

Abstract
Toxoid vaccines—vaccines based on inactivated bacterial toxins— are routinely used to promote antitoxin immunity for the treatment and prevention of bacterial infections1–4. Following chemical or heat denaturation, inactivated toxins can be administered to mount toxin-specific immune responses. However, retaining faithful antigenic presentation while removing toxin virulence remains a major challenge and presents a trade-off between efficacy and safety in toxoid development. Here we show a nanoparticle-based toxin-detainment strategy that safely delivers non-disrupted pore-forming toxins for immune processing. Using erythrocyte membrane-coated nanoparticles and staphylococcal α-haemolysin, we demonstrate effective virulence neutralization via spontaneous particle entrapment. As compared to vaccination with heat-denatured toxin, mice vaccinated with the nanoparticle-detained toxin showed superior protective immunity against toxin adverse effects. We find that the non-disruptive detoxification approach benefited the immunogenicity and efficacy of toxoid vaccines. We anticipate the reported study to open new possibilities in the preparation of antitoxin vaccines against the many virulence factors that threaten public health.
Immunization against bacterial pore-forming toxins (PFTs) has much clinical relevance as these membrane-damaging proteins underlie the virulence mechanisms in numerous public health threats, including infections by pathogenic Escherichia coli, Helicobacter pylori, and Staphylococcus aureus5–7. Toward maximizing PFT vaccine efficacy, a major challenge lies in establishing non-toxic toxoids that preserve the antigenic epitopes of the toxin proteins. Conventional toxoid preparation via protein denaturation possesses significant shortfalls that can lead to inadequate vaccine potency and poor quality control8. Chemical- and heat-mediated detoxification processes are difficult to fine-tune, and they are known to disrupt a protein’s tertiary structure, causing altered antigenic presentation and compromised immunogenicity9, 10. Although immunostimulatory adjuvants have been applied to raise the potency of denatured antigens, risk of reactogenicity and other adverse effects may occur and thus render the option less desirable11.
Efforts to improve vaccine potency and safety have given rise to alternative toxin-inactivation strategies that subvert a toxin’s virulence while preserving its native structure. For instance, non-virulent toxin mutants, prepared from recombinant protein engineering, have shown strong therapeutic efficacy in animal models and have entered human clinical trials12–15. These encouraging results suggest that toxoid preparation may benefit from minimally disruptive detoxification methods that better preserve a toxin’s epitopic expression.
To inactivate PFTs without protein denaturation, we neutralise toxins' membrane-damaging activity using a red blood cell (RBC) membrane-coated nanoparticle system16,17. The particle-stabilized biomembranes serve to anchor PFTs without compromising the toxins’ structural integrity (Fig. 1a,b and Supplementary Fig. 1). Using staphylococcal α-haemolysin (Hla) as a model toxin and mixing it with preformed RBC membrane-coated nanoparticles, we first demonstrated the facile preparation of the Hla-loaded nanotoxoids, denoted as nanotoxoid(Hla). Upon removal of the unbound toxin, Hla retention within the nanotoxoids was examined using western blotting (Fig. 1c). The results indicate that 200 µg of the particle vectors was sufficient to absorb 3 µg of Hla, translating to an Hla-to-particle ratio ≈ 40:1 (Supplementary Note). At this toxin loading level no observable changes in the particle’s size, structure, and zeta potential were detected (Supplementary Fig. 2) and endotoxin was determined to be undetectable (Supplementary Fig. 3). A release kinetics study further demonstrated that no subsequent toxin release from the nanotoxoid(Hla) occurred over a period of 48 h (Fig. 1d), indicating that the Hla was safely locked into the particle vector. By labelling Hla with a fluorescent dye, it was observed that the nanotoxoid(Hla) enabled uptake of toxins by immune cells. Upon incubation with mouse dendritic cells, fluorescence microscopy revealed the nanotoxoid(Hla) as distinct features within the cells (Fig. 1e), which is consistent with the pattern of endocytic uptake frequently observed with nanoparticle vectors18, 19. Through direct engulfment of Hla into the digestive endolysosomal compartments, the nanoparticle-facilitated cellular endocytosis precludes the toxin’s perforating attack on cellular membranes and thus allows non-disrupted toxin to be delivered for immune processing. Subcutaneous injection of the nanotoxoid(Hla) to mice showed lymphatic drainage of the particles over time, suggesting the ability of the particle vector to deliver Hla efficiently to the immune system in vivo (Fig. 1f).
Figure 1. Schematic and in vitro characterizations.
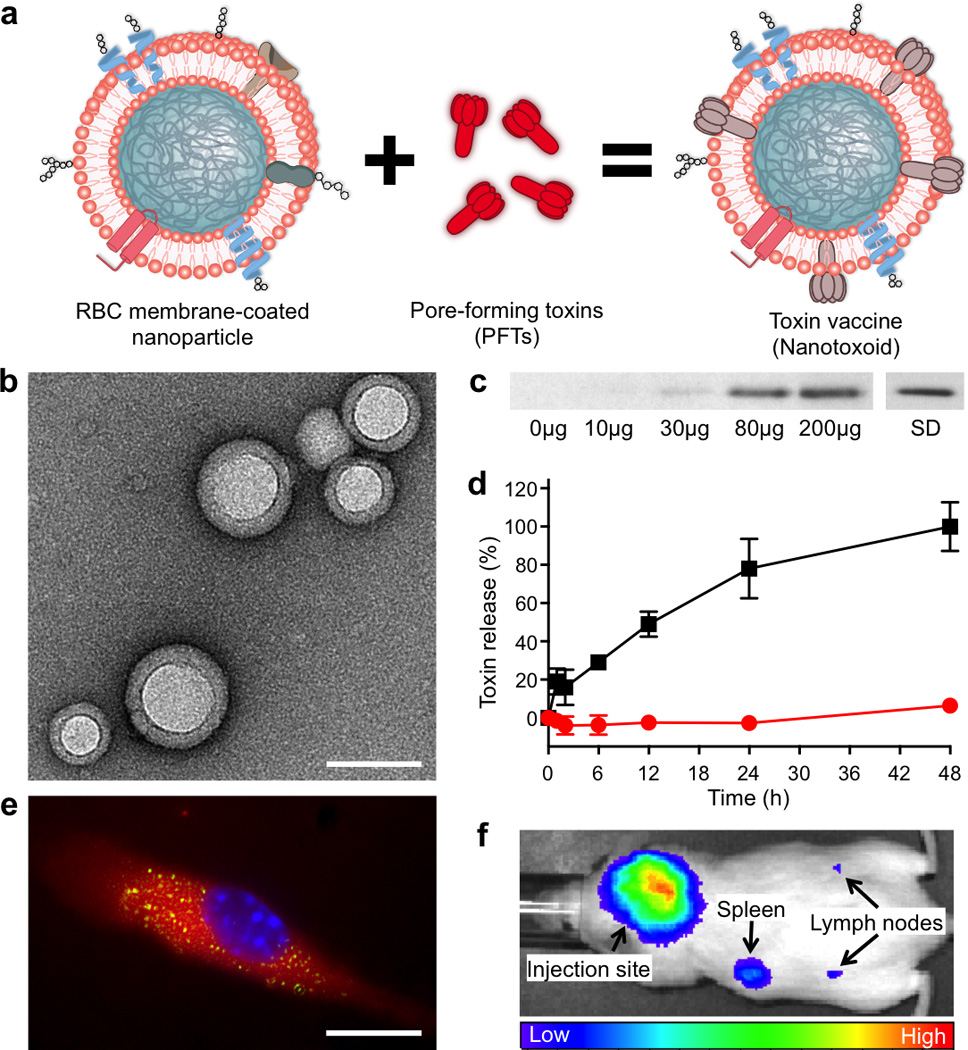
(a) Schematic preparation of nanoparticle-detained toxins, denoted as nanotoxoid, consisting of substrate-supported RBC membranes into which pore-forming toxins (PFTs) can spontaneously incorporate. (b) TEM visualization of the particle vectors with uranyl acetate staining (scale bar = 80 nm). (c) Western blotting results to verify the retention of 3 µg of staphylococcal α-hemolysin (Hla) by varying amounts of the particle vectors using 3 µg of free Hla as a standard (SD). (d) Release of toxin from the Hla-loaded nanotoxoids, denoted as nanotoxoid(Hla), over time in PBS buffer. Red circles indicate nanotoxoid(Hla) and black squares indicate free Hla. Error bars represent standard deviations of the mean. (e) Uptake of nanotoxoid(Hla) by a mouse dendritic cell (scale bar = 10 µm). The cell is membrane stained with DMPE-rhodamine B (red) and nuclei stained with DAPI (blue). FITC-labelled Hla (green) was used to monitor the toxin uptake. (f) Live, whole-body fluorescent imaging of nanotoxoid(Hla) at 1 h after subcutaneous administration.
To assess the toxin inactivation in the nanotoxoid, 200 µg of nanotoxoid(Hla) detaining 3 µg of Hla was injected into the superficial dorsal skin of mice. Untreated free Hla, Hla heated at 70°C for 30 min, and Hla heated at 70°C for 60 min were tested in parallel at an equivalent Hla dose. 24 h following the injections, the skin was sectioned to evaluate the toxicity of the different formulations using both TUNEL assay and haematoxylin and eosin (H&E) assay (Fig. 2a). It was revealed that untreated Hla caused a significant level of cellular apoptosis and observable lesions in the skin. Toxin neutralization by heat was shown to be time-dependent, as Hla heated for 30 min remained damaging to the skin, whereas 60 min of heating removed the toxin virulence. For the skin injected with the nanotoxoid(Hla), the epithelial structure remained intact and no cellular apoptosis was observed outside of hair follicles. Visual examination of mice subcutaneously administered with the nanotoxoid(Hla) also showed no observable lesions 48 h following the injections (Supplementary Fig. 4). This lack of toxin damage was observed consistently in 10 mice per test group. In vivo imaging of nanotoxoid(Hla) showed that the particles were eventually cleared over time as there was no trace of the particles after 2 weeks (Supplementary Fig. 5). To further confirm that the nanotoxoid can safely present the toxin antigens to antigen-presenting cells, an in vitro cytotoxicity test was conducted on mouse dendritic cells. Upon 48 h of incubation in 15 µg/mL of Hla content, untreated Hla resulted in 70% decrease in cell viability, whereas both heat-denatured Hla (60 min treatment) and nanotoxoid(Hla) showed no reduction (Fig. 2b). Flow cytometric analysis showed that the nanotoxoid(Hla) did not induce any additional underlying cellular apoptosis compared to untreated cells over a 72 h period (Fig. 2c and Supplementary Fig. 6). These results confirm the safety and reliability of the nanotoxoid-based toxin inactivation, which allows non-denatured toxin antigens to interact with tissues and immune cells with the same level of safety as those treated with extended heating.
Figure 2. Nanotoxoid(Hla) neutralises Hla virulence.
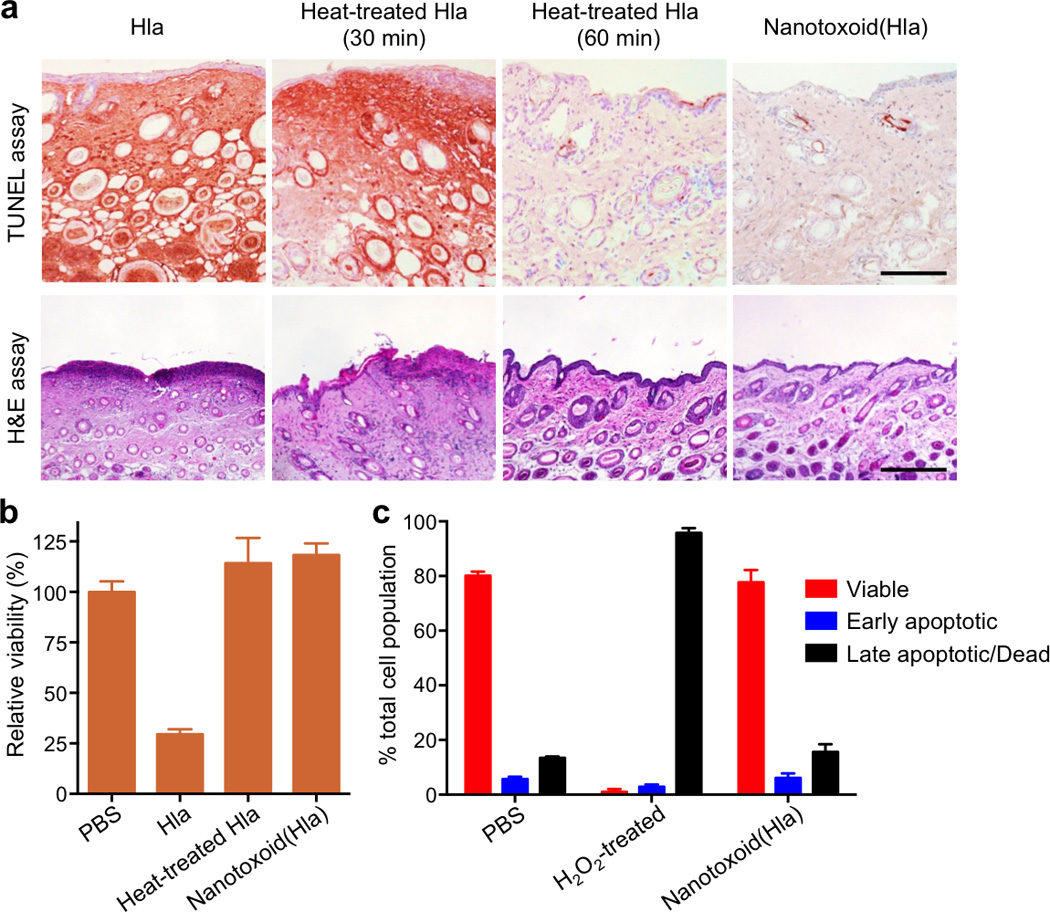
(a) Free Hla, heat-treated Hla (30 min), heat-treated Hla (60 min), and nanotoxoid(Hla) were injected into the superficial dorsal skin of mice. 24 h following the injections, the skin was removed and examined for apoptosis using a TUNEL assay. Histological analyses were performed with H&E stained skin 48 h following the injections (Scale bar = 400 µm). (b) Toxicity of different Hla formulations against dendritic cells derived from mice. The cells were incubated for 48 h with Hla, heat-treated Hla (60 min) and nanotoxoid(Hla) at 15 µg/mL Hla concentration. Cellular viability was assessed using an MTT assay (n=6). (c) Induction of dendritic cell apoptosis by nanotoxoid(Hla) at 60 µg/mL Hla concentration 72 h after initial incubation. Propidium iodide and Annexin V staining were analysed by flow cytometry (n=6). All error bars represent standard deviations of the mean.
Next, immunization studies were conducted to examine the vaccine potential of the nanotoxoid(Hla). An emphasis was placed on the elicitation of neutralizing antibodies, which are the hallmark of antitoxin immunity. Two vaccination schedules were performed: a prime only on day 0, and a prime on day 0 followed by two booster vaccinations on day 7 and day 14. To verify that the nanotoxoid(Hla) could indeed elicit Hla-specific antibodies, the plasma from 8 mice immunized with nanotoxoid(Hla) on a prime-boost schedule was pooled together and used as the primary immunostain for western blotting analysis. The results confirmed the presence of anti-Hla IgG (Fig 3a). It is important to note that this cocktail plasma showed no detectable cross-reactivity with the protein content on the blank particle vectors, denoted as nanotoxoid(-). To further confirm the lack of an autoimmune response, we demonstrated that mice immunized with nanotoxoid(Hla) exhibited no serum immunoglobulin M, immunoglobulin A, or immunoglobulin G antibodies against RBC proteins (Supplementary Fig. 7). RBC counts were at a similar level as compared to unvaccinated mice, indicating no induction of autoimmune anaemia (Fig. 3b). The results indicate that the RBC membranes provide a non-immunogenic substrate for toxin-detainment, enabling the cargo antigens to be processed selectively without raising potential complications associated with anti-vector immunity20, 21.
Figure 3. Nanotoxoid(Hla) vaccinations elicit strong Hla-specific antibody responses.
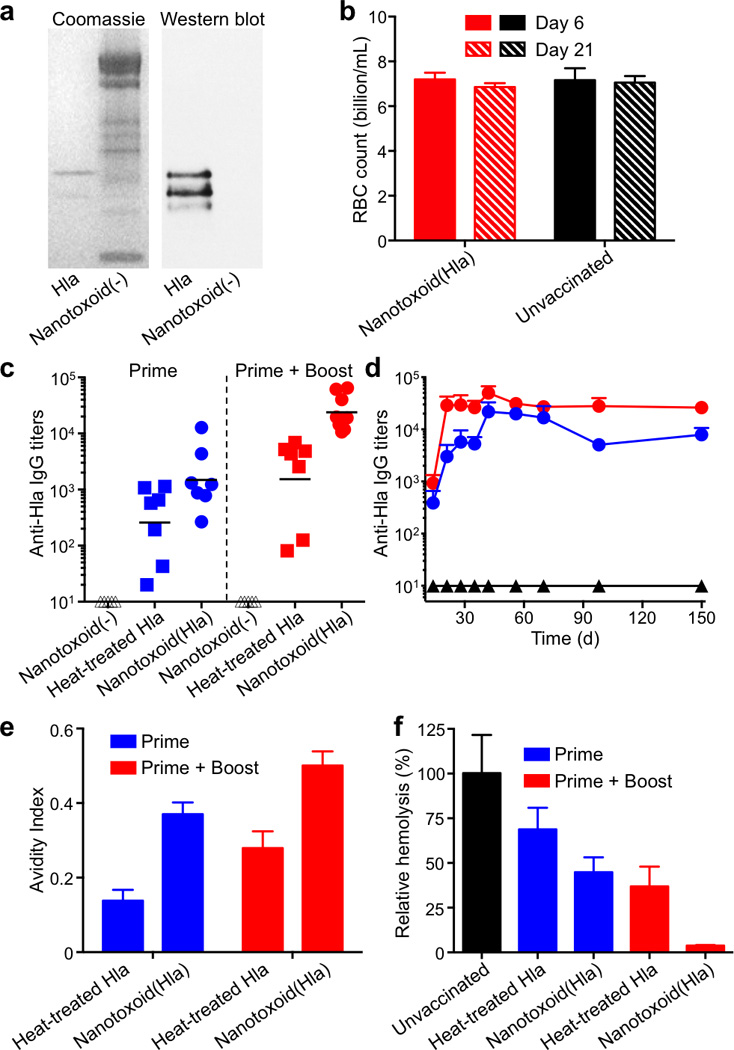
(a) Hla-specific antibody responses were verified in the nanotoxoid(Hla)-vaccinated mice through coomassie staining (left panel) and western blotting (right panel). Blank particle vector, denoted as nanotoxoid(-), was used as a control. (b) RBC counts of mice immunized with nanotoxoid(Hla) (n=6). (c) Anti-Hla IgG titres at day 21 (n=7). Black lines indicate geometric means. Anti-Hla titres from mice vaccinated with non-toxin loaded particle vectors (nanotoxoid(-)) were monitored as controls (triangle). (d) Time course of anti-Hla IgG titres in unvaccinated mice (black triangle) and mice immunized with nanotoxoid(Hla) (prime + boost; red circle) or nanotoxoid(Hla) (prime only; blue circle) (n=7). (e) Avidity index of the anti-sera from immunized mice binding to Hla toxin was quantified (n=7). (f) An RBC haemolysis assay was performed to verify the presence of functional titres (n=7). All error bars represent standard deviations of the mean.
The ability of the nanotoxoid(Hla) to elicit anti-Hla antibodies was then quantified. Determination of antibody responses on day 21 showed that the nanotoxoid(Hla) induced significantly higher Hla-specific antibody titres as compared to the heat-treated Hla (60 min treatment). Enhancements by 7- and 15-fold (geometric mean) under the prime only (p = 0.0951, n=7) and prime-boost vaccinations (p=0.0077, n=7), respectively (Fig. 3c). The increased titre level of nanotoxoid(Hla) was sustainable in a time course study over 150 days (Fig. 3d). Because previous report22 and our own study (Supplementary Fig. 8) have indicated that adjuvants do not significantly boost titre responses for immunizations using denatured Hla, no adjuvant was used in this study in order to best highlight the nanotoxoid(Hla) platform as an effective mode of attenuating toxicity while preserving immunogenicity of toxins. The nanotoxoid(-) vector alone did not induce a detectable antibody response (Fig. 3c), and the vector mixture with heat-treated Hla showed negligible enhancement in titre levels as compared to heat-treated Hla alone (Supplementary Fig. 9). These results highlight the challenge in raising immunogenicity of denatured toxins and demonstrate the benefit of using detained, but undenatured antigens for vaccination.
The nanotoxoid(Hla) also helped to improve antibody affinity to the targeted toxin, as was evidenced by the increased avidity of the antibody titres (Fig. 3e). To confirm that the antibody titres were capable of neutralizing Hla, an RBC haemolysis assay was conducted. Hla was mixed with the plasma from vaccinated mice and then incubated with purified mouse RBCs. The plasma from unvaccinated mice was used as a control and the haemolytic activity of Hla was determined by measuring the amount of released haemoglobin. The results showed that plasma from nanotoxoid(Hla)-vaccinated mice was more potent at neutralizing the toxin. For the prime-boost nanotoxoid(Hla) vaccination group, 25 µL of plasma was sufficient to completely inhibit the haemolytic activity of 1 µg of Hla (Fig. 3f). We speculate that the improved titre responses from nanotoxoid(Hla) were due to epitopic preservation of the undenatured toxin as well as to well documented benefits of particulate antigen vectors, which can enhance antigen uptake and processing by immune cells23, 24.
Finally, the protective immunity bestowed by the nanotoxoid(Hla) vaccine was evaluated by subjecting the vaccinated mice to both systemic and subcutaneous toxin administration. 21 days following the prime vaccination, the mice received a lethal bolus dose of Hla at 120 µg/kg through intravenous tail vein injection. This toxin dose resulted in 100% mortality within 2 h in the unvaccinated group. For the mice receiving the prime vaccination only, the benefit of the nanotoxoid(Hla) over the heat-treated Hla was evident as the survival rate increased from 10% to 50% (n=10). Moreover, nanotoxoid(Hla) boosters further improved the survival rate to 100% while a 90% survival rate was achieved by the heat-treated Hla vaccine with boosters (n=10) (Fig. 4a). Since Hla has also been identified as a key factor in necrotizing skin infections25, a subcutaneous toxin administration was also conducted to further evaluate the vaccine’s effectiveness in mounting immunity in skin tissue. Observation of skin damage following a 50 µL subcutaneous injection of 100 µg/mL Hla on the back region showed reduced lesion areas in all vaccinated mice, reflecting the presence of extravascular IgG that diminished the subcutaneous toxin threat. With the prime only vaccination, the nanotoxoid(Hla) vaccine conferred modestly stronger protection than the heat-treated Hla. Following booster vaccinations, however, the nanotoxoid(Hla) resulted in complete eradication of the toxin damage. In contrast, the mice vaccinated with the heat-treated Hla boosters remained susceptible to the necrotizing toxin (Fig. 4b and Supplementary Fig. 10).
Figure 4. Nanotoxoid(Hla) vaccinations bestow strong protective immunity.
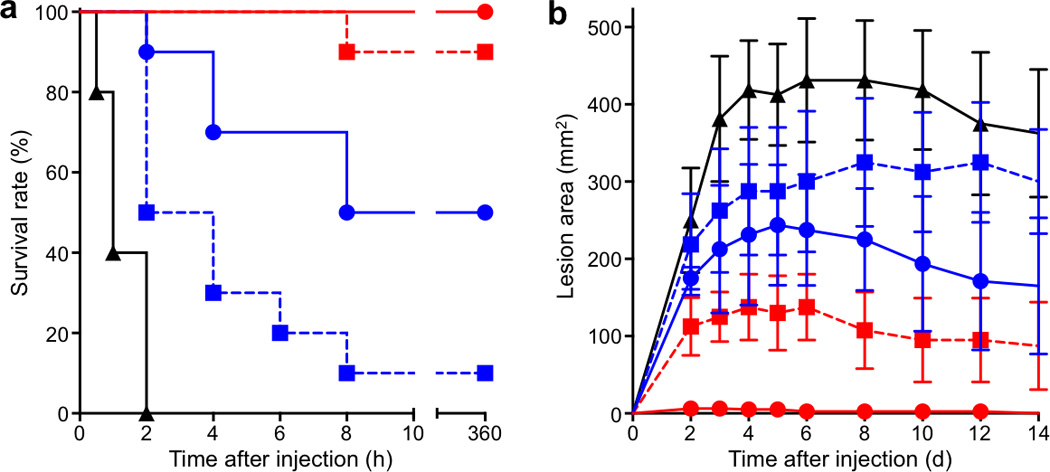
Unvaccinated mice (black) and mice vaccinated with heat-treated Hla (prime; blue square), nanotoxoid(Hla) (prime; blue circle), heat-treated Hla (prime + boost; red square), or nanotoxoid(Hla) (prime + boost; red circle) received intravenous or subcutaneous administration of Hla. (a) Survival rates of mice over a 15-day period following intravenous injections of 120 µg/kg Hla on day 21 via the tail vein (n=10). The unvaccinated mice were used as a negative control and mice vaccinated with heat-treated Hla served as positive controls. Both the prime only schedule and the prime-boost schedule were conducted. (b) Skin lesion size comparison following subcutaneous injections of 5 µg of Hla on day 21. The lesion size was measured for 14 days following the challenge. The means ± SD for each vaccination group were plotted (n=6).
Compared to the commonly used protein denaturation approach to achieve toxin vaccination, the non-disruptive preparation described here yielded a nanotoxoid with stronger immunogenicity and superior efficacy. Refinement of this approach can benefit from the unique strengths of nanoparticle-based immunoengineering26, 27, in which vaccine targeting to lymphoid organs and processing by antigen-presenting cells can be enhanced through nanovector designs24, 28, 29. Moving forward, this nanotoxoid platform can be generalized for other types of cellular membrane-coated particles30 and for the neutralization and delivery of other potent toxins to create a broad range of safe and effective antitoxin vaccines. For clinical test and use in humans, it can be applied either on the basis of patients’ blood types following a cross-match test or using donor blood from type O- individuals, as in the case of blood transfusion.
Methods
The nanotoxoid(Hla) were prepared by mixing Hla with RBC-membrane-coated nanoparticles, which were made by fusing RBC membrane vesicles onto preformed poly(lactic-co-glycolic acid) (PLGA) nanoparticles through an extrusion process16. The size of the resulting nanotoxoid was obtained from three dynamic light scattering (DLS) measurements. The morphology of the nanotoxoid(Hla) was observed using transmission electron microscopy (TEM). The retention of Hla by the nanoparticle vectors was quantified based on western blotting results using anti-Hla antibodies as an immunostain. The toxin release study was conducted by dialyzing the samples against phosphate buffered saline (PBS) using a Float-A-Lyzer G2 device with a molecular weight cut-off of 100 KDa. Cellular uptake of the nanotoxoid(Hla) was studied by incubating the particles with mouse dendritic cells derived from the bone marrow of Imprinting Control Region (ICR) mice, followed by fluorescent imaging. Live, whole-body imaging of the nanotoxoid(Hla) was conducted using a Xenogen IVIS 200 system.
The safety of the nanotoxoid(Hla) was assessed through histological examination of mouse skin and in vitro cellular viability and apoptosis studies with mouse dendritic cells. For autoimmune test, mice were immunized on the prime-boost schedule and autoimmune titres were analysed using RBC-coated plates. RBC counts of the whole blood were automatically analysed using a Drew Scientific Hemavet. The potency of the nanotoxoid(Hla) was examined through vaccinations in mice. Hla-specific humoral responses were studied through western blotting using cocktail plasma from vaccinated mice as the primary immunostain. Anti-Hla titres and titre avidity were assessed using an ELISA assay with Hla coated assay plates. Verification of functional anti-Hla titres was conducted through a haemolysis assay in which mice RBCs and Hla were mixed with purified plasma from vaccinated mice. The protective immunity bestowed by nanotoxoid(Hla) vaccinations was examined through systemic and subcutaneous injection of Hla in vaccinated mice followed by observation of survival and skin damages over time.
For more methodological details see Supplementary Methods.
Supplementary Material
Acknowledgements
This work is supported the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health under Award Number R01DK095168 and by the National Science Foundation Grant DMR-1216461. B.L. is supported by a National Institutes of Health R25CA153915 training grant from the National Cancer Institute.
Footnotes
Author contributions
L.Z. conceived and designed the experiments with C-M.H., R.F. and B.L; C-M.H., R.F. and B.L. performed all the experiments. All authors analysed and discussed the data and wrote the paper.
Additional information
Supplementary Information accompanies this paper at www.nature.com/naturenanotechnology. Reprints and permission information is available online at http://www.nature.com/reprints.
References
- 1.Kitchin NR. Review of diphtheria, tetanus and pertussis vaccines in clinical development. Expert Rev. Vaccines. 2011;10:605–615. doi: 10.1586/erv.11.60. [DOI] [PubMed] [Google Scholar]
- 2.Greenberg RN, Marbury TC, Foglia G, Warny M. Phase I dose finding studies of an adjuvanted Clostridium difficile toxoid vaccine. Vaccine. 2012;30:2245–2249. doi: 10.1016/j.vaccine.2012.01.065. [DOI] [PubMed] [Google Scholar]
- 3.Mortimer EA., Jr Immunization against infectious disease. Science. 1978;200:902–907. doi: 10.1126/science.347579. [DOI] [PubMed] [Google Scholar]
- 4.Holmgren J, et al. Development of improved cholera vaccine based on subunit toxoid. Nature. 1977;269:602–604. doi: 10.1038/269602a0. [DOI] [PubMed] [Google Scholar]
- 5.Gentschev I, Dietrich G, Goebel W. The E. coli alpha-hemolysin secretion system and its use in vaccine development. Trends Microbiol. 2002;10:39–45. doi: 10.1016/s0966-842x(01)02259-4. [DOI] [PubMed] [Google Scholar]
- 6.Cover TL, Blanke SR. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 2005;3:320–332. doi: 10.1038/nrmicro1095. [DOI] [PubMed] [Google Scholar]
- 7.Bubeck Wardenburg J, Schneewind O. Vaccine protection against Staphylococcus aureus pneumonia. J. Exp. Med. 2008;205:287–294. doi: 10.1084/jem.20072208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parish HJ, Cannon DA. Staphylococcal infection: antitoxic immunity. Br. Med. J. 1960;1:743–747. doi: 10.1136/bmj.1.5175.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Metz B, et al. Identification of formaldehyde-induced modifications in proteins: reactions with model peptides. J. Biol. Chem. 2004;279:6235–6243. doi: 10.1074/jbc.M310752200. [DOI] [PubMed] [Google Scholar]
- 10.Cryz SJ, Jr, Furer E, Germanier R. Effect of chemical and heat inactivation on the antigenicity and immunogenicity of Vibrio cholerae. Infect. Immun. 1982;38:21–26. doi: 10.1128/iai.38.1.21-26.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vogel FR. Improving vaccine performance with adjuvants. Clin. Infect. Dis. 2000;30(Suppl 3):S266–S270. doi: 10.1086/313883. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy AD, et al. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J. Infect. Dis. 2010;202:1050–1058. doi: 10.1086/656043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Adhikari RP, et al. Novel structurally designed vaccine for S. aureus alpha-hemolysin: protection against bacteremia and pneumonia. PLoS One. 2012;7:e38567. doi: 10.1371/journal.pone.0038567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jang SI, et al. Vaccination with Clostridium perfringens recombinant proteins in combination with Montanide ISA 71 VG adjuvant increases protection against experimental necrotic enteritis in commercial broiler chickens. Vaccine. 2012;30:5401–5406. doi: 10.1016/j.vaccine.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 15.Kirkham LA, et al. Construction and immunological characterization of a novel nontoxic protective pneumolysin mutant for use in future pneumococcal vaccines. Infect. Immun. 2006;74:586–593. doi: 10.1128/IAI.74.1.586-593.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu CM, et al. Erythrocyte membrane-camouflaged polymeric nanoparticles as a biomimetic delivery platform. Proc. Natl. Acad. Sci. U. S. A. 2011;108:10980–10985. doi: 10.1073/pnas.1106634108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu CM, Fang R, Copp J, Luk B, Zhang L. A biomimetic nanosponge that absorbs pore-forming toxins. Nat. Nanotechnol. 2013;8:336–340. doi: 10.1038/nnano.2013.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobrovolskaia MA, McNeil SE. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007;2:469–478. doi: 10.1038/nnano.2007.223. [DOI] [PubMed] [Google Scholar]
- 19.Moon JJ, et al. Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat. Mater. 2011;10:243–251. doi: 10.1038/nmat2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun C, et al. Circumventing anti-vector immunity using adenovirus-infected blood cells for repeated application of adenovirus vectored vaccines: proof-of-concept in rhesus macaques. J. Virol. 2012;86:11031–11042. doi: 10.1128/JVI.00783-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roberts DM, et al. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature. 2006;441:239–243. doi: 10.1038/nature04721. [DOI] [PubMed] [Google Scholar]
- 22.Skean JD, Overcast WW. Efficacy of staphylococcal vaccines to elicit antistaphylococcal alpha-hemolysin in dairy cows. J. Dairy Sci. 1968;51:1239–1242. doi: 10.3168/jds.S0022-0302(68)87164-4. [DOI] [PubMed] [Google Scholar]
- 23.Elamanchili P, Diwan M, Cao M, Samuel J. Characterization of poly(D,L-lactic-co-glycolic acid) based nanoparticulate system for enhanced delivery of antigens to dendritic cells. Vaccine. 2004;22:2406–2412. doi: 10.1016/j.vaccine.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 24.Moon JJ, et al. Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction. Proc. Natl. Acad. Sci. U. S. A. 2012;109:1080–1085. doi: 10.1073/pnas.1112648109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Inoshima I, et al. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat. Med. 2011;17:1310–1314. doi: 10.1038/nm.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moon JJ, Huang B, Irvine DJ. Engineering nano- and microparticles to tune immunity. Adv. Mater. 2012;24:3724–3746. doi: 10.1002/adma.201200446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Swartz MA, Hirosue S, Hubbell JA. Engineering approaches to immunotherapy. Sci. Transl. Med. 2012;4 doi: 10.1126/scitranslmed.3003763. 148rv149. [DOI] [PubMed] [Google Scholar]
- 28.Reddy ST, et al. Exploiting lymphatic transport and complement activation in nanoparticle vaccines. Nat. Biotechnol. 2007;25:1159–1164. doi: 10.1038/nbt1332. [DOI] [PubMed] [Google Scholar]
- 29.Hubbell JA, Thomas SN, Swartz MA. Materials engineering for immunomodulation. Nature. 2009;462:449–460. doi: 10.1038/nature08604. [DOI] [PubMed] [Google Scholar]
- 30.Parodi A, et al. Synthetic nanoparticles functionalized with biomimetic leukocyte membranes possess cell-like functions. Nat. Nanotechnol. 2013;8:61–68. doi: 10.1038/nnano.2012.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.