

Abstract
BACKGROUND
The transmembrane protein with epidermal growth factor and two follistatin motifs, TMEFF2, has been implicated in prostate cancer but its role in this disease is unclear. We recently demonstrated that the tumor suppressor role of TMEFF2 correlates, in part, with its ability to interact with sarcosine dehydrogenase (SARDH) and modulate sarcosine level. TMEFF2 overexpression inhibits sarcosine-induced invasion. Here, we further characterize the functional interaction between TMEFF2 and SARDH and their link with one-carbon (1-C) metabolism and invasion.
METHODS
RNA interference was used to study the effect of SARDH and/or TMEFF2 knockdown (KD) in invasion, evaluated using Boyden chambers. The dependence of invasion on 1-C metabolism was determined by examining sensitivity to methotrexate. Real-time PCR and western blot of subcellular fractions were used to study the effect of SARDH KD or TMEFF2 KD on expression of enzymes involved in one carbon (1-C) metabolism and on TMEFF2 expression and localization. Protein interactions were analyzed by mass-spectrometry. Cell viability and proliferation were measured by cell counting and MTT analysis.
RESULTS
While knocking down SARDH affects TMEFF2 subcellular localization, this effect is not responsible for the increased invasion observed in SARDH KD cells. Importantly, SARDH and/or TMEFF2 KD promote increased cellular invasion, sensitize the cell to methotrexate, render the cell resistant to invasion induced by sarcosine, a metabolite from the folate-mediated 1-C metabolism pathway, and affect the expression level of enzymes involved in that pathway.
CONCLUSIONS
Our findings define a role for TMEFF2 and the folate-mediated 1-C metabolism pathway in modulating cellular invasion.
Keywords: prostate cancer, cellular invasion, metabolism, tissue culture
INTRODUCTION
Cancer cells exhibit an altered metabolism that significantly differs from the metabolism of non-transformed cells. This metabolic reprogramming effect changes the uptake and use of nutrients that allow the cancer cell to maintain high rates of proliferation, growth and survival. Interest in tumor metabolism has increased in recent years as a number of reports have demonstrated that tumor suppressors and oncogenes regulate metabolism and that mutations in metabolic enzymes promote or suppress tumorigenesis [1, 2].
One carbon (1-C) metabolism is comprised of several connected metabolic pathways that promote the folate-mediated transfer of one-carbon units necessary for DNA synthesis and repair. Folate is also essential in its 5-methyl-tetrahydrofolate (THF) form as a methyl donor in the remethylation of homocysteine to methionine, which is then converted to S-adenosylmethionine (SAM), the universal methyl donor [3]. Although a definite link has not been established, evidence suggests a role for 1-C metabolism in tumorigenesis: i) variations in 1-C metabolism genes have been associated with risk of a number of cancers [4–9], ii) deregulation of protein methyltransferases, in charge of posttranslational methylation, has frequently been implicated in cancers [10–14], and iii) changes in DNA methylation patterns are associated with neoplasia [15–18]. In fact, a number of tumor types demonstrate a pattern of global DNA hypomethylation and gene-specific hypermethylation [19–21]. Other findings also support a role for 1-C metabolism in cancer. Recent data demonstrate that glycine decarboxylase (GLDC), an enzyme involved in the glycine/serine pathway, is overexpressed in lung and several other types of tumors [22]. Serine and glycine are two of the main 1-C donors and recent reports demonstrated that serine and glycine synthesis have a role in tumor cell proliferation [23–25]. In fact, overexpression of several other enzymes of the glycine/serine pathway can induce cellular transformation [22]. These data suggest that increased flux through the 1-C-metabolism interrelated pathways contributes to oncogenesis.
Sarcosine and dimethylglycine, the other two main 1-C donors, have also been associated with tumorigenesis, specifically with prostate cancer [26, 27]. Dimethylglycine is formed from betaine in a reaction catalyzed by betaine homocysteine methyltransferase and it is metabolized to sarcosine by the action of dimethylglycine dehydrogenase (DMGDH). Sarcosine donates a methyl group and is metabolized into glycine in a reaction catalyzed by SARDH. The reverse reaction is catalyzed by glycine N-methyl transferase (GNMT). Sarcosine has been recently identified as a key metabolite that is increased in prostate cancer and metastatic disease. Using prostate cancer cell lines, Sreekumar et al [27] demonstrated that the enzymes involved in sarcosine metabolism act as regulators of cell invasion and therefore as potential therapeutic targets for prostate cancer. The addition of sarcosine or knockdown of SARDH in benign prostate epithelial cells enhances invasion. Conversely, lowering the levels of GNMT reduces the invasiveness of DU145 cells [27]. Understanding the role of these enzymes in prostate cancer metabolism may provide valuable information on how prostate cancer is established and develops, and on the potential value of sarcosine as a biomarker.
TMEFF2 is expressed in the embryo [28, 29] and selectively in the adult brain and prostate [30–32]. A role for TMEFF2 in prostate cancer was suggested by studies indicating that TMEFF2 expression is altered in a significant fraction of primary and metastatic prostate tumors [28–33] but its role in this disease is not clear. We have recently described that TMEFF2 functions as a tumor suppressor and that this role correlates, at least in part, with its ability to interact with SARDH and modulate the cellular levels of sarcosine [34]. Using prostate cancer cell lines, we demonstrated that TMEFF2 overexpression in prostate epithelial RWPE cells blocked basal and sarcosine-induced cellular invasion and this effect was dependent on its ability to interact with SARDH and reduce cellular sarcosine levels. These results suggest that the role of TMEFF2 in prostate cancer may be mediated by its ability to modulate the 1-C metabolism and thus DNA synthesis and the methylation potential of the cell. An overview of the interaction of TMEFF2 with the -C metabolism is shown in figure 1.
Figure 1. TMEFF2 interaction with one-carbon metabolism.

The enzymes and metabolites discussed in this study are indicated in this schematic. TMEFF2 is shown interacting with SARDH. Changes in the expression of TMEFF2 affect the activity and/or expression of some of the enzymes of this pathway, and ultimately its balance, possibly affecting the methylation potential of the cell (see text). Negative regulation of GNMT by CH3-THF and of CH3-THF formation by SAM is indicated by dotted lines. SAH: S-adenosyl-homocysteine; Hy: homocysteine. Other abbreviations are as described throughout the manuscript.
The aim of this study was to further characterize the role of the TMEFF2-SARDH interaction in tumorigenesis and in the 1-C metabolism pathway. We demonstrate that reducing the expression of SARDH by sh_RNA promotes localization of TMEFF2 to the cytoskeleton and also results in increased cellular invasion. However, this effect was not altered by simultaneously reducing the expression of TMEFF2, supporting the notion that the effect of SARDH KD on invasion is not mediated by TMEFF2 relocalization. Importantly, while addition of sarcosine promoted increased invasion of prostate cancer 22Rv1 parent cells, it did not affect the invasive ability of the cells expressing sh_RNAs against TMEFF2, SARDH or both. These results indicate that sarcosine metabolism, not merely its concentration, and thus 1-C availability mediated by the function TMEFF2 and SARDH are responsible for the changes in invasion observed in 22Rv1 cells. Supporting this, we found that methotrexate specifically abrogates the increased invasion of 22Rv1 cells expressing sh_TMEFF2, further implicating the folate-mediated 1-C metabolism in the tumor suppressor function of TMEFF2.
MATERIALS AND METHODS
Cell Culture
HEK293T, 22Rv1, LNCaP and RWPE1 cells were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in DMEM, RPMI 1640 or KSF media (Life Technologies, Grand Island, NY). Both media were supplemented with 10% v/v FBS, 100 units/mL penicillin, 100 μg/mL streptomycin, and Amphotericin B (Life Technologies, Grand Island, NY). pLKO.1 vector containing shRNAs to TMEFF2, SARDH and scramble control were obtained from Open Biosystems (Lafayette, CO) and viral stocks were prepared using HEK293T and plasmids psPAX2 and VSV-G (Addgene, Cambridge, MA) for viral packaging. 22Rv1 cells were transduced with each shRNA viral stock and 6 μg/mL Polybrene (Millipore, Billerica, MA). After 48 hours, the transduced 22Rv1 cells were stably selected with 5 μg/mL puromycin (Millipore, Billerica, MA). For siRNA Transient Transfection ON-TARGET Plus siRNA to TMEFF2, SARDH and non-silencing control (Dharmacon, Lafayette, CO) were transiently transfected into 22Rv1 using DharmaFECT 1 (Dharmacon, Lafayette, CO).
Quantitative RT-PCR
Total RNA was extracted with RNeasy mini kit (Qiagen, Valencia, CA) and cDNA was synthesized with SuperScript III First-Strand synthesis system (Life Technologies, Grand Island, NY). Quantitative RT-PCR (qRT-PCR) was performed using the TaqMan Gene Expression Assay (Applied Biosystems, Carlsbad, CA) on the ABI 7300 Real Time PCR system. The following probes were used: a) TMEFF2: Hs01086906_m1 Probe: GAACAGATTATGCAGAGAATGCTAA; b) SARDH: Hs00990344_m1 Probe: TTCTGCCCG AGGAGCACAGGTCATT; c) DMGDH: Hs00203638_m1 Probe: TGAAGTTAAATAAGCCA GCAGACTT; d) GLDC: Hs01580591_m1 Probe: CAGACTCGAGCCAAATATACTGGAG; e) DHFR: Hs00758822_s1 Probe: AGGTCCTCCCGCTGCT GTCATGGTT.
Fractionation and Western Blot Analysis
22Rv1 cells were harvested and washed with phosphate buffered saline (PBS). Cells were lysed either with RIPA buffer or RIPA buffer containing 1% Triton X-100 and Protease inhibitor cocktail (Sigma, St. Louis, MO). After lysis, cells were centrifuged at 18,000xg for 30 minutes at 4°C and the resultant supernatant used for western analysis. For fractionation studies, cells lysed with RIPA buffer containing 1% Triton X-100 were subjected to centrifugation at 100,000xg for 1 hour at 4°C. The supernatant was collected for analysis and the Triton insoluble pellet fraction was resuspended directly in 2x Laemmli Sample Buffer (BIORAD, Hercules, CA). The ProteoExtract Subcellular Proteome Extraction Kit (Calbiochem, Mountain View, CA) was used as an alternative method for cellular fractionation. The proteins were separated on 10% SDS-PAGE gels and transferred to PVDF membranes (BIORAD, Hercules, CA). The blots were blocked in 5% w/v non-fat dry milk in PBS with 0.1% Tween 20 for 3 hours at RT with shaking. Blots were probed with either anti-TMEFF2 (1:1000) (Abcam, Cambridge, MA) or anti-β-Tubulin diluted in 5% PBS-T. Blots were probed with 1:5000 dilution of either goat anti-rabbit-IgG-HRP or goat anti-mouse-IgG-HRP (Santa Cruz Biotechnology, Santa Cruz, CA) in 5% PBS-T. All washes were performed with PBS containing 0.1% Tween 20. Bands were detected using SuperSignal West Pico Chemiluminescent substrate (Pierce, Rockford, IL) and exposure to X-ray film.
Invasion Assay
Cell invasion assays were performed in the presence of 2 μg/ml aphidicolin (Sigma, St. Louis, MO) to prevent proliferation, using 8 micron Matrigel invasion chambers (BD Biosciences, San Jose, CA) and 20% FBS in RPMI as a chemo-attractant. After 48 hours, cells attached to the bottom of the membrane were fixed with 70% ethanol, stained with 0.1% crystal violet, washed and photographed. The inserts were treated with 10% acetic acid and absorbance was measured at 562 nm. When specified, exogenous sarcosine (Sigma, St. Louis, MO) was added to the growth media at a concentration of 100 μM and the cells incubated for the indicated period of time. When specified, 0.5 μM methotrexate (Sigma, St. Louis, MO) was added to the cells in the upper chamber.
Sarcosine Assay
Cells were incubated for 24 hrs in 100 μM sarcosine and lysed in Sarcosine Assay buffer and analyzed for sarcosine levels with Sarcosine Assay Kit (BioVision, Milpitas, CA).
Immunofluorescence
Cells were plated at a density of 10,000 cells/well in a Lab-Tek 8 well chambered slide (Nalge Nunc International; Naperville, IL) and incubated overnight in a humidified 37°C incubator with 5% CO2. Cells were washed in PBS, fixed in 4% paraformaldehyde for 10 minutes and permeabilized with 0.1% Triton-X100 in PBS for 15 minutes. Cells were blocked in 5% goat serum in PBS-T (0.1% Tween 20) followed by incubation with a rabbit polyclonal anti-TMEFF2 antibody (Abcam; Cambridge, MA) diluted 1:50 in 5% goat serum in PBS-T (0.1% Tween 20). Secondary goat anti-rabbit IgG-AF488 (Invitrogen; Carlsbad, CA) antibody was added for 1 hr at a 1:500 dilution in 5% goat serum in PBS-T (0.1% Tween 20) for visualization. AF594 phalloidin to visualize actin was diluted 1:50 in PBS. Slide was preserved with Prolong Gold anti-fade mounting medium (Invitrogen; Carlsbad, CA). Confocal microscopy was utilized for imaging using a Carl Zeiss LSM 510 system.
Statistical Analysis
Data are expressed as mean ±SD. Differences were analyzed using paired, two-tailed t-tests. P values ≤ 0.05 (*) or ≤ 0.01 (**) were considered significant.
RESULTS
SARDH expression regulates TMEFF2 levels
We have recently reported a physical interaction between the TMEFF2 and SARDH proteins that results in reduced levels of cellular sarcosine and partly correlates with the ability of TMEFF2 to function as a tumor suppressor [34]. These results suggested that TMEFF2, together with SARDH, may participate in metabolism of sarcosine, a key intermediate in 1-C metabolism, and led us to propose a model by which the interaction with TMEFF2 affects the activity of SARDH and its ability to metabolize sarcosine.
To further characterize the interaction between SARDH and TMEFF2, we determined whether changes in SARDH levels could affect TMEFF2 expression. shRNA to SARDH (sh_SARDH) was utilized to stably reduce SARDH expression in 22Rv1 prostate cancer cells, and endogenous TMEFF2 expression was analyzed by qRT-PCR and/or western blot analysis. The results from the qRT-PCR analysis indicated that reducing expression of SARDH in 22Rv1 cells (22Rv1/sh_SARDH) had no significant effect on the level of TMEFF2 mRNA (Fig. 2A) while it decreased SARDH mRNA levels to 40% with respect to the results obtained using a scramble shRNA (sh_scramble) as negative control. As expected, shRNA to TMEFF2 (22Rv1/sh_TMEFF2) significantly decreased TMEFF2 mRNA levels to 10–20% of the sh_scramble (22Rv1/sh_scramble). Surprisingly, protein analysis by western blot of RIPA lysates from 22Rv1 cells expressing sh_SARDH revealed a three to four-fold increase in the level of TMEFF2 protein (Fig. 2B). Similar results were obtained using LNCaP cells, a prostate cancer cell line that also expresses detectable levels of endogenous TMEFF2 (Fig. 2B). Addition of exogenous sarcosine to 22Rv1 cells, while it promoted accumulation of intracellular sarcosine (Fig. 2C), it did not affect the amount of TMEFF2 protein as detected by western blot analysis (Fig. 2D). This result ruled out that the increase in TMEFF2 protein levels was due to accumulation of intracellular sarcosine levels as a result of the SARDH knock down --SARDH catalyzes the conversion of sarcosine into glycine and therefore reducing the levels of SARDH results in sarcosine accumulation--. These results indicate that SARDH KD results in increased TMEFF2 protein levels by a post-transcriptional mechanism.
Figure 2. Reducing the levels of SARDH by sh_RNA in 22Rv1 cells modulates TMEFF2 protein levels.

A: Relative mRNA expression of TMEFF2 and SARDH as determined by quantitative RT-PCR. Values were normalized to TATA Box Binding Protein (TBP) mRNA and then to the levels obtained for the corresponding mRNAs from cells expressing the sh_RNA scramble control, which was set to 1. B: Representative western blots indicating levels of TMEFF2 protein in RIPA lysates of 22Rv1/sh_SARDH or /sh_TMEFF2 cells (α= antibody). C: The relative levels of endogenous sarcosine in cells grown in the presence/absence of 100μM sarcosine were measured. A > 3-fold increase in endogenous sarcosine levels demonstrate that sarcosine was indeed internalized in 22Rv1/sh_scramble cells. D: Cells were grown in the presence of 100μM sarcosine and TMEFF2 levels analyzed by western blot (top). The results were quantified by densitometry (bottom) and represent the average of five independent repeats. A > 3-fold increase in endogenous sarcosine levels demonstrate that sarcosine was indeed internalized in those cells (right). E: Representative western blots indicating levels of TMEFF2 from lysates of 22Rv1/sh_SARDH or /sh_TMEFF2 cells obtained using RIPA buffer containing Triton X-100. β-tubulin was used as a loading control for A, B and D. Data shown are mean ± S.D. of at least two independent experiments with multiple replicates. *, p < 0.05, and **, p < 0.01.
Reducing SARDH levels promotes TMEFF2 localization to the cytoskeleton
The results described above indicate that the increase in TMEFF2 protein observed when SARDH levels are low occurs post-transcriptionally. Western blot analysis of lysates from 22Rv1 cells expressing sh_SARDH prepared in Triton X-100 containing RIPA buffer did not show an increase in TMEFF2 protein levels (Fig. 2E) that was observed when the lysates from the same cells were prepared in buffer lacking Triton-X100 (compare Fig. 2B and Fig. 2E), suggesting that a fraction of the TMEFF2 protein fractionates with the Triton X-100 insoluble fraction when SARDH protein levels are low. These results indicate that SARDH KD results in localization of a fraction of TMEFF2 to the cytoskeletal compartment, possibly affecting the stability and therefore the level of the TMEFF2 protein.
To further explore this possibility, we determined the localization of TMEFF2 in 22Rv1 cells expressing sh_SARDH, or the sh_scramble as control, using a commercially available cellular fractionation kit. Using this assay, we obtained four consecutive cellular fractions corresponding to the cytosolic, membrane, nuclear and cytoskeletal protein fractions and the presence/absence of TMEFF2 was analyzed by western blot. As shown in Figure 3A, in lysates from cells expressing the sh_scramble, TMEFF2 was found preferentially in the membrane and nuclear fractions however, in cells expressing the sh_SARDH, a portion of TMEFF2 was detected in the cytoskeletal fraction (Fig. 3A). Lack of reactivity in the previous fraction (nuclear) indicated that the TMEFF2 band detected in the cytoskeletal fraction was not due to cross-contamination of the fractions. Similar results were obtained when lysates were prepared in Triton X-100 containing RIPA buffer and separated into Triton soluble and insoluble fractions. A larger proportion of TMEFF2 was recovered in the Triton X-100 insoluble fraction when the cells expressed the sh_SARDH than from cells expressing the scramble shRNA (Fig. 3B).
Figure 3. A fraction of TMEFF2 localizes to the cytoskeleton in 22Rv1/sh_SARDH cells.

A: Lysates from 22Rv1 cells expressing sh_RNA to SARDH or sh_scramble as a control were subjected to fractionation. Subcellular fractions were analyzed by western blot using an antibody against TMEFF2. Note that localization of TMEFF2 changes in response to reduced levels of SARDH. B: Lysates from 22Rv1 cells expressing sh_RNA to SARDH, TMEFF2 or sh_scramble were prepared in RIPA buffer containing Triton X-100, fractionated via high speed centrifugation into supernatant and pellet fractions and analyzed by western blot analysis. C: Immunofluorescence staining of 22Rv1/sh_SARDH cells. Cells were fixed and stained with anti-TMEFF2 (green) and phalloidin to detect actin (red). Colocalization of TMEFF2 and actin is illustrated by yellow signal.
TMEFF2 interacts with actin and tubulin
The fact that reducing SARDH levels results in increased levels of TMEFF2 in the cytoskeletal fraction suggests that TMEFF2 interacts with components of the cytoskeleton and prompted us to test this notion. We have recently used mass spectrometry to identify TMEFF2 affinity complexes [34]. For this purpose, freestyle 293-F cells (Invitrogen) were stably transfected with TMEFF2-myc-his, a myc-his tagged form of TMEFF2 [34], and TMEFF2 complexes purified using a histidine affinity column, resolved in a polyacrylamide gel and subjected to MALDITOF/MS analysis. Binding to and elution of the TMEFF2 protein from the column was verified by western blot (See [34]). To identify specific TMEFF2 interactors, we compared TMEFF2 and empty vector affinity eluates and chose those bands that were mainly represented only in the TMEFF2 affinity eluates. Furthermore, to qualify as a specific interactor, a protein had to be identified in at least two out of four independent TMEFF2 affinity/MS analyses. β-actin was identified as a TMEFF2 interacting protein with a total of eight peptides displaying a probability based Mowse score of 70 [35]. Additional searches provided up to 10 different peptides corresponding to β-actin (Table I). α-tubulin was identified in three of the four analyses. A total of 10 peptides displaying a probability based Mowse score of 69 were identified for this protein and additional searches provided up to 11 peptides (Table II). The interaction between TMEFF2 and actin was confirmed by immunofluorescence co-localization studies of the endogenous proteins. 22Rv1 cells were incubated with polyclonal TMEFF2 antibody and the localization of the proteins was subsequently visualized by confocal microscopy using Alexa-Fluor 488 conjugated secondary antibody –to detect TMEFF2—and Alexa Fluor 594 Phalloidin for actin. Cells expressing TMEFF2 exhibited fluorescence concentrated at the plasma membrane but also in the cytoplasm in a punctate pattern. Co-localization of the proteins in the cytoplasm was evidenced by overlapping fluorescent signals (yellow; Fig. 3C). The interaction between TMEFF2 and α-tubulin was confirmed by co-immunoprecipitation (co-IP) analysis (Supplemental Fig. S1).
Table I.
β-actin peptides identified by mass spectrometry analysis of TMEFF2 affinity complexes
| Start - End | m/z | Peptide sequence |
|---|---|---|
| 51–61 | 1198.3 | DSYVGDEAQSK |
| 69–84 | 1960.2 | YPIEHGIVTNWDDMEK |
| 85–95 | 1516.1 | IWHHTFYNELR |
| 96–113 | 1953.1 | VAPEEHPVLLTEAPLNPK |
| 148–177 | 3182.9 | TTGIVMDSGDGVTHTVPIYEGYALPHAILR |
| 197–206 | 1132.1 | GYSFTTTAER |
| 239–254 | 1791.1 | SYELPDGQVITIGNER |
| 292–312 | 2214.0 | DLYANTVLSGGTTMYPGIADR |
| 336–359 | 2729.9 | KYSVWIGGSILASLSTFQQMWISK |
| 360–372 | 1500.1 | QEYDESGPSIVHR |
Table II.
α-tubulin peptides identified by mass spectrometry analysis of TMEFF2 affinity complexes
| Start - End | m/z | Peptide sequence |
|---|---|---|
| 65–79 | 1702.3 | AVFVDLEPTVIDEVR |
| 85–96 | 1410.3 | QLFHPEQLITGK |
| 97–112 | 1779.3 | EDAANNYARGHYTIGK |
| 113–121 | 1085.3 | EIIDLVLDR |
| 216–229 | 1718.2 | NLDIERPTYTNLNR |
| 244–264 | 2409.2 | FDGALNVDLTEFQTNLVPYPR |
| 265–280 | 1756.5 | IHFPLATYAPVISAEK |
| 312–320 | 1249.2 | YMACCLLYR |
| 340–352 | 1584.1 | SIQFVDWCPTGFK |
| 374–390 | 1865.1 | AVCMLSNTTAIAEAWAR |
| 403–422 | 2330.0 | AFVHWYVGEGMEEGEFSEAR |
Reducing TMEFF2 or SARDH levels have a similar and non-additive effect on cellular invasion
SARDH KD in benign prostate epithelial RWPE1 cells results in increased endogenous sarcosine levels with a concomitant increase in cellular invasion. Moreover, addition of exogenous sarcosine to those cells promotes cellular invasion. These results led to propose that increased levels of sarcosine promote invasion [27]. We examined whether SARDH KD also promotes increased invasion of prostate cancer 22Rv1 cells by comparing the invasive ability of 22Rv1/sh_SARDH and of 22Rv1/sh_scramble cells. The results indicated that reducing the levels of SARDH by shRNA increased the invasive ability of cells when compared to the cells expressing the sh_scramble control (Fig. 4A). The invasive ability of the 22Rv1 cells was also significantly increased when the level of TMEFF2 was reduced by shRNA (Fig. 4A) or bicalutamide treatment (Supplementary fig. S3), a result that was expected on the basis of the proposed tumor suppressor role of TMEFF2 [34, 36, 37]. To rule out possible dsRNA non-specific effects on gene expression due to activation of the interferon response in the cells expressing the target TMEFF2 or SARDH sh_RNAs, the above experiments were repeated using 22Rv1 cells in which expression of TMEFF2 or SARDH was transiently knocked down using siRNA (these cells are also stably transfected with the scramble non target control shRNA). Similar to the results presented above, siRNA to TMEFF2 or SARDH increased invasion of 22Rv1/scramble_ shRNA cells (Fig. 4B). The invasion experiments were conducted in the presence of aphidicolin to prevent proliferation; however, while we previously observed that TMEFF2 overexpression affects growth of HEK293T cells [34], using MTT assays we have determined that silencing TMEFF2 or SARDH in 22Rv1 cells does not have a significant effect on cell proliferation (Supplemental Fig. S2).
Figure 4. Reducing the levels of TMEFF2 and/or SARDH similarly affects cellular invasion.

A & B: The effect of reducing the levels of TMEFF2 or SARDH using sh_RNA (A) or si_RNA (B) was analyzed using Boyden chambers. Cells adhering to the bottom of the membrane were fixed, stained with crystal violet and photographed. Representative images (left) of at least three independent experiments with multiple replicates, and quantification of the results (right) are shown. C: The effect of reducing the levels of TMEFF2 and SARDH simultaneously using sh_RNA to silence SARDH and si_RNA to silence TMEFF2 was analyzed as described for A and B. 22Rv1 cells expressing the scramble sh_RNA scramble or a non-silencing (NS) si_RNA were used as controls. (*) p < 0.05.
Since SARDH KD in 22Rv1 cells promoted the localization of a fraction of TMEFF2 protein to the cytoskeletal compartment, it was possible that the increase in invasion was not due to increased sarcosine levels but due to cytoskeletal changes. To analyze this possibility, we determined the invasion ability of 22Rv1/sh_SARDH expressing si_RNA to TMEFF2 to simultaneously reduce the expression of TMEFF2 and SARDH. The results indicated that using siRNA to TMEFF2 did not further promote an increase in the invasive ability of the 22Rv1/sh_SARDH cells in comparison to the same cells expressing a non-silencing siRNA control (sh_SARDH + si_NS; Fig. 4C). These results suggest that relocalization of TMEFF2 to the cytoskeleton as a result of reducing SARDH levels does not account for the increase in the invasive phenotype. The fact that, although reducing the levels of either TMEFF2 or SARDH proteins results in increased invasion, reducing the level of both proteins simultaneously does not have an additive effect supports the notion that TMEFF2 and SARDH participate in the same pathway.
Sarcosine metabolism is required for the effects on cellular invasion mediated by changes in the TMEFF2 and SARDH levels
We next examined whether the effect of KD SARDH on cellular invasion was due to the increase in the endogenous sarcosine level that should result from blocking the conversion of sarcosine to glycine. For this purpose, we analyzed whether the addition of exogenous sarcosine would potentiate invasion of cells in which expression of SARDH has been reduced using sh_RNA or siRNA. As shown in figure 5, addition of exogenous sarcosine to the growth media, while it promoted increased invasion of the 22Rv1/sh_scramble or 22Rv1/si_NS control cells, it had no effect in 22Rv1 cells in which TMEFF2 or SARDH was knocked down (using shRNA or siRNA) when compared to the same cells grown in alanine, an isomer of sarcosine (Fig. 5A and 5B). The 22Rv1/sh_scramble cells grown in the presence of sarcosine were more invasive than the 22Rv1/sh_SARDH, ruling out the possibility that the invasive potential of 22Rv1/sh_SARDH had reached a limit. Lack of response to exogenous sarcosine was also observed in 22Rv1/sh_SARDH cells expressing si_RNA to TMEFF2 to simultaneously reduce the expression of TMEFF2 and SARDH. Importantly, addition of exogenous sarcosine to 22Rv1/sh_TMEFF2 or 22Rv1/sh_SARDH cells promoted a five-fold increase in the accumulation of intracellular sarcosine as measured using a Sarcosine Assay Kit (not shown) indicating that lack of response to exogenous sarcosine is not due to a defect in sarcosine uptake. All together these results suggest that metabolism of sarcosine, not solely sarcosine concentration, is required for sarcosine-induced cellular invasion of prostate cancer 22Rv1 cells and that TMEFF2 and SARDH play a role in the same pathway involving metabolism of sarcosine.
Figure 5. TMEFF2 and/or SARDH KD block sarcosine-induced invasion.

A & B: The effect of reducing the levels of TMEFF2 or SARDH on sarcosine induced invasion was analyzed using Boyden chambers. Cells expressing the specified sh_RNA (A) or si_RNA (B) were grown in the presence of 50 μM alanine or sarcosine. Cells adhering to the bottom of the membrane were fixed, stained with crystal violet and photographed. Representative images (left) of at least three independent experiments with multiple replicates, and quantification of the results (right) are shown. C: The effect of reducing the levels of TMEFF2 and SARDH simultaneously was analyzed as described for A and B. (*) p < 0.05.
We did not observe an increase in endogenous sarcosine in 22Rv1/sh_SARDH cells with respect to the 22Rv1/sh_scramble control cells. While it is possible that the changes in sarcosine levels upon SARDH knockdown are below the detection limit of our assay, it is also possible that the activity of SARDH in prostate cancer cells, in which sarcosine levels are known to be higher than in non malignant cells [27], is already compromised; or that knocking down SARDH in prostate cancer cells affects the level or activity of other enzymes involved in the metabolism of sarcosine (i.e. DMGDH) preventing a large increase in sarcosine. Thus while sarcosine steady state levels don’t change, the flux of methyl groups through this pathway is likely to be affected.
Changes in TMEFF2 and SARDH levels affect the expression of some of the enzymes involved in sarcosine metabolism
We next analyzed whether the expression of GNMT or other enzymes involved in sarcosine and the 1-C metabolism is modulated by knockdown of TMEFF2 or SARDH. For this purpose, we performed qRT-PCR expression analysis of the GNMT, DMGDH, GLDC and DHFR transcripts in 22Rv1 cells expressing shRNAs targeting TMEFF2 or SARDH. Reducing the levels of SARDH or TMEFF2 resulted in a significant reduction (30–50%) in the level of DMGDH mRNA. SARDH shRNA did not affect the levels of any of the other three mRNAs measured while reducing TMEFF2 levels significantly reduced GLDC levels by two-fold (Fig. 6). Since these enzymes are important contributors to the 1-C metabolism, these results suggest that 1-C metabolism modulates cellular invasion.
Figure 6. TMEFF2 or SARDH KD affects the expression of enzymes involved in folate mediated 1-C metabolism.
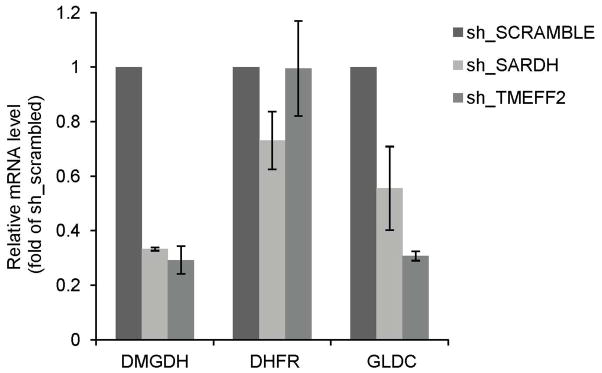
Relative mRNA expression of selected enzymes as determined by quantitative RT-PCR in 22Rv1 cells expressing the specified sh_RNA. Values were normalized to TBP mRNA and then to the levels obtained for the corresponding mRNAs from cells expressing the sh_RNA scramble control, which was set to 1. Data shown are mean ± S.D. of at least three independent experiments with multiple replicates. *, p < 0.05, and **, p < 0.01.
TMEFF2 modulates 1-C metabolism and cellular invasion
DMGDH, SARDH and GLDC are directly involved in reactions that donate 1-C units to drive formation of methyl-THF, important for nucleotide synthesis and for generation of SAM, the universal methyl donor. However, since TMEFF2 is not directly involved in these reactions we further tested its role in the 1-C metabolism by determining whether the production of methyl-THF is necessary for the role of TMEFF2 on modulating invasion. For this purpose, we tested whether the antifolate drug, methotrexate (MTX) could specifically abolish the increase on cellular invasion as a result of TMEFF2 knockdown, by reducing the levels of the THF cofactor. The results show that low doses of MTX specifically abolished invasion induced by TMEFF2 knockdown in 22Rv1 cells (22Rv1/sh_TMEFF2) but had no effect on control 22Rv1/sh_scramble cells (Fig. 7A). The MTX dose used did not differentially affect viability or proliferation of 22Rv1/sh_TMEFF2 or control cells indicating that the effect of MTX on invasion of 22Rv1/sh_TMEFF2 cells was not due to an effect of the drug on cell numbers (Fig. 7B). These results further support a link between TMEFF2, 1-C metabolism and invasion.
Figure 7. The anti-folate drug methotrexate inhibits invasion of 22Rv1 in which TMEFF2 or SARDH are knockdown.

A: Cells expressing sh_TMEFF2, sh_SARDH or sh_scramble as control, were placed in Boyden chambers and exposed to ± 0.5 μM methotrexate. After 48 hours, cells adhering to the bottom of the membrane were fixed, stained with crystal violet and photographed. Representative images (left) and quantification of the results (right) are shown. B: MTT assay of the same cells to determine proliferation rate in the presence/absence of methotrexate. C: Trypan Blue exclusion assay of the same cells grown on serum-starved media to determine viability under the conditions used for the Boyden chamber assay. For both assays, data shown are mean ± S.D. of at least three independent experiments with multiple replicates. *, p < 0.05, and **, p < 0.01.
*, p < 0.05, and **, p < 0.01.
DISCUSSION
We have previously reported that the tumor suppressor activity of TMEFF2 correlates, at least in part, with its ability to bind SARDH and modulate the levels of cellular sarcosine. Overexpression of TMEFF2 in benign prostate epithelial RWPE1 cells inhibits sarcosine-induced migration and invasion and results in decreased levels of sarcosine with respect to control cells [34]. However, the mechanism(s) underlying the effect of TMEFF2, SARDH and sarcosine on the invasive properties of the cells are not clear. The results presented here indicate that reducing expression of TMEFF2 or SARDH by sh_RNA or si_RNA or addition of sarcosine to prostate cancer 22Rv1 cells also promotes increased cellular invasion. However, addition of sarcosine to 22Rv1 cells in which TMEFF2 or SARDH expression has been reduced does not further induce cellular invasion. Moreover, the invasive ability of these cells is highly susceptible to the anti-folate methotrexate. These results suggest that sarcosine metabolism specifically, and 1-C availability in general, is central to the invasion phenotype mediated by changes in TMEFF2 and SARDH expression. Overall, the present study reveals a link between SARDH, TMEFF2 and 1-C metabolism in modulating cellular invasion.
Sarcosine, an intermediate in glycine metabolism, is being investigated as a biomarker for prostate cancer since increased levels were detected in urine and plasma of patients with prostate cancer, and its levels were even higher in metastatic disease [27,39]. Although its utility as a biomarker is controversial [38], in vitro studies indicated that sarcosine addition to benign RWPE1 prostate cells promoted increased cellular invasion. Similarly, feeding these cells with glycine resulted in increased cellular invasion that could be suppressed by si_RNA to GNMT [27], the enzyme that converts glycine to sarcosine and the primary source for sarcosine [40]. These results suggested that sarcosine accumulation in RWPE1 cells was sufficient to promote cellular invasion. The results presented here using 22Rv1 prostate cancer cells indicate that addition of sarcosine to cells in which expression of SARDH and/or TMEFF2 has been reduced does not promote increased invasion. This lack of response to sarcosine was not due to defects in sarcosine uptake and was dependent on reduction on the levels of SARDH or TMEFF2 as sarcosine addition to parental 22Rv1 cells resulted in increased invasion. These results indicate that sarcosine accumulation on its own does not promote increased invasion in 22Rv1 cells when the levels of TMEFF2 and/or SARDH are low. However, previous data from our lab indicate that RWPE1 cells are sensitive to sarcosine-induced invasion [34], even though the TMEFF2 protein levels in these cells are low. It is therefore possible that reducing the levels of TMEFF2 in 22Rv1, which normally expresses moderate levels of TMEFF2, renders cellular invasion more dependent on sarcosine metabolism (TMEFF2 addiction). Since sarcosine is an intermediate of 1-C metabolism, we hypothesized that these cells would also be more dependent on folate-mediated 1-C metabolism, and therefore more sensitive to anti-folate drugs. In fact, our data demonstrated that low doses of MTX specifically abrogate sarcosine-induced invasion in 22Rv1 cells in which TMEFF2 or SARDH has been knocked down, but had little effect on invasion of 22Rv1 cells. These results suggest that changes in SARDH and/or TMEFF2 may modify sarcosine and other 1-C metabolism intermediates fluxes to promote invasion. In support of this, a recent study has demonstrated that elevated levels of folic acid promote invasiveness of PC3, LNCaP and DU145 prostate cancer cells [41]. Interestingly, this effect was not correlated with increased levels of sarcosine as measured by gas chromatography and mass spectrometry supporting the idea that sarcosine accumulation is not responsible for the folate-induced invasion effect observed on these cells. Lack of correlation between sarcosine levels and invasion ability was also observed when different prostate cancer cell lines were compared [27].
Folate and other 1-C metabolism intermediates can influence prostate cancer through their role in 1-C metabolism pathway, which is necessary for DNA synthesis and methylation [42]. Early oncogenesis involves aberrant activation of cell proliferation and therefore increased DNA synthesis. This effect directs tumor cells to undergo a metabolic reprogramming which in some cases involves mutations/changes in the expression of enzymes of the folate-mediated 1-C metabolism pathway [43], sensitizing the cell to MTX. Our results showed that MTX treatment reduced the invasion of 22Rv1 cells in which TMEFF2 was knocked down independently of an effect on viability or proliferation and therefore of DNA synthesis. This suggests that the role of TMEFF2 in invasion is mediated through changes in methylation. In support of this, changes in protein and DNA methylation are known to affect tumor cell invasion [44, 45].
Interestingly, although the effects of SARDH and TMEFF2 KD on invasion are non-additive, the effect of MTX on invasion of 22Rv1 cells in which SARDH was knocked down was smaller than in the TMEFF2 KD cells, and was accompanied by a decrease on viability. These differences may reflect differential effects in the expression of other genes involved in the folate-mediated 1-C metabolism. In fact, as described here, TMEFF2 KD but not SARDH KD affects expression of GLDC. Changes in GLDC expression have been recently correlated with non-small cell lung, prostate and other cancers [22]. Finally, our results demonstrate that TMEFF2 KD or SARDH KD decreases the mRNA level of DMGDH, the enzyme that converts dimethylglycine into sarcosine. The most obvious consequence should be a decrease in invasion due to a decrease in sarcosine accumulation, contrary to our observations. It is possible that other effects mediated by the SARDH or TMEFF2 KD cancel out the effect of decreasing DMGDH levels, or that the contribution of the DMGDH catalyzed reaction to the total level of sarcosine is negligible (the GNMT reaction is the primary source of sarcosine [40]). Alternatively, it is possible that the effect of decreasing DMGDH, is cell specific, as is the case for other enzymes involved in sarcosine metabolism. In fact, results from Chinnaiyan’s lab indicated that while si_RNA to GNMT reduced invasion of DU145 cells, it did not have an effect on RWPE1 cells [27].
Our results demonstrate that SARDH KD in 22Rv1 cells results in TMEFF2 relocalization to the cytoskeleton and in fact, TMEFF2 interacts with some of the components of the cytoskeleton. However, simultaneous TMEFF2 KD does not alter the invasive phenotype of the SARDH KD cells suggesting that the altered localization of the TMEFF2 protein to the cytoskeleton is not responsible for the invasive phenotype seen in SARDH KD cells. In support of this interpretation, SARDH KD in other cells that express low levels or no TMEFF2 also promotes increased invasion (RWPE1 [27], and DU145, not shown). Our results indicate that SARDH KD in 22Rv1 also prevents localization of the TMEFF2 protein in the nucleus. Whether this effect can account for some of the phenotypes observed in the SARDH KD cells and the role of TMEFF2 in the nucleus are currently being investigated.
It is interesting to note that the TMEFF2 and SARDH proteins have distinct effects on each other. While overexpression of TMEFF2 affects SARDH activity resulting in decreased sarcosine levels and sarcosine-induced invasion that is dependent on their interaction, reducing SARDH levels promotes TMEFF2 relocalization to the cytoskeleton. Based on their ability to interact and their localization as determined by confocal microscopy, we have previously speculated that the SARDH and TMEFF2 interact in the Golgi apparatus during trafficking, thus modifying the activity of SARDH. The results presented here, suggest that in the presence of low levels or absence of SARDH, TMEFF2 is free to localize to other subcellular compartments. The consequence of TMEFF2 relocalization to the cytoskeleton in response to decreased levels of SARDH is not clear at this time.
CONCLUSIONS
In summary, our findings suggest a role for 1-C metabolism, and the enzymes involved in the pathway, in modulating cellular invasion. We have previously demonstrated that the ability of TMEFF2 to overcome sarcosine-induced invasion, correlates with its ability to interact with SARDH and modulate the levels of sarcosine [34]. Here we show that reducing the levels of TMEFF2 by sh_RNA or si_RNA also prevents sarcosine-induced invasion suggesting that an imbalance in TMEFF2 levels is enough to affect 1-C metabolism and consequently invasion. The susceptibility of TMEFF2 KD cells to MTX supports this conclusion. SARDH, GNMT and TMEFF2 are under transcriptional control mediated by the androgen receptor (AR). In addition, translation of TMEFF2 is increased by androgens/AR signaling [46]. These observations link AR signaling with the regulation of 1-C metabolism and invasion. Moreover, since AR expression, activity and function are methylation-dependent [47–49], it is possible that AR signaling-induced changes in the 1-C metabolism can function as a feed-forward mechanism to modulate its own activity.
The results presented here may offer important therapeutic implications. As described, the invasion, and therefore metastatic, ability of cells in which TMEFF2 expression has been reduced is highly susceptible to MTX, suggesting that targeting TMEFF2 in tumors that express this protein may increase their susceptibility to MTX or other anti-folates. Moreover, since TMEFF2 is hypermethylated and downregulated in glioma and several other cancers [50], this can potentially render the metastatic potential of the tumor more susceptible to anti-folate drugs.
Supplementary Material
Acknowledgments
This work was supported in part by a grant from the National Cancer Institute (1R15CA155873). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. No additional external funding received for this study.
We thank Greg Tipton and Dr. Ryan Overcash for technical assistance. We are grateful to the members of the Oncology department for helpful discussions.
Footnotes
The authors declare no conflict of interest.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fox JT, Stover PJ. Folate-mediated one-carbon metabolism. Vitam Horm. 2008;79:1–44. doi: 10.1016/S0083-6729(08)00401-9. [DOI] [PubMed] [Google Scholar]
- 4.Cebrian A, Pharoah PD, Ahmed S, Ropero S, Fraga MF, Smith PL, Conroy D, Luben R, Perkins B, Easton DF, Dunning AM, Esteller M, Ponder BA. Genetic variants in epigenetic genes and breast cancer risk. Carcinogenesis. 2006;27:1661–1669. doi: 10.1093/carcin/bgi375. [DOI] [PubMed] [Google Scholar]
- 5.Hubner RA, Muir KR, Liu JF, Sellick GS, Logan RF, Grainge M, Armitage N, Chau I, Houlston RS. Folate metabolism polymorphisms influence risk of colorectal adenoma recurrence. Cancer Epidemiol Biomarkers Prev. 2006;15:1607–1613. doi: 10.1158/1055-9965.EPI-06-0274. [DOI] [PubMed] [Google Scholar]
- 6.Kelemen LE, Sellers TA, Schildkraut JM, Cunningham JM, Vierkant RA, Pankratz VS, Fredericksen ZS, Gadre MK, Rider DN, Liebow M, Goode EL. Genetic Variation in the One-Carbon Transfer Pathway and Ovarian Cancer Risk. Cancer Res. 2008;68:2498–2506. doi: 10.1158/0008-5472.CAN-07-5165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lim U, Wang SS, Hartge P, Cozen W, Kelemen LE, Chanock S, Davis S, Blair A, Schenk M, Rothman N, Lan Q. Gene-nutrient interactions among determinants of folate and one-carbon metabolism on the risk of non-Hodgkin lymphoma: NCI-SEER case-control study. Blood. 2007;109:3050–3059. doi: 10.1182/blood-2006-07-034330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shen H, Wang L, Spitz MR, Hong WK, Mao L, Wei Q. A novel polymorphism in human cytosine DNA-methyltransferase-3B promoter is associated with an increased risk of lung cancer. Cancer Res. 2002;62:4992–4995. [PubMed] [Google Scholar]
- 9.Zhang Z, Shi Q, Liu Z, Sturgis EM, Spitz MR, Wei Q. Polymorphisms of methionine synthase and methionine synthase reductase and risk of squamous cell carcinoma of the head and neck: a case-control analysis. Cancer Epidemiol Biomarkers Prev. 2005;14:1188–1193. doi: 10.1158/1055-9965.EPI-04-0501. [DOI] [PubMed] [Google Scholar]
- 10.Bedford MT, Clarke SG. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Limm K, Ott C, Wallner S, Mueller DW, Oefner P, Hellerbrand C, Bosserhoff A. Deregulation of protein methylation in melanoma. Eur J Cancer. 2012 doi: 10.1016/j.ejca.2012.11.026. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe H, Soejima K, Yasuda H, Kawada I, Nakachi I, Yoda S, Naoki K, Ishizaka A. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell International. 2008;8:15. doi: 10.1186/1475-2867-8-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nature Reviews Cancer. 2012 doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 14.Yu Z, Chen T, Hebert J, Li E, Richard S. A mouse PRMT1 null allele defines an essential role for arginine methylation in genome maintenance and cell proliferation. Mol Cell Biol. 2009;29:2982–2996. doi: 10.1128/MCB.00042-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herman JG, Baylin SB. Gene Silencing in Cancer in Association with Promoter Hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 16.Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3:415–428. doi: 10.1038/nrg816. [DOI] [PubMed] [Google Scholar]
- 17.Momparler RL. Cancer epigenetics. Oncogene. 2003;22:6479–6483. doi: 10.1038/sj.onc.1206774. [DOI] [PubMed] [Google Scholar]
- 18.Qiu P, Zhang L. Identification of markers associated with global changes in DNA methylation regulation in cancers. BMC Bioinformatics. 2012;13 (Suppl 13) doi: 10.1186/1471-2105-13-S13-S7. S7-2105-13-S13-S7. Epub 2012 Aug 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deneberg S, Grovdal M, Karimi M, Jansson M, Nahi H, Corbacioglu A, Gaidzik V, Dohner K, Paul C, Ekstrom TJ, Hellstrom-Lindberg E, Lehmann S. Gene-specific and global methylation patterns predict outcome in patients with acute myeloid leukemia. Leukemia. 2010;24:932–941. doi: 10.1038/leu.2010.41. [DOI] [PubMed] [Google Scholar]
- 20.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 21.Feinberg AP, Ohlsson R, Henikoff S. The epigenetic progenitor origin of human cancer. Nat Rev Genet. 2006;7:21–33. doi: 10.1038/nrg1748. [DOI] [PubMed] [Google Scholar]
- 22.Zhang WC, Shyh-Chang N, Yang H, Rai A, Umashankar S, Ma S, Soh BS, Sun LL, Tai BC, Nga ME, Bhakoo KK, Jayapal SR, Nichane M, Yu Q, Ahmed DA, Tan C, Sing WP, Tam J, Thirugananam A, Noghabi MS, Pang YH, Ang HS, Mitchell W, Robson P, Kaldis P, Soo RA, Swarup S, Lim EH, Lim B. Glycine decarboxylase activity drives non-small cell lung cancer tumor-initiating cells and tumorigenesis. Cell. 2012;148:259–272. doi: 10.1016/j.cell.2011.11.050. [DOI] [PubMed] [Google Scholar]
- 23.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, Souza AL, Kafri R, Kirschner MW, Clish CB, Mootha VK. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science. 2012;336:1040–1044. doi: 10.1126/science.1218595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, Sasaki AT, Anastasiou D, Mullarky E, Vokes NI, Sasaki M, Beroukhim R, Stephanopoulos G, Ligon AH, Meyerson M, Richardson AL, Chin L, Wagner G, Asara JM, Brugge JS, Cantley LC, Vander Heiden MG. Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet. 2011;43:869–874. doi: 10.1038/ng.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, Chen WW, Barrett FG, Stransky N, Tsun ZY, Cowley GS, Barretina J, Kalaany NY, Hsu PP, Ottina K, Chan AM, Yuan B, Garraway LA, Root DE, Mino-Kenudson M, Brachtel EF, Driggers EM, Sabatini DM. Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature. 2011;476:346–350. doi: 10.1038/nature10350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Putluri N, Shojaie A, Vasu VT, Nalluri S, Vareed SK, Putluri V, Vivekanandan-Giri A, Byun J, Pennathur S, Sana TR, Fischer SM, Palapattu GS, Creighton CJ, Michailidis G, Sreekumar A. Metabolomic Profiling Reveals a Role for Androgen in Activating Amino Acid Metabolism and Methylation in Prostate Cancer Cells. PLoS One. 2011;6:e21417. doi: 10.1371/journal.pone.0021417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu J, Laxman B, Mehra R, Lonigro RJ, Li Y, Nyati MK, Ahsan A, Kalyana-Sundaram S, Han B, Cao X, Byun J, Omenn GS, Ghosh D, Pennathur S, Alexander DC, Berger A, Shuster JR, Wei JT, Varambally S, Beecher C, Chinnaiyan AM. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457:910–914. doi: 10.1038/nature07762. [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 28.Heanue TA, Pachnis V. Expression profiling the developing mammalian enteric nervous system identifies marker and candidate Hirschsprung disease genes. Proc Natl Acad Sci US A. 2006;103:6919–6924. doi: 10.1073/pnas.0602152103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Uchida T, Wada K, Akamatsu T, Yonezawa M, Noguchi H, Mizoguchi A, Kasuga M, Sakamoto C. A novel epidermal growth factor-like molecule containing two follistatin modules stimulates tyrosine phosphorylation of erbB-4 in MKN28 gastric cancer cells. Biochem Biophys Res Commun. 1999;266:593–602. doi: 10.1006/bbrc.1999.1873. [DOI] [PubMed] [Google Scholar]
- 30.Afar DE, Bhaskar V, Ibsen E, Breinberg D, Henshall SM, Kench JG, Drobnjak M, Powers R, Wong M, Evangelista F, O’Hara C, Powers D, DuBridge RB, Caras I, Winter R, Anderson T, Solvason N, Stricker PD, Cordon-Cardo C, Scher HI, Grygiel JJ, Sutherland RL, Murray R, Ramakrishnan V, Law DA. Preclinical validation of anti-TMEFF2-auristatin E-conjugated antibodies in the treatment of prostate cancer. Mol Cancer Ther. 2004;3:921–932. [PubMed] [Google Scholar]
- 31.Gery S, Koeffler HP. Repression of the TMEFF2 promoter by c-Myc. J Mol Biol. 2003;328:977–983. doi: 10.1016/s0022-2836(03)00404-2. [DOI] [PubMed] [Google Scholar]
- 32.Glynne-Jones E, Harper ME, Seery LT, James R, Anglin I, Morgan HE, Taylor KM, Gee JM, Nicholson RI. TENB2, a proteoglycan identified in prostate cancer that is associated with disease progression and androgen independence. Int J Cancer. 2001;94:178–184. doi: 10.1002/ijc.1450. [DOI] [PubMed] [Google Scholar]
- 33.Mohler JL, Morris TL, Ford OH, 3rd, Alvey RF, Sakamoto C, Gregory CW. Identification of differentially expressed genes associated with androgen-independent growth of prostate cancer. Prostate. 2002;51:247–255. doi: 10.1002/pros.10086. [DOI] [PubMed] [Google Scholar]
- 34.Chen X, Overcash R, Green T, Hoffman D, Asch AS, Ruiz-Echevarria MJ. The tumor suppressor activity of the transmembrane protein with epidermal growth factor and two follistatin motifs 2 (TMEFF2) correlates with its ability to modulate sarcosine levels. J Biol Chem. 2011;286:16091–16100. doi: 10.1074/jbc.M110.193805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pappin DJ, Hojrup P, Bleasby AJ. Rapid identification of proteins by peptide-mass fingerprinting. Curr Biol. 1993;3:327–332. doi: 10.1016/0960-9822(93)90195-t. [DOI] [PubMed] [Google Scholar]
- 36.Elahi A, Zhang L, Yeatman TJ, Gery S, Sebti S, Shibata D. HPP1-mediated tumor suppression requires activation of STAT1 pathways. Int J Cancer. 2008;122:1567–1572. doi: 10.1002/ijc.23202. [DOI] [PubMed] [Google Scholar]
- 37.Gery S, Sawyers CL, Agus DB, Said JW, Koeffler HP. TMEFF2 is an androgen-regulated gene exhibiting antiproliferative effects in prostate cancer cells. Oncogene. 2002;21:4739–4746. doi: 10.1038/sj.onc.1205142. [DOI] [PubMed] [Google Scholar]
- 38.Cao DL, Ye DW, Zhu Y, Zhang HL, Wang YX, Yao XD. Efforts to resolve the contradictions in early diagnosis of prostate cancer: a comparison of different algorithms of sarcosine in urine. Prostate Cancer Prostatic Dis. 2011;14:166–172. doi: 10.1038/pcan.2011.2. [DOI] [PubMed] [Google Scholar]
- 39.Khan AP, Rajendiran TM, Ateeq B, Asangani IA, Athanikar JN, Yocum AK, Mehra R, Siddiqui J, Palapattu G, Wei JT, Michailidis G, Sreekumar A, Chinnaiyan AM. Neoplasia. 2013;15:491–501. doi: 10.1593/neo.13314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luka Z, Mudd SH, Wagner C. Glycine N-methyltransferase and regulation of S-adenosylmethionine levels. J Biol Chem. 2009;284:22507–22511. doi: 10.1074/jbc.R109.019273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Petersen LF, Brockton NT, Bakkar A, Liu S, Wen J, Weljie AM, Bismar TA. Elevated physiological levels of folic acid can increase in vitro growth and invasiveness of prostate cancer cells. BJU Int. 2011;109:788–795. doi: 10.1111/j.1464-410X.2011.10437.x. [DOI] [PubMed] [Google Scholar]
- 42.Choi SW, Mason JB. Folate and carcinogenesis: an integrated scheme. J Nutr. 2000;130:129–132. doi: 10.1093/jn/130.2.129. [DOI] [PubMed] [Google Scholar]
- 43.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldwin RM, Morettin A, Paris G, Goulet I, Côté J. Alternatively spliced protein arginine methyltransferase 1 isoform PRMT1v2 promotes the survival and invasiveness of breast cancer cells. Cell Cycle. 2012;11:4597–4612. doi: 10.4161/cc.22871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhuang J, Jones A, Lee SH, Ng E, Fiegl H, Zikan M, Cibula D, Sargent A, Salvesen HB, Jacobs IJ, Kitchener HC, Teschendorff AE, Widschwendter M. The dynamics and prognostic potential of DNA methylation changes at stem cell gene loci in women’s cancer. PLoS Genet. 2012;8:e1002517. doi: 10.1371/journal.pgen.1002517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Overcash R, Chappell V, Green T, Geyer C, Asch A, Ruiz-Echevarría M. Androgen signaling promotes translation of TMEFF2 in prostate cancer cells via phosphorylation of the alpha subunit of the translation initiation factor 2 (eIF2α) PLoS One. 2013;8:e55257. doi: 10.1371/journal.pone.0055257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kinoshita H, Shi Y, Sandefur C, Meisner LF, Chang C, Choon A, Reznikoff CR, Bova GS, Friedl A, Jarrard DF. Methylation of the androgen receptor minimal promoter silences transcription in human prostate cancer. Cancer Res. 2000;60:3623–3630. [PubMed] [Google Scholar]
- 48.Gaughan L, Stockley J, Wang N, McCracken SRC, Treumann A, Armstrong K, Shaheen F, Watt K, McEwan IJ, Wang C, Pestell RG, Robson CN. Regulation of the androgen receptor by SET9-mediated methylation. Nucleic Acids Res. 2011;39:1266–1279. doi: 10.1093/nar/gkq861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ko S, Ahn J, Song CS, Kim S, Knapczyk-Stwora K, Chatterjee B. Lysine methylation and functional modulation of androgen receptor by Set9 methyltransferase. Mol Endocrinol. 2011;25:433–444. doi: 10.1210/me.2010-0482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lin K, Taylor JR, Jr, Wu TD, Gutierrez J, Elliott JM, Vernes JM, Koeppen H, Phillips HS, de Sauvage FJ, Meng YG. TMEFF2 is a PDGF-AA binding protein with methylation-associated gene silencing in multiple cancer types including glioma. PLoS One. 2011;6:e18608. doi: 10.1371/journal.pone.0018608. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.