

Abstract
In high concentrations of fresh nonimmune human serum, Mycobacterium tuberculosis activates the alternative pathway of complement and binds C3 protein, resulting in enhanced phagocytosis by complement receptors on human alveolar macrophages. Yet in the lung, the alternative pathway of complement is relatively inactive compared to the classical pathway. To begin to determine whether C3 opsonophagocytosis of M. tuberculosis by alveolar macrophages can occur in the lung of the immunologically naive host, we characterized the binding of C3 to M. tuberculosis in different concentrations of fresh nonimmune human serum and concentrated human bronchoalveolar lavage fluid. Here we show that in human serum, C3 binding to M. tuberculosis is rapid, initiated by either the alternative pathway or the classical pathway, depending on the concentration of serum, and occurs by covalent linkages between the bacterial surface and the C3 cleavage products, C3b or C3bi. Human bronchoalveolar lavage fluid contains C3 protein and functional classical pathway activity that mediates the binding of C3 to the surface of M. tuberculosis. These studies provide evidence that when M. tuberculosis is first inhaled into the lungs of the human host, the bacterium is opsonized by C3 cleavage via classical pathway activation within the alveolus, providing a C3-dependent entry pathway into resident alveolar macrophages.
Mycobacterium tuberculosis is an obligate human pathogen that is inhaled on droplet nuclei into the lungs and deposits in the terminal bronchioles and distal alveoli. These host-adapted bacteria have evolved the ability to survive in mononuclear phagocytes, and it is felt that entry into the alveolar macrophage is important for a successful initial infection (25).
Prior studies have established that M. tuberculosis is phagocytosed by the macrophage complement receptors (CR) CR1, CR3, and CR4 and the mannose receptor (16, 40, 41). Additional receptors for M. tuberculosis likely exist, but their precise role in the phagocytosis of M. tuberculosis is not clear (for a review, see reference 10). M. tuberculosis phagocytosis is enhanced in the presence of low concentrations of nonimmune serum as a result of binding of complement protein C3 to the surface of the bacteria and increased ligation of CR, although nonopsonic binding of M. tuberculosis to CR3 is also important in phagocytosis (8, 41, 45, 46).
In contrast to blood monocytes, human alveolar macrophages are reported to have more CR4 than CR1 or CR3 (31). Consistent with these data, antibodies to CR4 blocked phagocytosis of M. tuberculosis to a greater extent in human alveolar macrophages than did antibodies to CR1 and CR3, whereas the opposite was true for phagocytosis by blood monocytes (16). Just as the expression of CR differ with respect to cell type, the ligands that bind CR also differ. For example, CR1 binds complement components C1q, C4b, and C3b, and CR3 and CR4 bind primarily C3bi, as well as other noncomplement microbial ligands (22, 38). Therefore, it is likely that specific proteins of the complement system as well as their proteolytic forms that are bound to M. tuberculosis may affect the subsequent fate of the organism by directing the pathogen to particular receptors and altering phagocytosis.
Proteolytic cleavage forms of the complement proteins are generated upon activation of the complement cascade. Three activation pathways have been described: the classical pathway, the alternative pathway, and the lectin pathway. Each of these pathways is initiated by specific stimuli and serves distinct but not exclusive roles in innate immunity (27, 33, 34, 53, 54). Although CR and the complement system have been studied in some detail, less is known about the mechanisms of complement protein binding to M. tuberculosis or its role in the initial pathogenesis of the disease. The first encounter of M. tuberculosis with its human host is in the airspaces of the lung, where interactions between the bacteria and airway surface components may affect the initial pathogenesis of the disease. Alveolar cells (macrophages and epithelial cells) produce several proteins of the classical and alternative pathways of complement (6, 47), and complement proteins have been detected in the bronchoalveolar lavage (BAL) fluid from several different mammalian species including humans (7, 13, 23, 50). Prior studies indicate that functional complement activity is present in the lung, although C3 activation is reduced when compared to the level of activity in serum, possibly due to the presence of an inhibitor (13). Additionally, classical pathway activity is more readily detected than alternative pathway activity, possibly due to very low levels of alternative pathway components in lavage fluid (50). Surfactant protein A, a lung collectin with a structure similar to that of C1q, is reported to regulate complement activity in the alveolus (51).
It is believed that C3 opsonization of M. tuberculosis in the lung is important for the pathogenesis of tuberculosis during the initial interaction of the bacteria with the human host. Since M. tuberculosis is phagocytosed by CR that are expressed on human alveolar macrophages and since the classical pathway of complement appears to be more active than the alternative pathway in the lung, we hypothesized that M. tuberculosis can activate the classical complement pathway in the lung and fix C3 protein for opsonophagocytosis. To begin to address this hypothesis, here we characterize C3 binding to M. tuberculosis in different concentrations of fresh nonimmune human serum (NHS) and determine if BAL fluid from healthy humans contains functional C3 protein that binds to M. tuberculosis. In these studies we show that the binding of C3 to M. tuberculosis occurs rapidly, is mediated by covalent bonds between C3 cleavage proteins and M. tuberculosis surface molecules, and occurs by both the alternative pathway and the C1q-dependent classical pathway, depending on the concentration of serum. We further show that BAL fluid from healthy humans contains classical complement activity that can lead to C3 deposition on the surface of M. tuberculosis.
MATERIALS AND METHODS
Sera, complement proteins, buffers, and antibodies.
NHS was collected from healthy tuberculin-negative human volunteers who had no known exposure to tuberculosis, according to a protocol that has been approved by the Internal Review Board, College of Medicine, University of Iowa. The serum was separated, filtered, stored, and handled so as to preserve complement activity (18). Pooled human serum, C1q-depleted serum (C1qD), factor B-depleted serum (FBD), purified C1q protein, and purified factor B protein (FB) were purchased from Advanced Research Technologies (San Diego, Calif.), stored, and handled per the manufacturer's instructions. Veronal-buffered saline containing gelatin (GVBS) and GVBS containing calcium and magnesium ions (GVBS++) were purchased from Advanced Research Technologies. Goat anti-human C3 immunoglobulin G (IgG) conjugated to horseradish peroxidase (HRP) and goat anti-human IgG plus IgA plus IgM (anti-IgGAM) conjugated to HRP were purchased from Cappel Research products (Durham, N.C.). Mouse anti-human C3bi neoantigen monoclonal antibody (MAb) was purchased from Quidel (San Diego, Calif.). Goat anti-mouse IgG conjugated to HRP was purchased from Bio-Rad (Hercules, Calif.). Rabbit anti-acetone-dried Erdman M. tuberculosis serum was produced by immunizing a New Zealand White rabbit with 50 mg of an acetone-dried Erdman M. tuberculosis preparation in incomplete Freund’s adjuvant two times at an interval of 1 month, followed by two boosts with 25 mg of the preparation 3 and 5 months after the first injection.
BAL.
Tuberculin-negative healthy human volunteers with no known exposure to tuberculosis and no smoking history underwent BAL with approximately 200 ml of saline according to a previously described procedure (20) that has been approved by the Internal Review Board, College of Medicine, University of Iowa. After cellular material was removed, EDTA was added to the lavage fluid to a final concentration of 2 mM, and the mixture kept at <4°C during manipulations to prevent complement activation. The BAL fluid was passed through a 0.22-μm-pore-size filter and then was concentrated (cBAL fluid) 20- to 30-fold by using a Centriprep 10 concentrator (Millipore, Bedford, Mass). BAL and cBAL fluids were stored at −70°C until the day of use. Prior to experimentation, the BAL and cBAL fluids were dialyzed at 4°C by using a membrane with a 10,000-molecular-weight cutoff for 2 h against Dulbecos phosphate-buffered saline (PBS) containing calcium and magnesium ions (PBS++) at a 4,000/1 (vol/vol) ratio. The protein concentration was determined by using a bicinchoninic acid assay against bovine serum albumin standards (Pierce, Rockford, Ill.).
Bacteria preparation and treatments.
Lyophilized M. tuberculosis strain Erdman (ATCC 35801) was obtained from the American Tissue Culture Collection (Rockville, Md.), reconstituted, grown on 7H11 agar, and harvested as described previously (41). The stock concentration of bacteria (1 × 108 to 2 × 108 bacteria/ml) and the degree of clumping (≤10%) were determined by counting in a Petroff-Hausser chamber (11).
To examine C3 binding to bacteria, approximately 2.5 × 107 M. tuberculosis bacilli in PBS++ were combined with 2.5 or 25% sera in the presence and absence of 10 mM EDTA, 10 mM EGTA with 7 mM MgCl2 (EGTA-Mg), or purified complement proteins as described in the text, and incubated at 37°C for 5 to 60 min. The samples were then placed on ice for 2 min and pelleted by centrifugation at 10,000 × g. The supernatant was removed, and then the pellets were washed three times in ice-cold PBS containing protease inhibitors (1 μg of pepstatin/ml, 10 μg of leupeptin/ml, 10 μg of soybean trypsin inhibitor/ml, 1 mM phenylmethylsulfonylfluoride, and 1 μg of aprotinin/ml). The bacterial pellets were then resuspended in PBS and prepared for enzyme-linked immunosorbent assay (ELISA), Western blotting, or hydroxylamine treatment as described below. In some experiments, M. tuberculosis bacilli were incubated with cBAL fluid as described above and prepared for Western blotting or ELISA.
Hydroxylamine treatment.
M. tuberculosis bacilli were incubated with sera as described above and then were treated with 1 M hydroxylamine in 0.05% sodium dodecyl sulfate [SDS]-20 mM Tris (pH 10.5) for 1 h at 37°C to cleave ester bonds (4). The bacteria were then pelleted by centrifugation at 10,000 × gAve, and the pellets were separated from the supernatants. The bacterial pellets were then washed three times in ice-cold PBS containing protease inhibitors, and the pellets and supernatants were prepared for Western blotting.
Western blotting.
Protein and bacterial samples were combined with sample buffer (0.6 M Tris-HCl [pH 6.8] containing 2% SDS, 10% glycerol, 0.025% bromphenol blue, and 5% 2-ME), heated to 100°C for 10 min, resolved by SDS-7.5% polyacrylamide gel electrophoresis (PAGE), and then transferred to nitrocellulose. The nitrocellulose membranes were blocked in PBS containing 5% nonfat dry milk and 0.05% Tween-20 for at least 1 h at room temperature or overnight at 4°C. The membranes were then incubated with HRP-conjugated primary antibody in blocking buffer for 5 h at room temperature or overnight at 4°C, washed three to five times in PBS, and developed by using enhanced chemiluminescence (Amersham, Buckinghamshire, England). For Western blotting with the MAb to human C3bi neoantigen or polyclonal antiserum to M. tuberculosis, samples were prepared as follows: after incubation with the primary antibody and washing, the membranes were incubated with the appropriate secondary antibody for 2 h at room temperature, washed, and developed as above.
ELISA.
The washed bacterial pellets were suspended in 320 μl of PBS and dispensed into triplicate wells (100 μl/well) of a 96-well tissue culture plate. The wells were allowed to evaporate to dryness and then were exposed to UV light for 2 h to kill viable bacteria (11). The wells were then blocked with 3% ovalbumin in PBS overnight at 4°C and incubated with a 1:10,000 dilution of an HRP-conjugated goat anti-human C3 antibody or HRP-anti-human IgGAM for 2 h at room temperature. The wells were then washed three times in PBS and developed with substrate (Bio-Rad). The reaction was stopped with 1% oxalic acid, and the absorbance units for each well were determined in a plate reader at 405 nm (A405). The background was defined as the A405 of wells that did not contain bacterial samples and was subtracted out in each case. The background as defined above was ≤0.1 A405 in all experiments.
Hemolytic assays.
Standard hemolytic assays were used to determine the activity of complement in serum and BAL fluid samples, where one unit of hemolytic activity is the amount of undiluted serum or cBAL fluid required to cause lysis of 50% of the red blood cells (RBCs) for the classical pathway (CH50) or the alternative pathway (AP50) (23, 50, 52). The results are reported as the hemolytic units contained in 1 ml of undiluted serum or cBAL fluid. For the CH50 assay, serial dilutions of serum or cBAL fluid in GVBS++ were mixed with 2 × 107 antibody-sensitized sheep RBCs in a total volume of 500 μl. The samples were then incubated for 30 min at 37°C and placed on ice. One milliliter of ice-cold saline was added to each tube, the RBCs were pelleted, and the supernatants were then assayed for hemoglobin at 412 nm. For the AP50 assays, serial dilutions of serum or cBAL fluid were mixed in a solution of 3 parts 5% dextrose in water and 1 part GVBS containing EGTA-Mg. The samples were mixed with 107 rabbit RBCs (200 μl), incubated at 37°C for 60 min, and then placed on ice. Two milliliters of ice-cold saline was added to each sample, the RBCs were pelleted, and the supernatants were assayed for hemoglobin at 412 nm. Controls to determine the levels of 0 and 100% lysis were included in all CH50 and AP50 experiments.
Statistics.
For some experiments, a two-tailed Student's t test was used to analyze the differences between test groups and control groups.
RESULTS
Human complement protein C3 binds to M. tuberculosis.
Prior studies have established that BCG activates the alternative complement pathway and that the virulent Erdman strain of M. tuberculosis activates complement and fixes C3 protein (36, 41). To better understand this process, we incubated M. tuberculosis in 25% NHS for increasing times at 37°C, followed by washing in protease inhibitors to prevent subsequent degradation of C3 protein, and then resolved the bacterial pellets by SDS-PAGE and Western blotting (Fig. 1). Multiple C3 immunoreactive species are visualized by Western blotting (lanes 2 to 6) under reducing conditions. The 75-kDa species seen in all lanes corresponds to the β-chain of C3 (C3β). C3β is not cleaved during activation of complement and is linked to the α-chain (C3α) via a disulfide bond. C3α is 120 kDa under reducing conditions (lane 1) and contains an internal reactive thiolester. Upon activation, a small fragment of the α-chain is cleaved and released, resulting in the reactive 110-kDa (reduced) protein, C3b. This activation results in the exposure of the reactive thiolester, which can then bind to targets via a covalent ester or amide linkage. Therefore, the apparent molecular mass of C3b bound to a molecule from M. tuberculosis would be expected to be greater than 110 kDa. The immunoreactive species that are ≥110 kDa visualized in lanes 2 to 6 and the virtual absence of the 120-kDa species in these lanes indicate that C3 cleavage fragments (C3b and possibly others) are bound to acceptor molecules on the surface of the bacteria.
FIG. 1.

C3 binds to M. tuberculosis. M. tuberculosis bacilli (approximately 2.5 × 107) were incubated in NHS for 5 to 60 min, cooled on ice, and then washed three times in ice-cold PBS containing protease inhibitors. Sample buffer was added to the washed bacterial pellets or purified C3 protein, the samples were heated to 100°C, resolved by SDS-7.5% PAGE, and transferred to nitrocellulose. Western blotting was performed with a polyclonal antibody to human C3 protein. Lane 1, 200 ng of purified C3 protein; lanes 2 to 6, M. tuberculosis bacilli incubated in NHS for 5, 10, 15, 30, and 60 min, respectively. The positions of C3α and C3β are shown on the left. The molecular mass markers are shown on the right. The results shown are representative of three independent experiments.
Densitometry (data not shown) of the stable C3β species indicates that C3 binding to M. tuberculosis is maximal within 5 min. Further analysis of the results shown in Fig. 1 reveals that with increases in the time of incubation in NHS, there is increased abundance of species of lower molecular mass (<75 kDa) and the relative intensity of the 150-kDa species increases. These data suggest that bound C3b is progressively cleaved into smaller forms and that the 150-kDa species is dependent upon progressive cleavage of the originally bound protein.
To determine whether C3 binding to M. tuberculosis is dependent on activation of the complement cascade (classical and/or alternative pathways), we performed Western blotting of bacteria after incubation in 25% serum in the presence and absence of either EDTA or EGTA-Mg (Fig. 2). The complete absence of binding in the presence of Ca2+ and Mg2+ chelation by EDTA (lane 2) indicates that the binding of C3 to the bacteria occurs through the activation of complement, which is dependent on divalent cations. In the presence of selective calcium chelation by EGTA-Mg (lane 3), which abolishes the classical pathway, no decrease in C3 binding is observed, indicating that the alternative pathway of complement activation is sufficient to fix C3 on the surface of the bacteria in 25% serum.
FIG. 2.
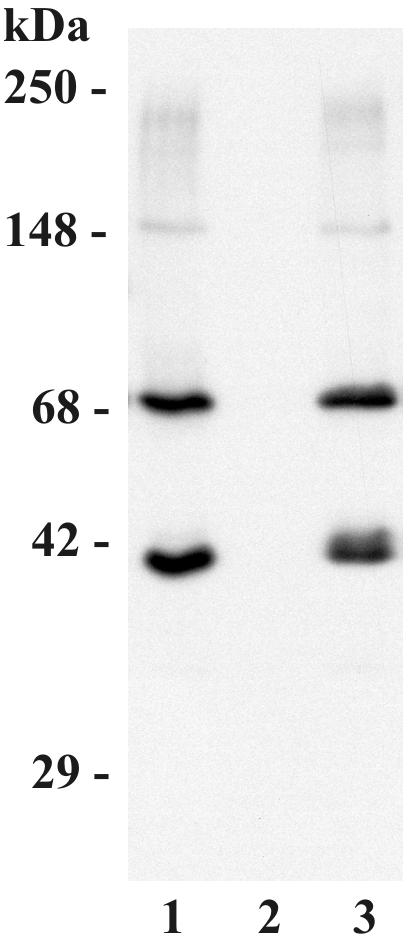
C3 binds to M. tuberculosis via the alternative pathway in 25% NHS. M. tuberculosis bacilli (2.5 × 107) were incubated in NHS in the presence or absence of 10 mM EDTA or EGTA-Mg for 30 min at 37°C. The bacterial pellets were then washed and processed for Western blotting as described in the legend of Fig. 1. Lane 1, control condition in the presence of NHS without EDTA or EGTA; lane 2, EDTA; lane 3, EGTA-Mg. The molecular mass indicators are shown on the left. The results shown are representative of three independent experiments.
C3b binds to acceptor molecules via covalent linkages between the reactive thiolester and an acceptable target containing a hydroxyl or amine group. To investigate for the presence of a covalent bond mediating the interaction between C3 and the surface of the bacteria and to determine the forms of bound C3 protein, we used a hydroxylamine treatment to release bound C3, followed by Western blotting (Fig. 3). Hydroxylamine cleaves ester bonds but leaves amide bonds intact (4). Hydroxylamine treatment markedly reduces the amount of C3 immunoreactive species associated with the bacterial pellet (lane 3). However, in the hydroxylamine treatment supernatant, immunoreactive species are readily apparent at 110 and 68 kDa, indicating C3b and C3bi, respectively, that have been released from the bacteria (lane 4). These experiments demonstrate that C3 binds covalently to its acceptor primarily via ester bonds, although amide bonds are also likely to be present. To confirm the presence of C3bi, Western blotting of bacterial lysates was also performed with a MAb specific for C3bi (Fig. 4, lane 3). Multiple immunoreactive species are observed, including a band that colocalizes with the 150-kDa species seen in association with M. tuberculosis. Taken together, these data indicate that the C3 protein binds covalently to M. tuberculosis primarily via ester bonds and is bound in the forms of C3b and C3bi; the data also suggest that the prominent 150-kDa species is C3bi bound to an acceptor molecule from M. tuberculosis. It is likely that other M. tuberculosis acceptor molecules also exist.
FIG. 3.
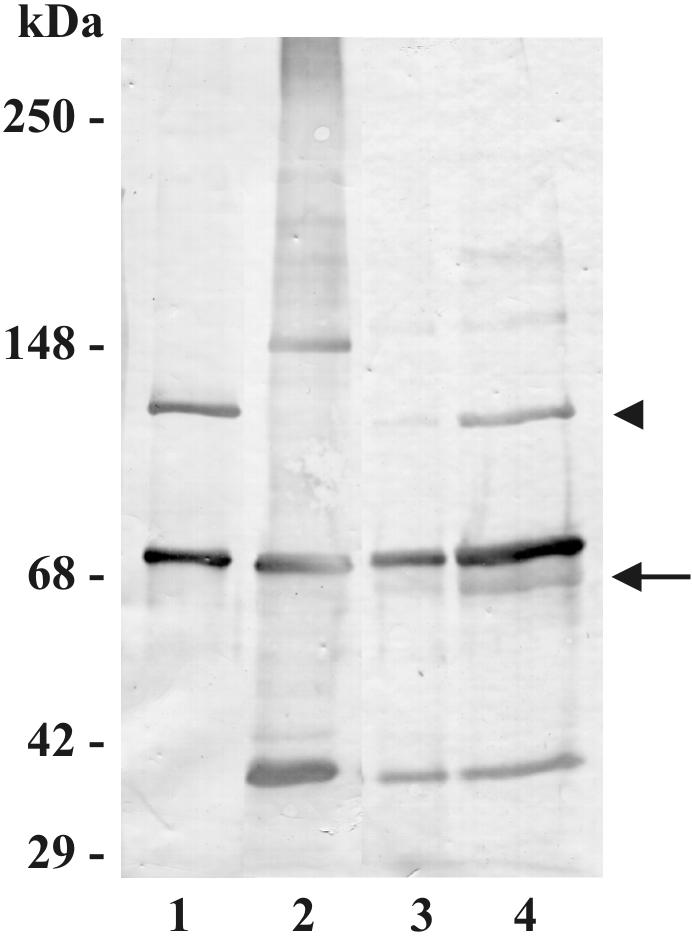
Hydroxylamine releases bound C3 from M. tuberculosis bacilli. M. tuberculosis bacilli (2.5 × 107) were incubated in NHS at 37°C for 30 min and then washed three times in ice-cold PBS containing protease inhibitors. M. tuberculosis pellets were then treated with hydroxylamine as described in Materials and Methods. The bacterial pellets and hydroxylamine supernatants were then resolved by SDS-7.5% PAGE under reducing conditions and subjected to Western blotting with an anti-human C3 antibody. Lane 1, 200 ng of purified C3 protein; lane 2, NHS-incubated M. tuberculosis bacilli without hydroxylamine treatment; lane 3, M. tuberculosis pellet after hydroxylamine treatment; lane 4, hydroxylamine supernatant from the M. tuberculosis pellet in lane 3. The molecular mass indicators are shown on the left. The positions of C3b (arrowhead) and C3bi (arrow) are shown on the right. The results shown are representative of four independent experiments.
FIG. 4.
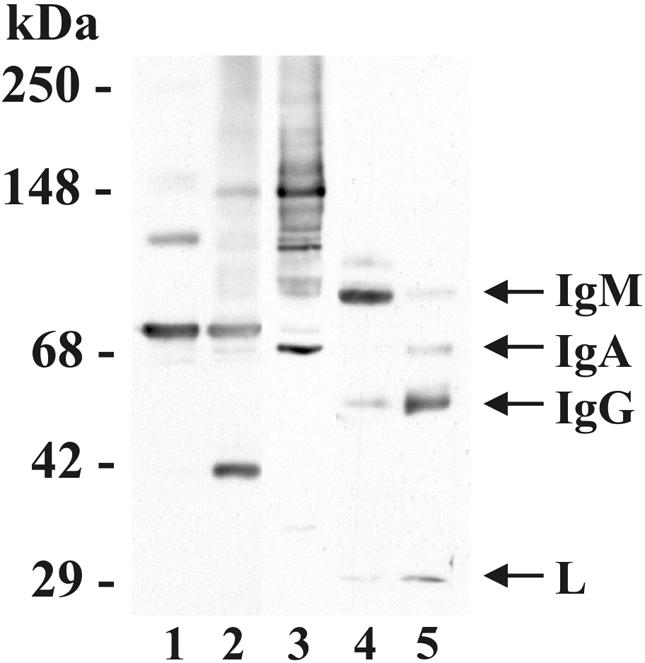
C3 binds multiple targets on M. tuberculosis. Purified C3 protein (200 ng; lane 1), washed NHS-incubated M. tuberculosis pellets (lanes 2 to 4), or 20 μl of NHS diluted 1/1,000 (lane 5), was resolved by SDS-7.5% PAGE under reducing conditions and then transferred to nitrocellulose. The lanes were divided, incubated in anti-C3 antibody (lanes 1 to 2), monoclonal anti-C3bi (lane 3), or anti-IgGAM antibody (lanes 4 to 5) and then the appropriate secondary antibody. The molecular mass markers are shown on the left. The positions of IgM, IgA, IgG, and light (L) chain are shown on the right. The results shown are representative of three independent experiments.
To examine the possibility that a fragment of C3 could be covalently bound to Ig associated with the M. tuberculosis pellet, we performed Western blotting with a polyclonal antibody that recognizes human IgG, IgA, and IgM (Fig. 4). Anti-IgGAM readily detects heavy and light chain Ig molecules from NHS (lane 5) and recognizes Ig that is associated with M. tuberculosis bacilli (lane 4). An additional faint band at ≈104 kDa is observed that is either a nonreduced Ig complex or an Ig-serum protein complex. However, clear colocalization is not observed with C3 bands from the M. tuberculosis pellets. These data suggest that IgM and IgG bind to M. tuberculosis bacilli, but there appears to be no clear Ig-C3 complex associated with M. tuberculosis.
Since the majority of C3 protein bound to the M. tuberculosis pellet is in the form of C3bi and C3b, we next used a polyclonal anti-M. tuberculosis antiserum to determine whether there are C3-M. tuberculosis colocalizing bands above 68 kDa, the molecular mass of C3bi (Fig. 5). Colocalization of C3 and the M. tuberculosis protein(s) is suggested at an approximate molecular weight of 200. In addition, there are M. tuberculosis-immunoreactive bands observed in the presence of serum (lane 3) but not in the presence of serum and EDTA (lane 4), suggesting that complement activation and binding results in a molecular weight shift of M. tuberculosis molecules. These data provide further evidence that C3 binds to acceptor sites on the surface of M. tuberculosis.
FIG. 5.
C3 binds to M. tuberculosis proteins. M. tuberculosis bacilli (2.5 × 107) were incubated with NHS in the absence (lanes 1 and 3) or presence (lanes 2 and 4) of 10 mM EDTA, washed, resolved by SDS-PAGE under reducing conditions, and transferred to nitrocellulose. The lanes were divided and then incubated in anti-C3 antibody (lanes 1 and 2), or anti-M. tuberculosis antiserum (lanes 3 and 4). The results shown are representative of three independent experiments. The molecular mass markers are shown on the left.
C3 protein binds to M. tuberculosis via the alternative and classical pathways.
The studies above and a prior study have determined that M. tuberculosis activates the alternative pathway of complement in relatively high concentrations of serum (41). However, the first exposure of this pathogen to its human host is in the lung, where the concentration of complement components is believed to be low relative to serum (7, 50). In low concentrations of complement components, the classical pathway of activation is expected to predominate, and the alternative pathway is relatively inactive (12). In order to determine if the complement cascade could serve a role in the innate immune response to M. tuberculosis in the lung, we performed a C3 binding ELISA to determine if M. tuberculosis could activate the classical pathway of complement and fix C3 protein in a low concentration of NHS (2.5%) in the presence and absence of 10 mM EDTA (chelates Ca2+ and Mg2+) or 10 mM EGTA and 7 mM Mg2+ (chelates Ca2+). In 25% NHS, EDTA inhibited C3 binding to the bacterium by 96.4% ± 2.4% (mean ± standard error of the mean; n = 5, P < 0.05), whereas EGTA-Mg inhibited C3 binding by only 4.5% ± 4.3% (n = 5). These data indicate that at 25% serum, the alternative pathway is sufficient for depositing C3 on the bacteria, which is consistent with our Western blotting data. In contrast, in 2.5% serum, the inhibition of C3 binding to M. tuberculosis by EDTA and EGTA-Mg were equivalent (inhibition by EDTA, 96.0% ± 4.3% versus inhibition by EGTA-Mg, 93.0% ± 3.4%; n = 8, P < 0.05). Thus, there is a greater contribution of the classical pathway in C3 binding in low concentrations of complement components, akin to the concentrations likely to be present in the lung.
To confirm these findings, we performed experiments to determine the contribution of both the classical and alternative pathways by using 2.5% sera that is deficient in either complement C1q of the classical pathway or FB of the alternative pathway, respectively (Fig. 6). C3 binding was significantly reduced in the absence of C1q and was restored to control values in serum reconstituted with functional C1q. These data confirm that at low concentrations of serum, the C1q-dependent classical pathway activation of complement occurs and results in the binding of C3 protein to M. tuberculosis. C3 binding to M. tuberculosis in the absence of FB was also reduced, but the data did not reach statistical significance. These data support the hypothesis that M. tuberculosis activates the classical and alternative pathways of complement, although at low concentrations of complement proteins, the classical pathway appears to have a greater role in the deposition of C3 protein on the bacterial surface.
FIG. 6.

C3 binding to M. tuberculosis is reduced in the absence of C1q. M. tuberculosis bacilli (2.5 × 107) were incubated in 2.5% pooled human serum (PHS), 2.5% C1qD, 2.5% C1qD repleted with 200 μg of C1q protein per ml of serum (C1qD + C1q), FBD, or FBD repleted with 200 μg of FB per ml of serum (FBD + FB), for 30 min at 37°C. The bacterial pellets were washed, and C3 binding was detected by ELISA. Shown are the means ± standard errors of the means of triplicate determinations of four to seven independent experiments. *, P < 0.05.
M. tuberculosis activates the classical pathway and fixes C3 protein in BAL fluid.
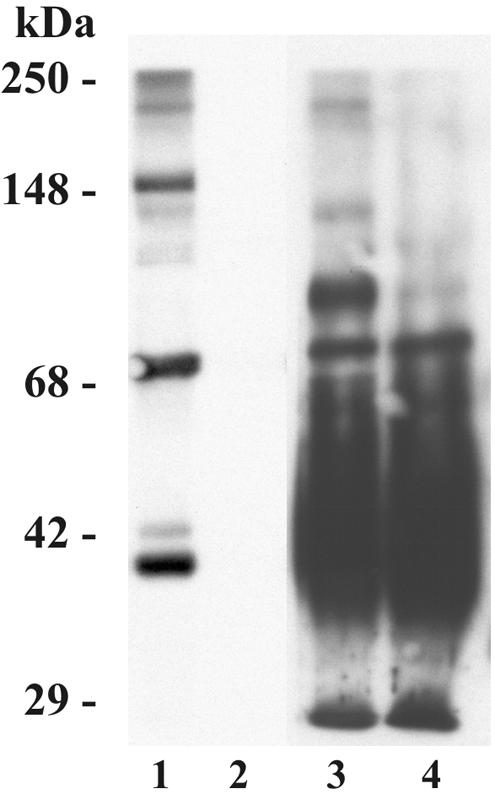
To provide more direct evidence that complement activation and C3 deposition on M. tuberculosis could occur in the lung, we performed Western blotting to determine if BAL fluid from healthy humans contains C3 protein (Fig. 7). As previously demonstrated by Watford et al. (50), C3 protein is readily detected in lavage fluids, particularly in cBAL fluid. Experiments to determine if cBAL fluid contains functional complement activity demonstrate that the classical pathway is active in cBAL fluid (Table 1). Alternative pathway activity in cBAL fluid was detected but at a very low level. Even when undiluted cBAL fluid was used, the lysis of the rabbit RBCs did not reach 50%.
FIG. 7.

C3 protein is detected in BAL fluid. Purified C3 protein (200, 100, 50, or 25 ng in lanes 1 to 4, respectively), 20 μl of BAL fluid (lane 5), or 20 μl of twofold serial dilutions of cBAL fluid (lanes 6 to 8) was resolved by SDS-PAGE under reducing conditions and subjected to Western blotting as described in the legend of Fig. 1. The molecular mass indicators are shown on the right. The positions of C3α and C3β are shown on the left. The results shown are representative of three independent experiments.
TABLE 1.
Classical and alternative pathway activity in cBAL fluid and NHSa
| Donor and sample type | Total protein (mg/ml) | CH50/mg of total protein | AP50/mg of total protein |
|---|---|---|---|
| Donor 1 | |||
| NHS | 67.0 | 7.5 | 4.8 |
| cBAL fluid | 4.1 | 10.7 | 2.4 |
| Donor 2 | |||
| NHS | 68.0 | 7.6 | 8.7 |
| cBAL fluid | 2.8 | 9.2 | 0.3 |
| Donor 3 | |||
| NHS | 62.0 | 8.6 | 8.4 |
| cBAL fluid | 2.1 | 5.9 | NDb |
Total protein, CH50/ml of sample, and AP50/ml of sample were determined for matched cBAL fluid and NHS samples as described in Materials and Methods. The values for CH50/ml and AP50/ml were then normalized to the total protein concentration for each donor. Results are given as the mean hemolytic units contained in 1 ml of sample ± SEM. CH50 is 516.4 ± 9.6 per ml of serum and 27.3 ± 9.1 per ml of cBAL (n = 3). AP50 is 478.7 ± 82.2 per ml of serum and 3.6 ± 3.2 per ml of cBAL (n = 3).
ND, not detected.
Since cBAL fluid contains easily detectable amounts of C3 protein and functional hemolytic activity, we hypothesized that C3 protein in lung lining fluid could bind to M. tuberculosis. The ELISA results shown in Fig. 8 demonstrate that C3 protein from cBAL fluid binds to M. tuberculosis. This binding is nearly abolished in the presence of EGTA-Mg, providing evidence that C3 binding results from complement activation and that this is due to the classical pathway.
FIG. 8.
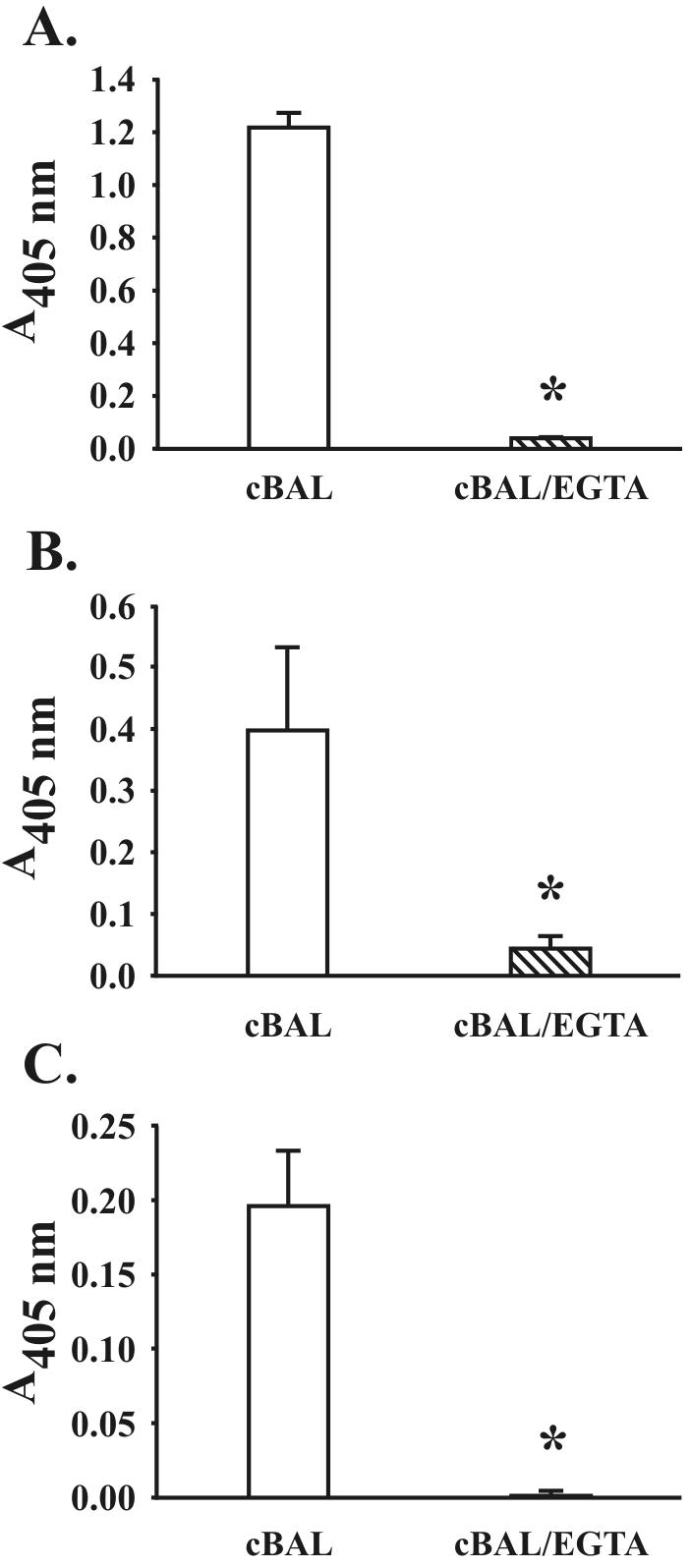
M. tuberculosis fixes C3 protein in BAL fluid. M. tuberculosis bacilli (2.5 × 107) were incubated with 220 μl of cBAL fluid in the presence and absence of 10 mM EDTA or 10 mM EGTA-Mg for 30 min at 37°C, washed in ice-cold PBS containing protease inhibitors, and dispensed into triplicate wells of a 96-well microtiter plate. C3 binding was then determined by ELISA. The background (<0.05 A405) was equal to the results obtained in the presence of EDTA and was subtracted from the results shown. Shown are the means ± standard deviations of triplicate determinations from the three different donor cBAL fluid samples reported in Table 1 (where A is donor 1, B is donor 2, and C is donor 3). *, P < 0.05.
Classical pathway activation can be initiated by Ig binding followed by formation of functional C1 or may be independent of Ig (28). To determine if Ig in normal BAL fluid and NHS could bind M. tuberculosis, we incubated M. tuberculosis in cBAL fluid or NHS. We then detected Ig binding by ELISA. The results shown in Fig. 9 demonstrate that in both cBAL fluid and NHS from healthy donors, Ig binds to M. tuberculosis bacilli in a concentration-dependent manner. Taken together with our Western blotting data (Fig. 4), these results provide evidence that Ig in the alveolar surface material from healthy humans has the capacity to bind to M. tuberculosis bacilli. Whether Ig binding in the airspace modulates complement opsonization of the bacilli in this location is currently not known.
FIG. 9.

M. tuberculosis binds Ig in BAL fluid and NHS. M. tuberculosis bacilli (2.5 × 107) were incubated with increasing amounts of cBAL fluid (A) or NHS (B) in a final volume of 500 μl as indicated for 30 min at 37°C, washed in ice-cold PBS containing protease inhibitors, and dispensed into triplicate wells of a 96-well microtiter plate and evaporated to dryness. The wells were then blocked, incubated in HRP anti-human IgGAM antibody, and developed with peroxidase substrate. The A405 was determined for each well, and the background (wells that did not contain M. tuberculosis; A405 ≤ 0.05) was subtracted out in each case. Shown are the means ± standard deviations of triplicate determinations from one experiment representative of three.
DISCUSSION
M. tuberculosis is an obligate human pathogen that has evolved the ability to avoid some host defense mechanisms that typically kill other pathogens. For example, the phagocytosis of M. tuberculosis by human mononuclear phagocytes occurs without stimulating an oxidant burst, and the subsequent intracellular trafficking of the bacteria leads to limited fusion of the phagosome with lysosomes (2, 9). In some cases M. tuberculosis is able to use host defense mechanisms to its advantage. Interactions between M. tuberculosis and C3 appear to be advantageous to the bacteria, since C3 opsonization results in enhanced phagocytosis of the bacteria by human alveolar and monocyte-derived macrophages that are known to allow for growth of M. tuberculosis in vitro (9, 16, 41). In order for complement to play a role in the phagocytosis of M. tuberculosis in vivo, the tissue site where M. tuberculosis resides must support opsonization of the bacterium with C3.
Studies performed with human serum have shown that M. tuberculosis and M. bovis BCG can activate the alternative pathway of complement and fix C3 to its surface (36, 41). In addition, the complement component C2a can interact with the surface of pathogenic mycobacteria and form a classic-like C3 convertase, which participates in C3b opsonization of the bacteria (43). Recently, the heparin-binding hemagglutinin from the surface of M. tuberculosis has been reported as a C3 acceptor molecule (29). Apart from these studies, our knowledge regarding the requirements for and regulation of C3 binding to M. tuberculosis, particularly in the lung of the naïve host, is limited.
In the present studies, we demonstrate for the first time that in serum C3 binding to M. tuberculosis is rapid, initiated by activation of the classical as well as the alternative pathways, and occurs via covalent linkages to multiple bacterial surface targets in the forms of C3b and C3bi. In addition, our studies indicate that the classical pathway mediates C3 binding to M. tuberculosis from lavage fluid. Thus, these studies provide evidence that M. tuberculosis can activate complement and bind C3 protein in the airspace of the naive host.
We found that M. tuberculosis activates the classical pathway, in addition to the alternative pathway, in human serum. When the concentration of serum was 25%, the alternative pathway was sufficient for binding C3 since inhibition of the classical pathway by selective calcium chelation (EGTA-Mg) had no effect on the amount of C3 binding. However, when the concentration of serum was reduced to 2.5%, inhibition of the classical pathway by selective calcium chelation reduced total C3 binding by 88.7%. In low concentrations of serum, involvement of the classical pathway was confirmed by experiments showing that depletion of C1q from serum significantly reduced C3 binding and that addition of purified C1q to the depleted serum returned C3 binding to the control values. These findings are of interest, since the classical C1q-dependent mechanism is dependent on specific Ig bound to the target. In this regard, sera from patients with active tuberculosis disease contain circulating immune complexes (39) and mediate classical pathway activation and C3 binding by BCG (15). Complement activation correlates with the amount of anti-lipoarabinomannan antibody in sera from tuberculosis patients compared to healthy controls (15). In contrast to these studies, our experiments were performed in sera that came from healthy donors. At least two possible mechanisms may explain our findings. First, naturally occurring, low-titer, specific antibody to several mycobacteria is present in nonimmune serum (3). This low-titer antibody can participate in the classical pathway cascade (42). Second, antibody-independent C1q binding to microorganisms has been described that also has the capacity to activate the classical pathway (26, 28). Our present studies indicate that Ig from nonimmune serum and BAL fluid binds to M. tuberculosis (Fig. 4 and 9), although we have not determined if Ig binding to M. tuberculosis is necessary for activation of the classical pathway.
Based on our experiments where selective calcium chelation nearly abolished C3 binding to M. tuberculosis, we expected to observe little to no binding of C3 in 2.5% C1q-depleted serum. However, low-level C3 binding activity remains (Fig. 6). These data suggest that there is a C1q-independent pathway that is further reduced in the absence of calcium. Calcium chelation does not affect the alternative pathway. However, we have not ruled out the possibility that the lectin pathway may play a role in the deposition of C3 to the bacterial surface. The lectin pathway is not dependent on C1q or calcium per se, but binding of mannose-binding lectin (MBL) to microbial surfaces is calcium dependent, and this binding activates complement through the actions of two MBL-associated serine proteases, MASP-1 and MASP-2 (32, 48, 49, 54). Thus, calcium chelation would reduce MBL binding and consequently lectin pathway activation. We have not determined whether MBL interacts with M. tuberculosis, but binding of MBL to Mycobacterium avium has been reported (35), and certain allelic differences in the MBL gene are associated with protection against tuberculosis meningitis in children (17). Likewise, the C2a pathway for virulent mycobacteria is not dependent on C1q, although C2a is not known to exist in human serum in the absence of the activation of C2 (43). However, C2a is generated by activation of the classical pathway (30) and may be present at sites of inflammation including the lung (7, 14). Therefore, it remains possible that the C2a pathway may amplify classical-pathway-mediated C3 binding to M. tuberculosis.
Taken together, our data demonstrate that the alternative pathway is sufficient for depositing C3 on the surface of M. tuberculosis in high concentrations of complement components, but as the concentration of complement decreases, the classical pathway plays a relatively greater role. This situation may more closely resemble that present in the lung, particularly during primary infection.
Our data show that C3 binds via ester and amide linkages to M. tuberculosis. The internal reactive thiolester of C3 becomes available for covalent binding to hydroxyl or amine groups after the protein is activated by proteases (24, 44). The preference for ester versus amide bond formation is dependent on the protein and the available target. As opposed to C4, C3 preferentially forms ester bonds if the appropriate hydroxyl group is available (24). Our data indicate that C3 binds covalently to M. tuberculosis primarily via ester bonds, although amine-containing epitopes on the surface of M. tuberculosis may also play a role.
The molecular mass range of the C3 immunoreactive species observed in our Western blots and the apparent colocalization of M. tuberculosis-C3 immunoreactive species suggest that C3 binding occurs to multiple acceptor molecules on the surface of M. tuberculosis. These observations are supported by Western blotting with the MAb to C3bi, which reveals multiple species at molecular masses greater than 68 kDa. Importantly, our data fail to demonstrate binding of C3 to an M. tuberculosis-Ig complex. The bacterial determinants responsible for complement activation and C3 binding for M. tuberculosis are not known. However, the heparin-binding hemagglutinin of M. tuberculosis has been identified as one C3 acceptor molecule (29). Based on our data, it is probable that multiple C3 acceptor molecules exist on M. tuberculosis.
Our studies demonstrate that the C3 protein bound to M. tuberculosis is in the forms of C3b and C3bi. These data are important since intact C3b is necessary for the propagation of the complement cascade toward the terminal components and membrane attack complex, while C3bi cannot participate in this reaction. Additionally, C3b is a ligand for CR1, whereas C3bi is a ligand for CR3 and CR4 (38). C3bi is formed from C3b when the latter is cleaved by factor I in association with proteins that serve as cofactors, such as factor H or CR1 (12). The efficiency of the conversion of C3b to C3bi on the surfaces of microorganisms depends on a competition for binding C3b by factors H and B. C3b associated with factor B cannot associate with factor I. Conversely, C3b associated with factor H does not interact with factor D of the alternative pathway (for a review, see reference 12). Although the details of these reactions are not known for M. tuberculosis, conversion of C3b to C3bi may be important in that most C3bi-coated organisms would be phagocytosed by CR3 and CR4 rather than CR1. In contrast, intact C3b on the surface of M. tuberculosis may allow continuation of the complement cascade, producing inflammatory mediators that may be important in host defense against M. tuberculosis infection (1, 21).
To provide evidence that the complement system in the lung could support C3 opsonization of M. tuberculosis, we concentrated human BAL fluid, characterized the CH50 and AP50 activities of this fluid, and studied the ability of the fluid to support C3 binding to the bacteria. Our data are consistent with other studies in that the classical pathway appears to be more active in cBAL fluid than the alternative pathway, since lysis of rabbit RBCs is minimal under conditions of our assay (50). Our data also show that when cBAL fluid is used as a complement source, C3 binds to M. tuberculosis, and this binding is abolished in the presence of EDTA and EGTA-Mg. These results provide evidence that C3 binding to M. tuberculosis from lavage fluid is mediated by the classical pathway. Although we have not directly ruled out lectin- or C2a-pathway-mediated binding of C3, MBL and C2a have not been described in the lung in the absence of inflammation (14, 37). Possibly, the existence of the C2a or lectin pathway in the lung may enhance the activation of C3, functionally replacing the minimally active alternative pathway.
The lung complement system has long been believed to be both helpful and detrimental to the host. On the one hand, a well-regulated highly localized complement response to inhaled pathogens has the potential to greatly increase host defense. On the other hand, unregulated complement activation in the lung could result in tissue damage and significant toxicity. In vivo data for the role of C3 and CR in the pathogenesis of tuberculosis are not conclusive. The C3−/− mouse has been studied to examine the role of C3 in the pathogenesis of M. avium (5). In the studies reported, 104 or 106 bacteria were administered via retro-orbital injection, rather than by low-dose aerosol, making it difficult to draw clear conclusions concerning the respiratory pathogen M. tuberculosis. Similarly, 2 ×105 CFU of M. tuberculosis given by tail vein injection failed to demonstrate a susceptibility difference between CR3−/− mice and wild-type mice (19).
The data presented here provide evidence that the complement system likely plays a role in the pathogenesis of tuberculosis during the innate immune response by opsonizing M. tuberculosis with specific C3 cleavage products for phagocytosis into the alveolar macrophage through activation of the classical complement pathway in the lung. We speculate that during the innate immune response when then initial inoculum of bacteria is believed to be low, survival of M. tuberculosis bacilli in the lungs (hence, successful infection) could be enhanced by a relatively small increase in phagocytosis by the alveolar macrophage. Thus, complement opsonization of M. tuberculosis likely plays a critically important role during the first encounter of the microbe with the human host.
Acknowledgments
This work was supported by NIH grants HL03885 (J.S.F.) and AI33004 (L.S.S.), and by grant M01-RR-59 from the National Center for Research Resources, General Clinical Research Centers Program, NIH.
The authors thank Thomas M. Kaufman, Department of Internal Medicine, the Iowa City Veterans Affairs Medical Center and the University of Iowa, Iowa City, for his expert technical assistance.
Editor: S. H. E. Kaufmann
REFERENCES
- 1.Actor, J. K., E. Breij, R. A. Wetsel, H. Hoffmann, R. L. Hunter, Jr., and C. Jagannath. 2001. A role for complement C5 in organism containment and granulomatous response during murine tuberculosis. Scand. J. Immunol. 53:464-474. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong, J. A., and P. D. Hart. 1971. Response of cultured macrophages to Mycobacterium tuberculosis, with observations on fusion of lysosomes with phagosomes. J. Exp. Med. 134:713-740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bardana, E. J. J., J. K. McClatchy, R. S. Farr, and P. Minden. 1973. Universal occurrence of antibodies to tubercle bacilli in sera from non-tuberculous and tuberculous individuals. Clin. Exp. Immunol. 13:65-77. [PMC free article] [PubMed] [Google Scholar]
- 4.Berger, M. 1990. Third component of human complement: C3. Methods Enzymol. 184:619-628. [DOI] [PubMed] [Google Scholar]
- 5.Bohlson, S. S., J. A. Strasser, J. J. Bower, and J. S. Schorey. 2001. Role of complement in Mycobacterium avium pathogenesis: in vivo and in vitro analyses of the host response to infection in the absence of complement component C3. Infect. Immun. 69:7729-7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole, F. S., W. J. J. Matthews, T. H. Rossing, D. J. Gash, N. A. Lichtenberg, and J. E. Pennington. 1983. Complement biosynthesis by human bronchoalveolar macrophages. Clin. Immunol. Immunopathol. 27:153-159. [DOI] [PubMed] [Google Scholar]
- 7.Coonrod, J. D., and K. Yoneda. 1981. Complement and opsonins in alveolar secretions and serum of rats with pneumonia due to Streptococcus pneumoniae. Rev. Infect. Dis. 3:310-322. [DOI] [PubMed] [Google Scholar]
- 8.Cywes, C., N. L. Godenir, H. C. Hoppe, R. R. Scholle, L. M. Steyn, R. E. Kirsch, and M. R. Ehlers. 1996. Nonopsonic binding of Mycobacterium tuberculosis to human complement receptor type 3 expressed in Chinese hamster ovary cells. Infect. Immun. 64:5373-5383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Douvas, G. S., E. M. Berger, J. E. Repine, and A. J. Crowle. 1986. Natural mycobacteriostatic activity in human monocyte-derived adherent cells. Am. Rev. Respir. Dis. 134:44-48. [DOI] [PubMed] [Google Scholar]
- 10.Ernst, J. D. 1998. Macrophage receptors for Mycobacterium tuberculosis. Infect. Immun. 66:1277-1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferguson, J. S., D. R. Voelker, F. X. McCormack, and L. S. Schlesinger. 1999. Surfactant protein D binds to Mycobacterium tuberculosis bacilli and lipoarabinomannan via carbohydrate-lectin interactions resulting in reduced phagocytosis of the bacteria by macrophages. J. Immunol. 163:312-321. [PubMed] [Google Scholar]
- 12.Figueroa, J. E., and P. Densen. 1991. Infectious diseases associated with complement deficiencies. Clin. Microbiol. Rev. 4:359-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giclas, P. C., T. E. King, S. L. Baker, J. Russo, and P. M. Henson. 1987. Complement activity in normal rabbit bronchoalveolar fluid. Description of an inhibitor of C3 activation. Am. Rev. Respir. Dis. 135:403-411. [DOI] [PubMed] [Google Scholar]
- 14.Greffard, A., J. J. Bourgarit, S. le Maho, and C. R. Lambre. 1987. Determination of the complement component C2 by ELISA in human serum and bronchoalveolar lavage fluids. Immunol. Lett. 15:145-151. [DOI] [PubMed] [Google Scholar]
- 15.Hetland, G., H. G. Wiker, K. Hogasen, B. Hamasur, S. B. Svenson, and M. Harboe. 1998. Involvement of antilipoarabinomannan antibodies in classical complement activation in tuberculosis. Clin. Diagn. Lab. Immunol. 5:211-218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirsch, C. S., J. J. Ellner, D. G. Russell, and E. A. Rich. 1994. Complement receptor-mediated uptake and tumor necrosis factor-alpha-mediated growth inhibition of Mycobacterium tuberculosis by human alveolar macrophages. J. Immunol. 152:743-753. [PubMed] [Google Scholar]
- 17.Hoal-Van Helden, E. G., J. Epstein, T. C. Victor, D. Hon, L. A. Lewis, N. Beyers, D. Zurakowski, A. B. Ezekowitz, and P. D. Van Helden. 1999. Mannose-binding protein B allele confers protection against tuberculous meningitis. Pediatr. Res. 45:459-464. [DOI] [PubMed] [Google Scholar]
- 18.Horwitz, M. A., and S. C. Silverstein. 1980. Influence of the Escherichia coli capsule on complement fixation and on phagocytosis and killing by human phagocytes. J. Clin. Investig. 65:82-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu, C., T. Mayadas-Norton, K. Tanaka, J. Chan, and P. Salgame. 2000. Mycobacterium tuberculosis infection in complement receptor 3-deficient mice. J. Immunol. 165:2596-2602. [DOI] [PubMed] [Google Scholar]
- 20.Hunninghake, G. W., J. E. Gadek, O. Kawanami, V. J. Ferrans, and R. G. Crystal. 1979. Inflammatory and immune processes in the human lung in health and disease: evaluation by bronchoalveolar lavage. Am. J. Pathol. 97:149-206. [PMC free article] [PubMed] [Google Scholar]
- 21.Jagannath, C., H. Hoffmann, E. Sepulveda, J. K. Actor, R. A. Wetsel, and R. L. Hunter. 2000. Hypersusceptibility of A/J mice to tuberculosis is in part due to a deficiency of the fifth complement component (C5). Scand. J. Immunol. 52:369-379. [DOI] [PubMed] [Google Scholar]
- 22.Klickstein, L. B., S. F. Barbashov, T. Liu, R. M. Jack, and A. Nicholson-Weller. 1997. Complement receptor type 1 (CR1, CD35) is a receptor for C1q. Immunity 7:345-355. [DOI] [PubMed] [Google Scholar]
- 23.Kolb, W. P., L. M. Kolb, R. A. Wetsel, W. R. Rogers, and J. O. Shaw. 1981. Quantitation and stability of the fifth component of complement (C5) in bronchoalveolar lavage fluids obtained from non-human primates. Am. Rev. Respir. Dis. 123:226-231. [DOI] [PubMed] [Google Scholar]
- 24.Law, S. K., N. A. Lichtenberg, and R. P. Levine. 1979. Evidence for an ester linkage between the labile binding site of C3b and receptive surfaces. J. Immunol. 123:1388-1394. [PubMed] [Google Scholar]
- 25.Leemans, J. C., N. P. Juffermans, S. Florquin, N. van Rooijen, M. J. Vervoordeldonk, A. Verbon, S. J. van Deventer, and T. van der Poll. 2001. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J. Immunol. 166:4604-4611. [DOI] [PubMed] [Google Scholar]
- 26.Leist-Welsh, P., and A. B. Bjornson. 1982. Immunoglobulin-independent utilization of the classical complement pathway in opsonophagocytosis of Escherichia coli by human peripheral leukocytes. J. Immunol. 128:2643-2651. [PubMed] [Google Scholar]
- 27.Matsushita, M., M. Kuraya, N. Hamasaki, M. Tsujimura, H. Shiraki, and T. Fujita. 2002. Activation of the lectin complement pathway by H-ficolin (Hakata antigen). J. Immunol. 168:3502. [DOI] [PubMed] [Google Scholar]
- 28.Mintz, C. S., P. I. Arnold, W. Johnson, and D. R. Schultz. 1995. Antibody-independent binding of complement component C1q by Legionella pneumophila. Infect. Immun. 63:4939-4943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mueller-Ortiz, S. L., A. R. Wanger, and S. J. Norris. 2001. Mycobacterial protein HbhA binds human complement component C3. Infect. Immun. 69:7501-7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller-Eberhard, H. J., M. J. Polley, and M. A. Calcott. 1967. Formation and functional significance of a molecular complex derived from the second and the fourth component of human complement. J. Exp. Med. 125:359-380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Myones, B. L., J. G. Dalzell, N. Hogg, and G. D. Ross. 1988. Neutrophil and monocyte cell surface p150,95 has iC3b-receptor (CR4) activity resembling CR3. J. Clin. Investig. 82:640-651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neth, O., D. L. Jack, A. W. Dodds, H. Holzel, N. J. Klein, and M. W. Turner. 2000. Mannose-binding lectin binds to a range of clinically relevant microorganisms and promotes complement deposition. Infect. Immun. 68:688-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pangburn, M. K. 1986. The alternative pathway, p. 45-62. In G. D. Ross (ed.), Immunobiology of the complement system: an introduction for research and clinical medicine. Academic Press, Inc., Orlando, Fla.
- 34.Petersen, S. V., S. Thiel, L. Jensen, T. Vorup-Jensen, C. Koch, and J. C. Jensenius. 2000. Control of the classical and the MBL pathway of complement activation. Mol. Immunol. 37:803-811. [DOI] [PubMed] [Google Scholar]
- 35.Polotsky, V. Y., J. T. Belisle, K. Mikusova, R. A. Ezekowitz, and K. A. Joiner. 1997. Interaction of human mannose-binding protein with Mycobacterium avium. J. Infect. Dis. 175:1159-1168. [DOI] [PubMed] [Google Scholar]
- 36.Ramanathan, V. D., J. Curtis, and J. L. Turk. 1980. Activation of the alternative pathway of complement by mycobacteria and cord factor. Infect. Immun. 29:30-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reading, P. C., L. S. Morey, E. C. Crouch, and E. M. Anders. 1997. Collectin-mediated antiviral host defense of the lung: evidence from influenza virus infection of mice. J. Virol. 71:8204-8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ross, G. D. 1986. Opsonization and membrane complement receptors, p. 87-114. In G. D. Ross (ed.), Immunobiology of the complement system: an introduction for research and clinical medicine. Academic Press, Inc., Orlando, Fla.
- 39.Sai Baba, K. S., K. D. Moudgil, R. C. Jain, and L. M. Srivastava. 1990. Complement activation in pulmonary tuberculosis. Tubercle 71:103-107. [DOI] [PubMed] [Google Scholar]
- 40.Schlesinger, L. S. 1993. Macrophage phagocytosis of virulent but not attenuated strains of Mycobacterium tuberculosis is mediated by mannose receptors in addition to complement receptors. J. Immunol. 150:2920-2930. [PubMed] [Google Scholar]
- 41.Schlesinger, L. S., C. G. Bellinger-Kawahara, N. R. Payne, and M. A. Horwitz. 1990. Phagocytosis of Mycobacterium tuberculosis is mediated by human monocyte complement receptors and complement component C3. J. Immunol. 144:2771-2780. [PubMed] [Google Scholar]
- 42.Schlesinger, L. S., and M. A. Horwitz. 1994. A role for natural antibody in the pathogenesis of leprosy: antibody in nonimmune serum mediates C3 fixation to the Mycobacterium leprae surface and hence phagocytosis by human mononuclear phagocytes. Infect. Immun. 62:280-289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schorey, J. S., M. C. Carroll, and E. J. Brown. 1997. A macrophage invasion mechanism of pathogenic mycobacteria. Science 277:1091-1093. [DOI] [PubMed] [Google Scholar]
- 44.Sim, R. B., T. M. Twose, D. S. Paterson, and E. Sim. 1981. The covalent-binding reaction of complement component C3. Biochem. J. 193:115-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stokes, R. W., I. D. Haidl, W. A. Jefferies, and D. P. Speert. 1993. Mycobacteria-macrophage interactions. Macrophage phenotype determines the nonopsonic binding of Mycobacterium tuberculosis to murine macrophages. J. Immunol. 151:7067-7076. [PubMed] [Google Scholar]
- 46.Stokes, R. W., L. M. Thorson, and D. P. Speert. 1998. Nonopsonic and opsonic association of Mycobacterium tuberculosis with resident alveolar macrophages is inefficient. J. Immunol. 160:5514-5521. [PubMed] [Google Scholar]
- 47.Strunk, R. C., D. M. Eidlen, and R. J. Mason. 1988. Pulmonary alveolar type II epithelial cells synthesize and secrete proteins of the classical and alternative complement pathways. J. Clin. Investig. 81:1419-1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tan, S. M., M. C. Chung, O. L. Kon, S. Thiel, S. H. Lee, and J. Lu. 1996. Improvements on the purification of mannan-binding lectin and demonstration of its Ca(2+)-independent association with a C1s-like serine protease. Biochem. J. 319:329-332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thiel, S., T. Vorup-Jensen, C. M. Stover, W. Schwaeble, S. B. Laursen, K. Poulsen, A. C. Willis, P. Eggleton, S. Hansen, U. Holmskov, K. B. Reid, and J. C. Jensenius. 1997. A second serine protease associated with mannan-binding lectin that activates complement. Nature 386:506-510. [DOI] [PubMed] [Google Scholar]
- 50.Watford, W. T., A. J. Ghio, and J. R. Wright. 2000. Complement-mediated host defense in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 279:L790-L798. [DOI] [PubMed] [Google Scholar]
- 51.Watford, W. T., J. R. Wright, C. G. Hester, H. Jiang, and M. M. Frank. 2001. Surfactant protein A regulates complement activation. J. Immunol. 167:6593-6600. [DOI] [PubMed] [Google Scholar]
- 52.Whaley, K. 1985. Measurement of complement, p. 77-139. In K. Whaley (ed.), Methods in complement for clinical immunologists. Churchill Livingstone, New York, N.Y.
- 53.Wong, N. K. H., M. Kojima, J. Dobo, G. Ambrus, and R. B. Sim. 1999. Activities of the MBL-associated serine proteases (MASPs) and their regulation by natural inhibitors. Mol. Immunol. 36:853-861. [DOI] [PubMed] [Google Scholar]
- 54.Zhang, Y., C. Suankratay, X. H. Zhang, T. F. Lint, and H. Gewurz. 1999. Lysis via the lectin pathway of complement activation: minireview and lectin pathway enhancement of endotoxin-initiated hemolysis. Immunopharmacology 42:81-90. [DOI] [PubMed] [Google Scholar]