cell proliferation via the mTOR pathway. J Immunol. 2008;180:2357–2366. doi: 10.4049/jimmunol.180.4.2357. [DOI] [PubMed] [Google Scholar]
- 10.Jindra PT, Jin YP, Jacamo R, Rozengurt E, Reed EF. MHC class I and integrin ligation induce Erk activation via an mTORC2-dependent pathway. Biochem Biophys Res Commun. 2008;369:781–787. doi: 10.1016/j.bbrc.2008.02.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamakuchi M, Kirkiles-Smith NC, Ferlito M, Cameron SJ, Bao C, Fox-Talbot K, Wasowska BA, Baldwin WM, III, Pober JS, Lowenstein CJ. Antibody to human leukocyte antigen triggers endothelial exocytosis. Proc Natl Acad Sci U S A. 2007;104:1301–1306. doi: 10.1073/pnas.0602035104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uehara S, Chase CM, Cornell LD, Madsen JC, Russell PS, Colvin RB. Chronic cardiac transplant arteriopathy in mice: Relationship of alloantibody, c4d deposition and neointimal fibrosis. Am J Transplant. 2007;7:57–65. doi: 10.1111/j.1600-6143.2006.01599.x. [DOI] [PubMed] [Google Scholar]
- 13.Hirohashi T, Uehara S, Chase CM, DellaPelle P, Madsen JC, Russell PS, Colvin RB. Complement independent antibody-mediated endarteritis and transplant arteriopathy in mice. Am J Transplant. 2010;10:510–517. doi: 10.1111/j.1600-6143.2009.02958.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jindra PT, Hsueh A, Hong L, Gjertson D, Shen XD, Gao F, Dang J, Mischel PS, Baldwin WM, III, Fishbein MC, Kupiec-Weglinski JW, Reed EF. Anti-MHC class I antibody activation of proliferation and survival signaling in murine cardiac allografts. J Immunol. 2008;180:2214–2224. doi: 10.4049/jimmunol.180.4.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fehlmann M, Peyron JF, Samson M, Van Obberghen E, Brandenburg D, Brossette N. Molecular association between major histocompatibility complex class I antigens and insulin receptors in mouse liver membranes. Proc Natl Acad Sci U S A. 1985;82:8634–8637. doi: 10.1073/pnas.82.24.8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramalingam TS, Chakrabarti A, Edidin M. Interaction of class I human leukocyte antigen (HLA-I) molecules with insulin receptors and its effect on the insulin-signaling cascade. Mol Biol Cell. 1997;8:2463–2474. doi: 10.1091/mbc.8.12.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harel-Bellan A, Krief P, Rimsky L, Farrar WL, Mishal Z. Flow cytometry resonance energy transfer suggests an association between low-affinity interleukin 2 binding sites and HLA class I molecules. Biochem J. 1990;268:35–40. doi: 10.1042/bj2680035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shatz CJ. MHC class I: An unexpected role in neuronal plasticity. Neuron. 2009;64:40–45. doi: 10.1016/j.neuron.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miranti CK, Brugge JS. Sensing the environment: A historical perspective on integrin signal transduction. Nat Cell Biol. 2002;4:E83–E90. doi: 10.1038/ncb0402-e83. [DOI] [PubMed] [Google Scholar]
- 20.Abdel-Ghany M, Cheng HC, Elble RC, Pauli BU. Focal adhesion kinase activated by β4 integrin ligation to mCLCA1 mediates early metastatic growth. J Biol Chem. 2002;277:34391–34400. doi: 10.1074/jbc.M205307200. [DOI] [PubMed] [Google Scholar]
- 21.Giancotti FG. Targeting integrin β4 for cancer and anti-angiogenic therapy. Trends Pharmacol Sci. 2007;28:506–511. doi: 10.1016/j.tips.2007.08.004. [DOI] [PubMed] [Google Scholar]
- 22.Uemura T, Shiozaki K, Yamaguchi K, Miyazaki S, Satomi S, Kato K, Sakuraba H, Miyagi T. Contribution of sialidase NEU1 to suppression of metastasis of human colon cancer cells through desialylation of integrin β4. Oncogene. 2009;28:1218–1229. doi: 10.1038/onc.2008.471. [DOI] [PubMed] [Google Scholar]
- 23.Homan SM, Mercurio AM, LaFlamme SE. Endothelial cells assemble two distinct α6β4-containing vimentin-associated structures: Roles for ligand binding and the β4 cytoplasmic tail. J Cell Sci. 1998;111:2717–2728. doi: 10.1242/jcs.111.18.2717. [DOI] [PubMed] [Google Scholar]
- 24.Costes SV, Daelemans D, Cho EH, Dobbin Z, Pavlakis G, Lockett S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys J. 2004;86:3993–4003. doi: 10.1529/biophysj.103.038422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zohar O, Reiter Y, Bennink JR, Lev A, Cavallaro S, Paratore S, Pick CG, Brooker G, Yewdell JW. Cutting edge: MHC class I–Ly49 interaction regulates neuronal function. J Immunol. 2008;180:6447–6451. doi: 10.4049/jimmunol.180.10.6447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lyons AB. Analysing cell division in vivo and in vitro using flow cytometric measurement of CFSE dye dilution. J Immunol Methods. 2000;243:147–154. doi: 10.1016/s0022-1759(00)00231-3. [DOI] [PubMed] [Google Scholar]
- 27.Nikolopoulos SN, Blaikie P, Yoshioka T, Guo W, Giancotti FG. Integrin β4 signaling promotes tumor angiogenesis. Cancer Cell. 2004;6:471–483. doi: 10.1016/j.ccr.2004.09.029. [DOI] [PubMed] [Google Scholar]
- 28.Figueiredo C, Seltsam A, Blasczyk R. Class-, gene-, and group-specific HLA silencing by lentiviral shRNA delivery. J Mol Med. 2006;84:425–437. doi: 10.1007/s00109-005-0024-2. [DOI] [PubMed] [Google Scholar]
- 29.Chen CH, Su CY, Chien CY, Huang CC, Chuang HC, Fang FM, Huang HY, Chen CM, Chiou SJ. Overexpression of β2-microglobulin is associated with poor survival in patients with oral cavity squamous cell carcinoma and contributes to oral cancer cell migration and invasion. Br J Cancer. 2008;99:1453–1461. doi: 10.1038/sj.bjc.6604698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Gall S, Heard JM, Schwanz O. Analysis of Nef-induced MHC-I endocytosis. Res Virol. 1997;148:43–47. doi: 10.1016/s0923-2516(97)81912-7. [DOI] [PubMed] [Google Scholar]
- 31.Singh RK, Lau D, Noviello CM, Ghosh P, Guatelli JC. An MHC-I cytoplasmic domain/HIV-1 Nef fusion protein binds directly to the μ subunit of the AP-1 endosomal coat complex. PLoS One. 2009;4:e8364. doi: 10.1371/journal.pone.0008364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Capps GG, Van Kampen M, Ward CL, Zúñiga MC. Endocytosis of the class I major histocompatibility antigen via a phorbol myristate acetate-inducible pathway is a cell-specific phenomenon and requires the cytoplasmic domain. J Cell Biol. 1989;108:1317–1329. doi: 10.1083/jcb.108.4.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Basha G, Lizée G, Reinicke AT, Seipp RP, Omilusik KD, Jefferies WA. MHC class I endosomal and lysosomal trafficking coincides with exogenous antigen loading in dendritic cells. PLoS One. 2008;3:e3247. doi: 10.1371/journal.pone.0003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gur H, el-Zaatari F, Geppert TD, Wacholtz MC, Taurog JD, Lipsky PE. Analysis of T cell signaling by class I MHC molecules: The cytoplasmic domain is not required for signal transduction. J Exp Med. 1990;172:1267–1270. doi: 10.1084/jem.172.4.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339–362. doi: 10.1146/annurev.immunol.021908.132554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merdek KD, Yang X, Taglienti CA, Shaw LM, Mercurio AM. Intrinsic signaling functions of the β4 integrin intracellular domain. J Biol Chem. 2007;282:30322–30330. doi: 10.1074/jbc.M703156200. [DOI] [PubMed] [Google Scholar]
- 37.Jin YP, Singh RP, Du ZY, Rajasekaran AK, Rozengurt E, Reed EF. Ligation of HLA class I molecules on endothelial cells induces phosphorylation of Src, paxillin, and focal adhesion kinase in an actin-dependent manner. J Immunol. 2002;168:5415–5423. doi: 10.4049/jimmunol.168.11.5415. [DOI] [PubMed] [Google Scholar]
- 38.Coupel S, Leboeuf F, Boulday G, Soulillou JP, Charreau B. RhoA activation mediates phosphatidylinositol 3-kinase-dependent proliferation of human vascular endothelial cells: An alloimmune mechanism of chronic allograft nephropathy. J Am Soc Nephrol. 2004;15:2429–2439. doi: 10.1097/01.ASN.0000138237.42675.45. [DOI] [PubMed] [Google Scholar]
- 39.Lepin EJ, Zhang Q, Zhang X, Jindra PT, Hong LS, Ayele P, Peralta MV, Gjertson DW, Kobashigawa JA, Wallace WD, Fishbein MC, Reed EF. Phosphorylated S6 ribosomal protein: A novel biomarker of antibody-mediated rejection in heart allografts. Am J Transplant. 2006;6:1560–1571. doi: 10.1111/j.1600-6143.2006.01355.x. [DOI] [PubMed] [Google Scholar]
- 40.Shaw LM, Rabinovitz I, Wang HHF, Toker A, Mercurio AM. Activation of phosphoinositide 3-OH kinase by the α6β4 integrin promotes carcinoma invasion. Cell. 1997;91:949–960. doi: 10.1016/s0092-8674(00)80486-9. [DOI] [PubMed] [Google Scholar]
- 41.Rasmuson T, Grankvist K, Ljungberg B. Serum β2-microglobulin and prognosis of patients with renal cell carcinoma. Acta Oncol. 1996;35:479–482. doi: 10.3109/02841869609109926. [DOI] [PubMed] [Google Scholar]
- 42.Ueda Y, Ishikawa K, Shiraishi N, Yokoyama S, Kitano S. Clinical significance of HLA class I heavy chain expression in patients with gastric cancer. J Surg Oncol. 2008;97:451–455. doi: 10.1002/jso.20985. [DOI] [PubMed] [Google Scholar]
- 43.Nomura T, Huang WC, Zhau HE, Wu D, Xie Z, Mimata H, Zayzafoon M, Young AN, Marshall FF, Weitzmann MN, Chung LW. β2-Microglobulin promotes the growth of human renal cell carcinoma through the activation of the protein kinase A, cyclic AMP–responsive element-binding protein, and vascular endothelial growth factor axis. Clin Cancer Res. 2006;12:7294–7305. doi: 10.1158/1078-0432.CCR-06-2060. [DOI] [PubMed] [Google Scholar]
- 44.Ramnath N, Tan D, Li Q, Hylander BL, Bogner P, Ryes L, Ferrone S. Is down-regulation of MHC class I antigen expression in human non-small cell lung cancer associated with prolonged survival? Cancer Immunol Immunother. 2006;55:891–899. doi: 10.1007/s00262-005-0085-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Madjd Z, Spendlove I, Pinder SE, Ellis IO, Durrant LG. Total loss of MHC class I is an independent indicator of good prognosis in breast cancer. Int J Cancer. 2005;117:248–255. doi: 10.1002/ijc.21163. [DOI] [PubMed] [Google Scholar]
- 46.Van der Zee CE, Kreft M, Beckers G, Kuipers A, Sonnenberg A. Conditional deletion of the Itgb4 integrin gene in Schwann cells leads to delayed peripheral nerve regeneration. J Neurosci. 2008;28:11292–11303. doi: 10.1523/JNEUROSCI.3068-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Oliveira AL, Thams S, Lidman O, Piehl F, Hökfelt T, Karre K, Lindå H, Cullheim S. A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc Natl Acad Sci U S A. 2004;101:17843–17848. doi: 10.1073/pnas.0408154101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sabha M, Jr, Emirandetti A, Cullheim S, De Oliveira AL. MHC I expression and synaptic plasticity in different mice strains after axotomy. Synapse. 2008;62:137–148. doi: 10.1002/syn.20475. [DOI] [PubMed] [Google Scholar]
- 49.Parham P, Barnstable CJ, Bodmer WF. Use of a monoclonal antibody (W6/32) in structural studies of HLA-A,B,C antigens. J Immunol. 1979;123:342–349. [PubMed] [Google Scholar]
- 50.Navab M, Hough GP, Stevenson LW, Drinkwater DC, Laks H, Fogelman AM. Monocyte migration into the subendothelial space of a coculture of adult human aortic endothelial and smooth muscle cells. J Clin Invest. 1988;82:1853–1863. doi: 10.1172/JCI113802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harris PE, Bian H, Reed EF. Induction of high affinity fibroblast growth factor receptor expression and proliferation in human endothelial cells by anti-HLA antibodies: A possible mechanism for transplant atherosclerosis. J Immunol. 1997;159:5697–5704. [PubMed] [Google Scholar]
- 52.Jin YP, Fishbein MC, Said JW, Jindra PT, Rajalingam R, Rozengurt E, Reed EF. Anti-HLA class I antibody–mediated activation of the PI3K/Akt signaling pathway and induction of Bcl-2 and Bcl-xL expression in endothelial cells. Hum Immunol. 2004;65:291–302. doi: 10.1016/j.humimm.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 53.Backhaus R, Zehe C, Wegehingel S, Kehlenbach A, Schwappach B, Nickel W. Unconventional protein secretion: Membrane translocation of FGF-2 does not require protein unfolding. J Cell Sci. 2004;117:1727–1736. doi: 10.1242/jcs.01027. [DOI] [PubMed] [Google Scholar]
- 54.Chung J, Yoon SO, Lipscomb EA, Mercurio AM. The Met receptor and α6β4 integrin can function independently to promote carcinoma invasion. J Biol Chem. 2004;279:32287–32293. doi: 10.1074/jbc.M403809200. [DOI] [PubMed] [Google Scholar]
- 55.Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 56.Bell L, Madri JA. Effect of platelet factors on migration of cultured bovine aortic endothelial and smooth muscle cells. Circ Res. 1989;65:1057–1065. doi: 10.1161/01.res.65.4.1057. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Immunoprecipitation with an integrin β1–specific antibody fails to pull down integrin β4 and the HLA-I heavy chain in endothelial cells.
Fig. S2. Knockdown of integrin β4 blocks HLA-I–mediated phosphorylation of target proteins in endothelial cells.
Fig. S3. The cytoplasmic domain of the HLA-I heavy chain is required for HLA-I–mediated cell proliferation.
Fig. S4. Knockdown of the HLA-I heavy chain inhibits the phosphorylation of ERK.