

Graphical abstract

Keywords: Cyclopropanes, Organocatalysis, Phase-transfer catalysis, Bifunctional, Cinchona alkaloids
Abstract
The first phase-transfer catalyzed cyclopropanation reaction of chalcones using bromomalonates as the nucleophiles in a Michael Initiated Ring Closing reaction (MIRC) was developed. Key to success was the use of a free OH-containing cinchona alkaloid ammonium salt catalyst and carefully optimized liquid/liquid reaction conditions. The reaction performed well for electron neutral and electron deficient chalcones giving the products in yields up to 98% and with enantiomeric ratios up to 91:9.
Introduction
Chiral cyclopropanes are highly versatile organic molecules. Their importance is a result of their unique reactivity which makes them outstanding synthons to access complex molecular scaffolds1 and because of their presence in different biologically active compounds.2 Although a variety of versatile strategies for the stereoselective syntheses of chiral cyclopropanes have been reported,1–3 the development of novel strategies to access these important compounds is a worthwhile goal. Asymmetric phase-transfer catalysis has proven its potential in numerous demanding applications4–7 and a variety of carefully fine-tuned catalysts have been reported in the past.4–9 Surprisingly, the use of (chiral) phase-transfer catalysts (PTCs) to facilitate (stereoselective) cyclopropanation reactions has so far been limited to a few examples only.10–12 Based on our recent success in developing novel TADDOL-derived PTCs9 and the knowledge gathered therein we have now undertaken systematic investigations concerning the PT-catalyzed Michael Initiated Ring Closing (MIRC) reaction of bromomalonates 1 with trans-chalcones (2) to furnish cyclopropanes 3 in the presence of a variety of known chiral quaternary ammonium salt PTCs (Scheme 1). The asymmetric cyclopropanation of chalcones with malonates is an unprecedented reaction and should therefore broaden the application scope of asymmetric phase-transfer catalysis toward such interesting chiral moieties.
Scheme 1.
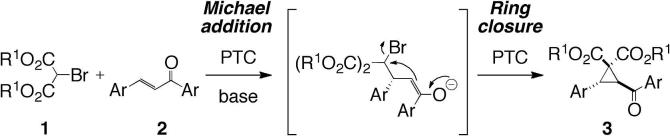
Targeted phase-transfer catalyzed MIRC-reaction of bromomalonates with chalcones.
Results and discussion
Table 1 gives an overview of the most significant results obtained in the course of a thorough and systematic screening. Initial studies were carried out using diethyl bromomalonate (1a) as the nucleophile in the presence of differently substituted cinchona alkaloid catalysts 4 and 5 under liquid/solid conditions (entries 1–9). The first challenge to be addressed was the identification of reaction conditions suppressing the rapid base-catalyzed dimerization of 1.13 Although the combination of different solvents with solid Cs2CO3 furnished the targeted product 3 in reasonable yields only very low selectivities were obtained. Out of different other solvent/solid base combinations only the use of K3PO4/toluene in combination with the free OH-containing quinidine-derived catalyst 4c furnished 3 in moderate selectivity at room temperature (entry 9).
Table 1.

| Entry | 1 | Cat. | Solv. | Base (equiv) | Concd. | Yielda (%) | er b (+/−) |
|---|---|---|---|---|---|---|---|
| 1 | 1a | 4a | CH2Cl2 | Cs2CO3 (4×) | Ac | 70 | 51:49 |
| 2 | THF | 77 | 43:57 | ||||
| 3 | Toluene | 30 | 44:56 | ||||
| 4 | 4b | THF | 45 | 46:54 | |||
| 5 | 4c | 40 | 44:56 | ||||
| 6 | 5a | 41 | 51:49 | ||||
| 7 | 5c | 14 | 52:48 | ||||
| 8 | 5d | 55 | 57:43 | ||||
| 9 | 4c | Toluene | K3PO4 (10×) | 45 | 31:69 | ||
| 10 | 4a | K3PO4 (50%) (10×) | 84 | 50:50 | |||
| 11 | 4b | 36 | 38:62 | ||||
| 12 | 4c | 48 | 26:74 | ||||
| 13 | 4d | 57 | 40:60 | ||||
| 14 | 4e | 44 | 31:69 | ||||
| 15 | 4f | 67 | 50:50 | ||||
| 16 | 5b | 30 | 70:30 | ||||
| 17 | 5d | 45 | 73:27 | ||||
| 18 | 6 | 57 | 47:53 | ||||
| 19 | 4c | Mesitylene | 43 | 24:76 | |||
| 20 | Toluene | Li2CO3 (50%) (10×) | 7 | 21:79 | |||
| 21 | K2CO3 (50%) (10×) | 35 | 24:76 | ||||
| 22 | Mesitylene | 54 | 22:78 | ||||
| 23 | 1b | n.r. | — | ||||
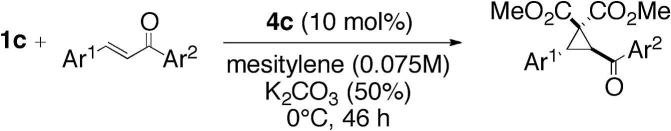
| 24 | 1c | 63 | 17:83 | ||||
| 25 | Bc | 10 | 17:83 | ||||
| 26 | Cc | 54 | 15:85 | ||||
| 27 | Dc | 83 | 15:85 | ||||
| 28 | Ec | 82 | 13:87 | ||||
| 29 | Fc | 39 | 10:90 | ||||
| 30 | 5d | Ec | 61 | 75:25 |
Isolated yield.
Determined by HPLC using a chiral stationary phase.
A: 3 equiv 2, rt, 22 h, 0.15 M; B: 3 equiv 1a, RT, 22 h, 0.15 M; C: 3 equiv 2, rt, 46 h, 0.075 M; D: 6 equiv 2, rt, 46 h, 0.075 M; E: 6 equiv 2, 0 °C, 46 h, 0.075 M; F: 6 equiv 2, −20 °C, 46 h, 0.075 M.
During the course of our investigations the Adamo group reported the first PT-catalyzed cyclopropanation of highly electrophilic 4-nitro-5-styrylisoxazoles using cinchona alkaloid-derived PTCs under liquid/liquid (toluene/aq K3PO4) conditions.11 Interestingly, applying analogous conditions to our reaction (entries 10–18), the presence of the free sec-9-OH-group in the cinchona alkaloid catalyst was found to be crucial to achieve reasonable selectivities. Whereas O-alkylated derivatives (e.g., 4a) as well as our standard TADDOL-based catalyst 6 gave racemic 3 only, the bifunctional quinidine and quinine-derived catalysts 4c and 5d were found to be the most active ones, giving 3 with enantiomeric ratios up to 74:26 at room temperature (entries 12, 17). Compared to the corresponding cinchonine and cinchonidine based catalysts 4b and 5b the 6′-OMe group of 4c and 5d has a beneficial effect on yield and selectivity. In sharp contrast, a 9-O-protected free 6′-OH based catalyst (4f, entry 15) was found to be absolutely nonselective.14 Further investigations using 4c under liquid/liquid biphasic conditions (entries 19–22) allowed us to identify the combination of mesitylene and aqueous K2CO3 as the most promising one with respect to yield and enantioselectivity (entry 22). Testing the influence of different ester-groups we found that the t-butyl ester 1b did not give any product at all whereas the methyl ester 1c performed reasonably well at room temperature already (entry 24). To improve yield and enantioselectivity different ratios of reagents, temperature, reaction time, and dilution were investigated (entries 24–29). Initially we employed an excess of 2 which could easily be recovered after the reaction. As the dimerization of 1 was found to be the major yield-limiting side reaction,13 the use of an excess of 1 was tested next. However, the yield dropped significantly under a variety of conditions (e.g., entry 25) and besides vast amounts of the unwanted dimer various decomposition products were observed. Test reactions revealed that product 3 undergoes further (most presumably ring opening) reactions with an excess of 1 under the reaction conditions. Thus, ensuring a permanent excess of the Michael acceptor 2 was found to be crucial to warrant good yields (entry 27). In addition, higher dilution and prolonged reaction time combined with a slightly reduced reaction temperature allowed us to isolate (−)-3 in high yields and with good enantioselectivities up to 90:10 (entries 28 and 29).15 Surprisingly, where the pseudoenantiomeric 4c and 5d performed similarly under Adamo’s conditions (entries 12 and 17), the quinine-derived 5d was found to be less selective than 4c under our optimized conditions (entry 30 vs 28).
Having identified suitable high-yielding and selective conditions for the cyclopropanation of the parent chalcone 2, a series of differently substituted chalcone derivatives was screened next (Table 2). Where electron neutral and electron deficient chalcones were well-tolerated (no matter which aryl group was modified) (entries 1–7), more electron rich electrophiles did not undergo the cyclopropanation reaction. Instead the main product in these experiments was the dimerization of bromomalonate 1c, thus illustrating again the high dependence of this reaction on the electrophilicity of the Michael acceptor and seemingly small changes in the electronic nature of the reagents have a dramatic influence on the outcome of the reaction.
Table 2.
| Entry | Chalcone | Ar1 | Ar2 | Prod. | Yielda (%) | er b (+/−) |
|---|---|---|---|---|---|---|
| 1 | 2 | Ph | Ph | 3c | 82 | 13:87 |
| 2 | 7 | 4-ClC6H4 | Ph | 8 | 98 | 12:88 |
| 3 | 9 | Ph | 4-ClC6H4 | 10 | 91 | 11:89 |
| 4 | 11 | Ph | 4-FC6H4 | 12 | 72 | 13:87 |
| 5 | 13 | 3-NO2C6H4 | Ph | 14 | 59 | 9:91 |
| 6 | 15 | 4-NO2C6H4 | Ph | 16 | 58 | 10:90 |
| 7 | 17 | Ph | 4-MeC6H4 | 18 | 72 | 16:84 |
| 8 | 19 | 4-OH-C6H4 | Ph | 20 | Nrc | Nd |
| 9 | 21 | Ph | 4-OH-C6H4 | 22 | Nrc | Nd |
| 10 | 23 | 4-MeO-C6H4 | Ph | 24 | Nrc | Nd |
| 11 | 25 | Ph | 4-MeO-C6H4 | 26 | Nrc | Nd |
Isolated yield.
Determined by HPLC using a chiral stationary phase.
Only dimerization of 1c.
Conclusion
Summarizing, the first phase-transfer catalyzed cyclopropanation reaction of bromomalonates and chalcones proceeding via a Michael Initiated Ring Closing (MIRC) reaction has been developed. Key to success was the use of a free OH-containing cinchona alkaloid ammonium salt catalyst and carefully optimized liquid/liquid reaction conditions. The exact role of the free-OH group is not clear yet. However, based on recent reports two possible modes of action seem reasonable.5 The free OH-group can either cause an additional coordination of the bromomalonate nucleophile thus achieving a better control of the orientation of the nucleophile compared to the standard ion pair formation achieved with classical chiral PTCs or an activation/coordination of the chalcone, thus resulting in a highly ordered transition state during the reaction.5 The reaction performed well for electron neutral and electron deficient chalcones giving the products in yields up to 98% and with enantiomeric ratios up to 91:9. In contrast, electron rich chalcones could not be successfully employed yet due to the lower reactivity of these Michael acceptors. Further investigations to expand this methodology toward other starting materials are currently undertaken and will be reported in due course.
Experimental section
General procedure for the phase-transfer catalyzed cyclopropanation using bromomalonates and chalcones
Reactions were usually carried out using less than 0.5 mmol bromomalonate (1). First 10 equiv K2CO3 (50% aq solution) was added to a solution of catalyst (10 mol %) in mesitylene (assuring a dilution of 0.075 M based on the amount of 1 used in the reaction). After flushing the solution with Argon, the corresponding chalcone derivative (6 equiv) was added. The vigorously stirred solution (>1200 rpm) was cooled to 0 °C (Ar-atmosphere). Subsequently, the bromomalonate was added in 3 portions (3 × 0.33 equiv over 24 h). The biphasic mixture was stirred for a total of 46 h at 0 °C. After extraction with CH2Cl2/H2O, the combined organic phases were dried over Na2SO4, evaporated to dryness and purified by column chromatography using heptanes/EtOAc = 40:1–10:1 as the eluent. The excess chalcone could easily be recovered hereby.
Cyclopropane (−)-3c
Obtained as a colorless oil in 82% yield (136 mg, 0.40 mmol) and with er = 87:13 upon reacting chalcone 2 (615 mg, 2.96 mmol) with bromomalonate 1c (104 mg, 0.49 mmol) in 6.4 mL mesitylene with 0.82 mL aqueous K2CO3 solution (50% w/w). Analytical data are in accordance with those reported in the literature.16 −25.9 (c 0.44, CHCl3); 1H NMR (300 MHz, δ, CDCl3, 298 K): 3.55 (s, 3H), 3.72 (s, 3H), 3.88 (d, J = 7.7 Hz, 1H), 4.14 (d, J = 7.7 Hz, 1H), 7.27–7.34 (m, 5H), 7.48–7.56 (m, 2H), 7.59–7.66 (m, 1H), 8.07–8.13 (m, 1H) ppm; 13C NMR (75 MHz, δ, CDCl3, 298 K): 35.1, 36.6, 46.0, 53.0, 53.1, 127.8, 128.4, 128.5, 128.6, 128.8, 133.4, 133.8, 136.7, 166.1, 166.6, 193.9 ppm; IR (film): ν = 3064, 3032, 2995, 2953, 2916, 2846, 2358, 2341, 1735, 1678, 1597, 1581, 1449, 1437, 1352, 1267, 1219, 1178, 1117, 1024, 1010, 943, 916, 739 cm−1; The enantioselectivity was determined by HPLC (Chiralcel OD-R, eluent: H2O/AcN = 55:45, 0.7 mL/min, 10 °C, retention times: (+)-enantiomer 36.4 min, (−)-enantiomer 39.6 min); HRMS (ESI): m/z calcd for C20H18O5: 339.1227 [M+H]+; found: 339.1227.
Acknowledgments
This work was supported by the Austrian Science Funds (FWF): Project No. P22508-N17. Richard Herchl is recipient of a DOC-fellowship of the Austrian Academy of Sciences at the Institute of Organic Chemistry, JKU Linz. The used NMR spectrometers were acquired in collaboration with the University of South Bohemia (CZ) with the financial support from the European Union through the EFRE INTERREG IV ETC-AT-CZ programme (project M00146, ‘RERI-uasb’).
Footnotes
Supplementary data (analytical data and experimental details can be found in the supporting material) associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.tetlet.2013.02.095.
Supplementary data
Analytical data and experimental details can be found in the supporting material.
References and notes
- 1.(a) Reissig H.-U., Zimmer R. Chem. Rev. 2003;103:1151–1196. doi: 10.1021/cr010016n. For selected reviews and recent reports about the applications of cyclopropanes to access complex molecules see: [DOI] [PubMed] [Google Scholar]; (b) Baldwin J.E. Chem. Rev. 2003;103:1197–1212. doi: 10.1021/cr010020z. [DOI] [PubMed] [Google Scholar]; (c) Brandi A., Cicchi S., Cordero F.M., Goti A. Chem. Rev. 2003;103:1213–1270. doi: 10.1021/cr010005u. [DOI] [PubMed] [Google Scholar]; (d) Simone F.D., Waser J. Synthesis. 2009:3353–3374. [Google Scholar]; (e) Zhang D., Song H., Qin Y. Acc. Chem. Res. 2011;44:447–457. doi: 10.1021/ar200004w. [DOI] [PubMed] [Google Scholar]; (f) Tang D., Qin Y. Synthesis. 2012;44:2969–2984. [Google Scholar]; (g) Kaschel J., Schneider T.F., Kratzert D., Stalke D., Werz D.B. Angew. Chem., Int. Ed. 2012;51:11153–11156. doi: 10.1002/anie.201205880. [DOI] [PubMed] [Google Scholar]; (h) Kaschel J., Schmidt C.D., Mumby M., Kratzert D., Stalke D., Werz D.B. Chem. Commun. 2013 doi: 10.1039/c2cc37631h. [DOI] [PubMed] [Google Scholar]; (i) Benfatti F., de Nanteuil F., Waser J. Chem. Eur. J. 2012;18:4844–4849. doi: 10.1002/chem.201103971. [DOI] [PubMed] [Google Scholar]; (j) Benfatti F., de Nanteuil F., Waser J. Org. Lett. 2012;14:386–389. doi: 10.1021/ol203144v. [DOI] [PubMed] [Google Scholar]
- 2.(a) Pietruszka J. Chem. Rev. 2003;103:1051–1070. doi: 10.1021/cr010027g. [DOI] [PubMed] [Google Scholar]; (b) Reichelt A., Martin S.F. Acc. Chem. Res. 2006;39:433–442. doi: 10.1021/ar030255s. [DOI] [PubMed] [Google Scholar]; (c) Waser M., Moher E.D., Borders S.S.K., Hansen M.M., Hoard D.W., Laurila M.E., LeTourneau M.E., Miller R.D., Phillips M.L., Sullivan K.A., Ward J.A., Xie C., Bye C.A., Leitner T., Herzog-Krimbacher B., Kordian M., Müllner M. Org. Process Res. Dev. 2011;15:1266–1274. [Google Scholar]
- 3.(a) Lebel H., Marcoux J.-F., Molinaro C., Charette A.B. Chem. Rev. 2003;103:977–1050. doi: 10.1021/cr010007e. [DOI] [PubMed] [Google Scholar]; (b) Gaunt M.J., Johansson C.C.C. Chem. Rev. 2007;107:5596–5605. doi: 10.1021/cr0683764. [DOI] [PubMed] [Google Scholar]; (c) Li A.-H., Dai L.-X., Aggarwal V.K. Chem. Rev. 1997;97:2341–2372. doi: 10.1021/cr960411r. [DOI] [PubMed] [Google Scholar]
- 4.(a) Maruoka K. WILEY-VCH; Weinheim: 2008. Asymmetric Phase Transfer Catalysis. For reviews about asymmetric phase-transfer catalysis: [Google Scholar]; b O’Donnell M.J. In: Catalytic Asymmetric Syntheses. 2nd ed. Ojima I., editor. WILEY-VCH; New York: 2000. pp. 727–755. [Google Scholar]; (c) Maruoka K., Ooi T. Chem. Rev. 2003;103:3013–3028. doi: 10.1021/cr020020e. [DOI] [PubMed] [Google Scholar]; (d) O’Donnell M.J. Acc. Chem. Res. 2004;37:506–517. doi: 10.1021/ar0300625. [DOI] [PubMed] [Google Scholar]; (e) Ooi T., Maruoka K. Angew. Chem., Int. Ed. 2007;46:4222–4266. doi: 10.1002/anie.200601737. [DOI] [PubMed] [Google Scholar]
- 5.Novacek J., Waser M. Eur. J. Org. Chem. 2013:637–648. For a review about bifunctional chiral quaternary ammonium salt catalysts see: [Google Scholar]
- 6.Enders D., Nguyen T.V. Org. Biomol. Chem. 2012;10:5327–5331. doi: 10.1039/c2ob25823d. For a review about phosphonium-based catalysts see: [DOI] [PubMed] [Google Scholar]
- 7.(a) Liu Y., Provencher B.A., Bartelson K.J., Deng L. Chem. Sci. 2011;2:1301–1304. doi: 10.1039/c1sc00137j. For recent impressive examples see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Provencher B.A., Bartelson K.J., Liu Y., Foxman B.M., Deng L. Angew. Chem., Int. Ed. 2011;50:10565–10569. doi: 10.1002/anie.201105536. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Maciver E.E., Knipe P.C., Cridland A.P., Thompson A.L., Smith M.D. Chem. Sci. 2012;3:537–540. [Google Scholar]; (d) Bernardi L., Indrigo E., Pollicino S., Ricci A. Chem. Commun. 2012;48:1428–1430. doi: 10.1039/c0cc05777k. [DOI] [PubMed] [Google Scholar]; (e) Shirakawa S., Liu K., Ito H., Maruoka K. Chem. Commun. 2011;47:1515–1517. doi: 10.1039/c0cc04447d. [DOI] [PubMed] [Google Scholar]; (f) Kano T., Yamamoto A., Song S., Maruoka K. Chem. Commun. 2011;47:4358–4360. doi: 10.1039/c0cc05786j. [DOI] [PubMed] [Google Scholar]; (g) Shirakawa S., Terao S.J., He R., Maruoka K. Chem. Commun. 2011;47:10557–10559. doi: 10.1039/c1cc14043d. [DOI] [PubMed] [Google Scholar]; (h) Lan Q., Wang X., Shirakawa S., Maruoka K. Org. Process Res. Dev. 2010;14:684–686. [Google Scholar]; (i) Shirakawa S., Liu K., Maruoka K. J. Am. Chem. Soc. 2012;134:916–919. doi: 10.1021/ja211069f. [DOI] [PubMed] [Google Scholar]; (j) Shibuguchi T., Mihara H., Kuramochi A., Ohshima T., Shibasaki M. Chem. Asian J. 2007;2:794–801. doi: 10.1002/asia.200700070. [DOI] [PubMed] [Google Scholar]; (k) Okada A., Shibuguchi T., Ohshima T., Masu H., Yamaguchi K., Shibasaki M. Angew. Chem., Int. Ed. 2005;44:4564–4567. doi: 10.1002/anie.200500927. [DOI] [PubMed] [Google Scholar]; (l) Johnson K.M., Rattley M.S., Sladojevich F., Barber D.M., Nunez M.G., Goldys A.M., Dixon D.J. Org. Lett. 2012;14:2492–2495. doi: 10.1021/ol300779x. [DOI] [PubMed] [Google Scholar]; (m) Moss T.A., Barber D.M., Kyle A.F., Dixon D.J. Chem. Eur. J. 2013;19:3071–3081. doi: 10.1002/chem.201203825. [DOI] [PubMed] [Google Scholar]
- 8.(a) Denmark S.E., Gould N.D., Wolf L.M. J. Org. Chem. 2011;76:4260–4336. doi: 10.1021/jo2005445. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Denmark S.E., Gould N.D., Wolf L.M. J. Org. Chem. 2011;76:4337–4357. doi: 10.1021/jo2005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Waser M., Gratzer K., Herchl R., Müller N. Org. Biomol. Chem. 2012;10:251–254. doi: 10.1039/c1ob06573d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gratzer K., Waser M. Synthesis. 2012;44:3661–3670. doi: 10.1055/s-0032-1316804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Arai S., Nakayama K., Ishida T., Shioiri T. Tetrahedron Lett. 1999;40:4215–4218. For the asymmetric phase-transfer catalyzed cyclopropanation by addition of nucleophiles to α-bromocycloalkenones see: [Google Scholar]
- 11.Fiandra C.D., Piras L., Fini F., Disetti P., Moccia M., Adamo M.F.A. Chem. Commun. 2012;48:3863–3865. doi: 10.1039/c2cc30401e. [DOI] [PubMed] [Google Scholar]
- 12.(a) McIntosh J.M., Khalil H. Can. J. Chem. 1978;56:2134–2138. For racemic phase-transfer catalyzed cyclopropanation reports see: [Google Scholar]; (b) Singh R.K., Danishefsky S. J. Org. Chem. 1975;40:2969–2970. [Google Scholar]; (c) Kozhushkov S.I., Leonov A., de Meijere A. Synthesis. 2003:956–958. [Google Scholar]; (d) Kryshtal G.V., Zhdankina G.M., Zlotin S.G. Russ. Chem. Bull. Int. Ed. 2011;60:2286–2290. [Google Scholar]
- 13.Base-catalysed dimerization of 1 is the only reaction taking place in the absence of any catalyst and was also the main side reaction in the course of all these investigations.
- 14.A free 6′-OH group was found to be crucial to obtain high selectivities employing cinchona alkaloid-derived PTCs for enantioselective Darzens reactions (Ref. 7a) and conjugate additions of cyanides (Ref. 7b).
- 15.Lowering the catalyst loading to 5 mol % did result in significantly reduced yields and selectivities.
- 16.(a) Kingsbury C.A., Durham D.L., Hutton R. J. Org. Chem. 1987;43:4696–4700. [Google Scholar]; (b) Ye Y., Zheng C., Fan R. Org. Lett. 2009;11:3156–3159. doi: 10.1021/ol9012102. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Analytical data and experimental details can be found in the supporting material.