

Abstract
Background
The pathogenesis of inflammatory bowel disease (IBD) is associated with a dysregulated mucosal immune response. Certain stimulators of innate immunity (CpG DNA or GM-CSF) are reported to be anti-inflammatory in IBD. Toll-like receptor-7 (TLR7) is an important regulator of innate immunity and its activation plays a key role in induction of type I IFN. The present study tests the hypothesis that TLR7 agonists, Imiquimod have therapeutic efficacy in IBD.
Methods
Acute colitis was induced in Balb/c mice by giving 5% DSS in drinking water for 7 days. Mice were treated with Imiquimod either orally or topically, and its therapeutic effects on diseases activity were examined. Isolated mouse CD11c+ dendritic cells and human intestinal epithelial cells (HT29 & HCT116) were treated with Imiquimod (10 μg/ml) and their susceptibility to intracellular Salmonella typhimurium infection was assessed by gentamicin protection assay.
Results
Oral administration of Imiquimod induced type I IFN expression in the gastrointestinal mucosa and ameliorated DSS-induced acute colitis as assessed by clinical parameters, histology and mRNA expression of proinflammatory cytokines. Topical administration of Imiquimod also ameliorated DSS-colitis by inducing the expression of type I IFN in the colonic mucosa. However, no evidences for a systemic IFN response were observed. Imiquimod treatments to both CD11c+ and intestinal epithelial cells significantly increased expression of antimicrobial peptides (AMPs) and reduced survival of intracellular Salmonella typhimurium.
Conclusions
Imiquimod induces type I IFN and AMP to ameliorate DSS-induced acute colitis and prevents Salmonella survival. Therefore, Imiquimod treatments provide a new therapeutic approach for IBD patients.
Keywords: innate immunity, inflammatory bowel diseases, mucosa, epithelial cells
Introduction
Crohn’s disease (CD) and ulcerative colitis (UC) are inflammatory bowel diseases in human gastrointestinal tract (GI). The etiology of IBD is thought to be multifactorial.1 Individuals who suffer from the disease use medications to ameliorate the symptoms. Current available treatments for IBD are empiric, nonspecific and mostly involve steroids, 5-aminosalacylic acid derivative, immunosuppressants, and antibodies against TNF-α.2, 3
Recent studies suggest that defects in mucosal immune function play a critical role in IBD pathogenesis.4 Members of the Toll-like receptor (TLR) family play a fundamental role in pathogen recognition and activation of innate immunity because they recognize pathogen-associated molecular patterns. These pathways rapidly direct the production of cytokines necessary for the development of downstream humoral and cell-mediated immunity. The various TLRs exhibit different patterns of expression: TLR9 is mainly expressed in plasmacytoid dendritic cells (PDCs) and responds to CpG DNA by directing the production of type I IFN. Studies have reported that CpG DNA has protective effects in experimental colitis.5-7 Similarly, TLR7 is highly expressed in PDCs and responds to stimulation, which is critical for an effective response to many viruses or other pathogens.8 Apart from PDCs, other cells also may express TLR7 with direct IFN expression. The imidazoquinolines, typified by Imiquimod and Resiquimod, are purine analogs and function as potent TLR7 activators. Imiquimod (1-[2-methylpropyl] -1H-imadazo[4,5c] quinoline-4-amine) is a low molecular weight compound that acts as a topical immune response modifier and shows antiviral and antitumor effects.9-13 Imiquimod is a topically applied drug used for the treatment of external genital and perianal warts, acitinic keratosis and superficial basal cell carcinoma14, 15 and has been shown to exert its effects through TLR7 stimulation, leading to release of IFN-α, TNF-α, IL6, IL12 and others.16-18 Imiquimod may also stimulate B cell proliferation, which may be mediated directly by TLR7 activation.19 These studies support a potential role of TLR7 in the regulation of the mucosal immune response. In this study, we examined the therapeutic effects of Imiquimod administered both orally and topically in a murine model of dextran sulfate sodium (DSS)-induced acute colitis. Imiquimod induced type I IFN and antimicrobial peptides (AMPs) resulting in the amelioration of DSS-induced colitis and reduced the survival of intracellular Salmonella typhimurium.
Materials and Methods
Animals
Pathogen-free 6-8 weeks old Balb/c female mice (Frederick Cancer Research and Development Center, Frederick, MD) were maintained on light/dark cycles of 12 hours. The Washington University Animal Studies Committee approved all experimental procedures.
Dextran Sulfate Sodium (DSS)-induced acute colitis
To induce colitis in mice, 5% DSS (USB Corporation, Cleveland, OH) was provided in drinking water for 7 days ad libitum. During the study period, daily oral gavage was performed for control, DSS treated mice with PBS and a group of DSS treated mice received Imiquimod (Invivogen, San Diego, CA; dose: 30 mg/kg body wt) diluted in PBS. Mice were monitored for weight loss, stool consistency and rectal bleeding (Seracult kit, Propper Manufacturing, Long Island city, NY) to assess the disease activity index (DAI).20 At day 7, mice were sacrificed following isoflurane (Aerrane, Baxter, IL) inhalation and cervical dislocation. Spleens were removed and weighed. Colons were excised from the cecum to the pelvic brim and their lengths were measured in centimeters. Sections from different parts of colon were snap-frozen and later used for myeloperoxidase assay, real-time reverse transcription polymerase chain reaction (RT-PCR) and histological analyses. In another experimental groups, daily topical administration of enema (0.5% carboxymethyl celllose, 0.4% polysorbate 80, 0.92% NaCl) was given with Imiquimod (30mg/kg body wt). Control and DSS treated mice received enema only. Clinical parameters and tissue collections were done as mentioned above.
Myeloperoxidase (MPO) Assay
Sections from distal colons of experimental mice were homogenized in 0.5% hexadecyltrimethyl-ammonium bromide (HTAB) in 10 mM 3-(N-Morpholino) propane sulfonic acid (MOPS) buffer. The homogenate was sonicated, freeze-thawed and centrifuged at 12000-15000 rpm for 15 minutes to collect supernatant. Myeloperoxidase activity was determined by previously described method.21
Real-time RT-PCR
Total RNA isolated from the GI tissues was subjected to mRNA purification using Oligotex mRNA mini kit (Qiagen, Valencia, CA). The purified mRNA was then reverse transcribed using SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA) in the presence of random hexamer primers (Invitrogen, Carlsbad, CA). The complimentary DNA (cDNA) was used for real-time RT-PCR analyses using Jumpstart Taq DNA polymerase (Sigma-Aldrich, St.Louis, MO) and SYBR green nucleic acid stain. Crossing threshold values for individual genes were normalized to β-Actin expression. Mouse primers used in analyses are shown in supplementary table I.
Microarray Analysis
Balb/c mice were orally treated with Imiquimod (30 mg/kg of body wt). After 6 and 18 hours, colons were removed and the total RNA was isolated. It was then purified using RNAeasy kit (Qiagen, Valencia, CA) and subjected to Agilent whole genome mouse array (Agilent Technologies, Santa Clara, CA).
CD11c Isolation, Stimulation and Bacterial Count
Pegylated murine recombinant GM-CSF produced by previously described methods was intraperitoneally injected in Balb/c mice for 4 days (5 μg/mouse).22 Spleens were removed, minced and treated with Collagenase type VIII (10,000 U/ml; Sigma-Aldrich, St.Louis, MO) solution. It was then passed through a 70 μm cell strainer using plungers and cells were counted. To obtain highly pure dendritic cells, Fc receptor blocking reagent (CD16/32 antibody: 1 μg/106, BD Pharmingen, San Jose, CA) was added to the cell suspension. Cells were then incubated with CD11c micro beads (100 μl/108 cells; Miltenyi Biotech, Auburn, CA) 15 minutes at 4-8 °C. Cells were washed and suspended with running buffer and passed through magnetic LS columns (Miltenyi Biotech, Auburn, CA) as per manufacturer’s instructions to isolate CD11c+ cells. These cells were seeded (5 × 106 cells / well) and cultured onto six-well plates and treated with Imiquimod (10μg/ml) for 24 hours. At termination, cell suspension was centrifuged, media supernatant was collected and the pellet containing CD11c+ cells was subjected to mRNA isolation followed by real-time RT-PCR analyses. The CD11c+ cells free media supernatant was inoculated with live S.typhimurium (STK 1028) at a multiplicity of infection (MOI) of 10 for 1 hour at 37 °C. Different dilutions of cell suspension were plated on Macconkey agar plates and after overnight incubation at 37 °C viable cell colonies were counted.
Intestinal epithelial cells stimulation and bacterial count
HT29 (ATCC: HTB-38) and HCT116 (ATCC: CCL-247) human epithelial cells were cultured in on T-75 flask. At confluency, cells were treated with Imiquimod (10 μg/ml) and TLR7 inhibitor chloroquine (100 μm) (Sigma Aldrich, St.Louis) for 24 hours. Cells were processed for RNA isolation followed by real-time RT-PCR analysis. Media supernatant was subjected to ELISA to quantify secreted Defensins 2 & 3 (Alpha Diagnostics, San Antonio, TX). Salmonella survival study was performed using media supernatant as shown in previously described CD11c experiment.
Gentamicin Protection Assay
HT29 and HCT116 human epithelial cells (1 × 106/well) were cultured in 6-well plate. Seven days after confluency, cells were treated with Imiquimod (10 μg/ml) for 12 hours. Cultures were inoculated with S. typhimurium or E. Coli (as negative control) at MOI of 10 and incubated for 1 hour at 37°C. Cells were then washed with Hank’s solution and incubated in a media containing 100 μg/ml Gentamicin (Sigma-Aldrich, St. Louis, MO) for 90 minutes to kill extracellular bacteria. Cells were lysed using 1% Triton-X, diluted with Luria-Bertani medium, and plated on Macconkey agar plate or LB agar plate for culture of S. typhimurium and E.coli, respectively. Following overnight incubation at 37°C viable colonies were counted.
Statistical Analysis
All data were expressed as mean ± SEM. Statistical analysis of significance was determined by Student’s ‘t’ test for unpaired data. P < .05 was considered significant.
Results
Oral Imiquimod Administration Induces Type I IFN Secretion in the gastrointestinal (GI) tract
Imiquimod is a known inducer of type I IFN especially IFN-α and the IFN-β gene family, as well as other proinflammatory cytokines.8, 23 In the present study, we examined specific IFN-α genes expressed in the GI tract. Following real-time RT-PCR analyses, the mRNA expression of IFN-α2, 9, 11, 12 & 14 was measured. IFN-α2 gene expression was selected as a marker of type I alpha IFN expression on the basis of very low but detectable baseline expression in the murine GI tract. In addition, the gene expression of IFN-β was also measured.
To study the systemic effect of Imiquimod in the GI tract, Balb/c mice were orally administered Imiquimod (30 mg/kg) by oral gavage. Gastrointestinal tissues were isolated at 2, 6 & 18 hours after Imiquimod administration. In stomach, there was no significant induction of type I IFN expression. In the small intestine (jejunum), Imiquimod treatment led to maximal increase in IFN-α2 (26 fold induction, P < 0.01) and IFN-β (8 fold, P < 0.05) expression at 2 hours. In the proximal colon, maximal induction of IFN-α2 (39 fold) and IFN-β (12 fold, P < 0.01) occured at 6 hours following Imiquimod administration. Similar results were observed in the distal colon. The kinetics of type I IFN differed, as Imiquimod-mediated increase in IFN-β expression was highest at 18 hours in the colon. In the spleen, maximal induction of IFN-α2 (15 fold, P < 0.01) and IFN-β (11 fold, P < 0.01) were observed at 18h after oral Imiquimod administration (Fig. 1). The mucosal expression of type I interferon was supported by an increase in known downstream interferon responsive gene biomarkers (Table 1). Increases in AMP gene expression were also observed (Table 1). These results suggest that oral administration of a TLR7 agonist stimulates expression of mucosal type I IFN and downstream genes in the small intestine, colon, and spleen.
Figure 1.
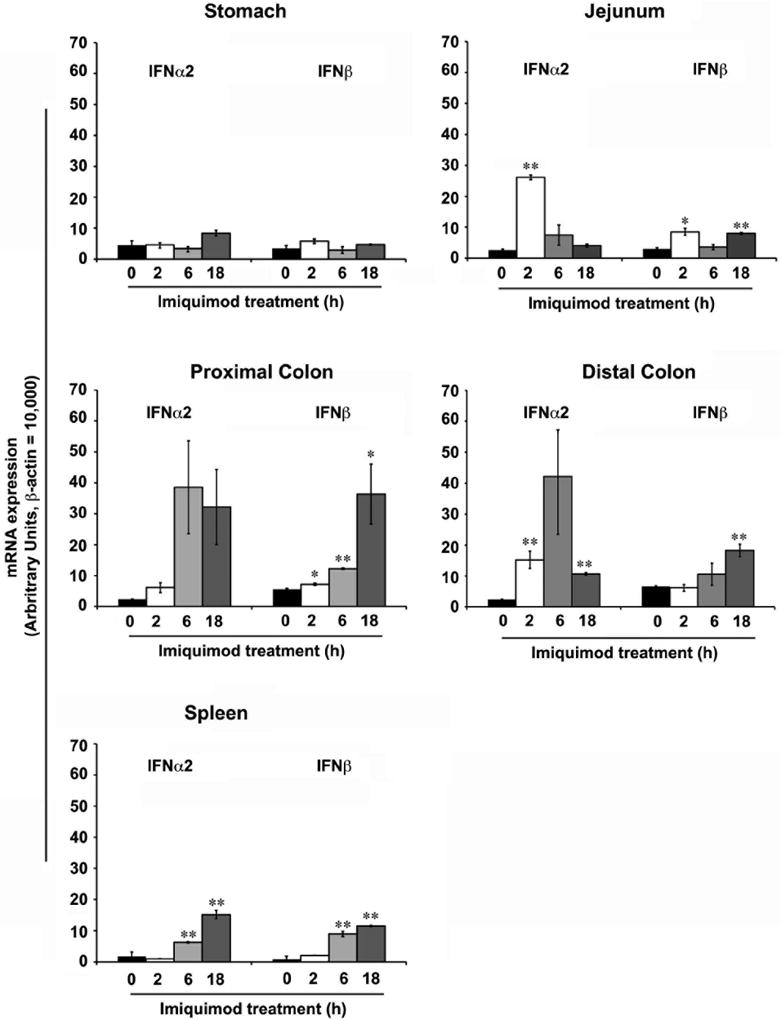
Imiquimod induces the expression of type I IFN in the gastrointestinal (GI) tract. Oral administration of Imiquimod (30 mg/kg body wt) to Balb/c mice exhibited differential induction in the expression of type I IFN (α, β) in various parts of the GI tract. In the stomach, no significant changes in the expression type I IFN were noted. In the jejunum, Imiquimod treatments led to significant increases in IFN-α2 and IFN-β expression at the 2 hours time point. In the proximal & distal colon treatments led to significant increases in both IFN-α2 and IFN-β expression at 6 &18 hours time points. Similar type I IFN induction was observed in spleen. N = 5; results represent the data from three individual experiments and expressed as the mean ± SEM (* P < .05; ** P < .01).
Table I.
List of Imiquimod-induced IFN and AMPs genes in mice colons
| Gene Name | Accesion no. | Gene Description | 6 h | 18 h |
|---|---|---|---|---|
| Oasl2 | NM_011854 | 2’-5’ oligoadenylate synthetase-like 2 | 13.6 | 12.3 |
| Oasl1 | NM_145209 | 2’-5’ oligoadenylate synthetase-like 1 | 6.3 | 4.2 |
| Oas3 | NM_145226 | 2’-5’ oligoadenylate synthetase 3 | 2.4 | 3.4 |
| Oas2 | NM_145227 | 2’-5’ oligoadenylate synthetase 2 | 11.7 | 12.8 |
| Oas1g | NM_011852 | 2’-5’ oligoadenylate synthetase 1G | 2.6 | 2.1 |
| Oas1f | NM_145153 | 2’-5’ oligoadenylate synthetase 1F | 3.5 | 3.8 |
| Oas1b | NM_011853 | 2’-5’ oligoadenylate synthetase 1B | 2.2 | 2.4 |
| Oas1a | NM_145211 | 2’-5’ oligoadenylate synthetase 1A | 4.3 | 4.7 |
| Isg20 | NM_020583 | interferon-stimulated protein | 2.1 | 1.7 |
| Isg20 | NM_020583 | interferon-stimulated protein | 1.8 | 1.8 |
| Irf7 | NM_016850 | interferon regulatory factor 7 | 2.7 | 5.0 |
| Irf1 | NM_008390 | interferon regulatory factor 1 | 1.6 | 1.6 |
| Ifrg15 | NM_022329 | interferon alpha responsive gene | 2.1 | 1.9 |
| Ifrg15 | NM_022329 | interferon alpha responsive gene | 2.0 | 1.7 |
| Ifitm7 | NM_028968 | interferon induced transmembrane protein 7 | 3.2 | 2.8 |
| Ifitm3 | NM_025378 | interferon induced transmembrane protein 3 | 3.1 | 2.6 |
| Ifit3 | NM_010501 | interferon-induced protein with tetratricopeptide repeats 3 | 5.2 | 3.5 |
| Ifit2 | NM_008332 | interferon-induced protein with tetratricopeptide repeats 2 | 6.9 | 3.2 |
| Ifit1 | NM_008331 | interferon-induced protein with tetratricopeptide repeats 1 | 37.0 | 12.4 |
| Ifih1 | NM_027835 | interferon induced with helicase C domain 1 | 2.7 | 2.0 |
| Ifi44 | NM_133871 | interferon-induced protein 44 | 15.8 | 16.4 |
| Ifi205 | NM_172648 | interferon activated gene 205 | 3.2 | 1.6 |
| Ifi204 | NM_008329 | interferon activated gene 204 | 3.8 | 2.2 |
| Hamp2 | NM_183257 | hepcidin antimicrobial peptide 2 | 1.5 | 1.3 |
| Hamp1 | NM_032541 | hepcidin antimicrobial peptide 1 | 2.5 | 1.2 |
| Defcr6 | NM_007852 | defensin related cryptdin 6 | 0.6 | 3.1 |
| Defcr4 | NM_010039 | defensin related cryptdin 4 | 0.5 | 3.9 |
| Defcr3 | NM_007850 | defensin related cryptdin 3 | 0.6 | 3.7 |
| Defcr23 | NM_001012307 | defensin-related cryptdin 23 | 0.6 | 3.7 |
| Defcr22 | NM_207658 | defensin-related cryptdin 22 | 0.7 | 2.3 |
| Defcr20 | NM_183268 | defensin related cryptdin 20 | 0.5 | 3.4 |
| Defcr-rs7 | NM_007848 | defensin related cryptdin related sequence 7 | 0.5 | 4.4 |
| Defcr-rs2 | NM_007847 | defensin related cryptdin related sequence 2 | 0.6 | 7.2 |
| Defcr-rs12 | NM_007846 | defensin related cryptdin related sequence 12 | 0.5 | 4.3 |
| Defcr-rs10 | NM_007845 | defensin related cryptdin related sequence 10 | 0.5 | 6.3 |
| Defcr-rs1 | NM_007844 | defensin related sequence cryptdin peptide (paneth cells) | 0.8 | 2.2 |
| Defb1 | NM_007843 | defensin beta 1 | 1.9 | 0.8 |
| Defa1 | NM_010031 | defensin alpha 1 | 0.6 | 3.4 |
Following microarray analysis, Imiquimod treatments increased a total of 116 gene expression in colon. A list of important IFN and AMPs genes are included in the table. The fold differences are based on 2 array experiments
Oral Administration of Imiquimod Ameliorates DSS-Induced Colitis
We next used murine model of DSS-induced acute colitis to evaluate the efficacy of oral Imiquimod treatments. DSS-treated animals exhibited a weight loss with a maximum of 17.1% at day 4 followed by 14.6%, 13.3%, 14.1% on days 5, 6 and 7 respectively (Fig. 2A upper left). Following Imiquimod administration, DSS-induced weight loss was reduced reaching 1.4%, 0.5%, 3.1%, and 6.9% on days 4,5, 6 and 7 respectively. Two additional clinical parameters (e.g. stool consistency and rectal bleeding) were also scored, and the total DAI was estimated in these mice. Imiquimod treatments resulted in a significant reduction in DAI score (1.9 vs. 0.53; P < 0.01) of DSS-treated mice. During necroscopy, colons were removed and their lengths were measured. Reduced colon length is an indication of fibrosis and inflammation. DSS-treated mice exhibited a marked shortening of the colon length (DSS: 7 cm vs. Control: 10 cm; P < 0.003). Imiquimod- treatment significantly ameliorated colon shortening (Imiquimod: 9 cm vs. DSS: 7 cm; P < 0.008) (Fig. 2A upper right). To quantify acute inflammation in the colon, polymorphic neutrophils (PMN) infiltration was assessed by MPO assay. Imiquimod treatment (Imiquimod+DSS) led to a significant decrease in MPO levels when compared to mice treated with DSS alone (Fig. 2A lower left). In addition, spleen size was also significantly increased with Imiquimod treatments when compared to DSS-treated mice (Fig. 2A lower right). Oral administration of Imiquimod expanded immune cell populations. This was analysed by flow-cytometry of the spleen cells to examine changing immune cell populations. The number of myeloid DCs (CD11c+/CD11b+/CD8-) in Imiquimod-treated mice exhibited a significant increase compared to the vehicle-treated group. Furthermore, the number of lymphoid DCs (CD11c+/CD11b-/CD8a+) and type I IFN producing plasmacytoid DCs (PDCA-1/ Ly6c) also exhibited an increase, but did not achieve statistical significance compared to control mice (Fig. 2B).
Figure 2.
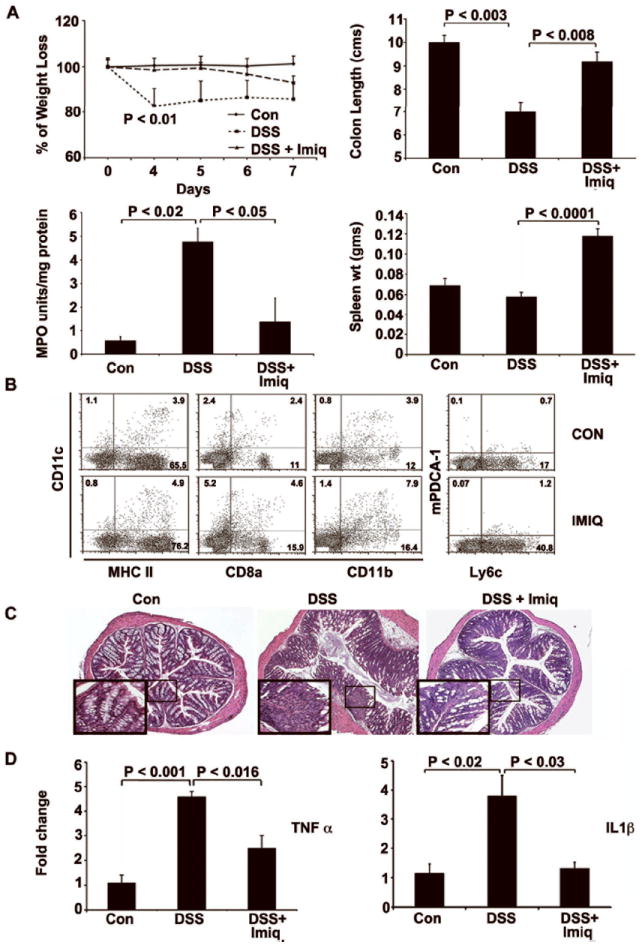
Imiquimod treatments ameliorate DSS-induced colitis. 5% DSS in drinking water was given to Balb/c mice for 7 days to induce acute colitis. Daily oral administration of Imiquimod (30 mg/kg body wt) during DSS treatment periods led to an improvement in clinical parameters associated with acute colitis. (A) Clinical parameters are listed as reduced weight loss, increased colon length, reduced MPO activity and increased spleen weight. (B) Following flow-cytometric analysis of the dendritic cell (DC) populations in the spleen, Imiquimod treatments significantly increased myeloid DC (CD11c+/CD11b+/CD 8-) population. (C) Following histological analyses of H&E-stained distal colon sections, Imiquimod treatment significantly reduced the crypt damage and inflammatory infiltrates in comparison to DSS-treated control mice. All histological sections are presented at 200× magnification and some specific areas of sections are magnified to show associated changes. (D) Following real-time RT-PCR analysis, Imiquimod treatments led to significant decreases in proinflammatory cytokines, including TNF-α and IL1-β in the distal colons. N= 7; results are representative of three individual experiments.
Following the experimental evidence of Imiquimod-mediated improvement in clinical markers associated with DSS-induced colitis, we next evaluated histopathological appearance of distal colon segments by H&E staining. In comparison to the control mice, more histological damage was observed in DSS-treated mice. The colon exhibited overt colitis with ulceration and acute inflammatory infiltrates. Imiquimod (DSS + Imiquimod) treated mice exhibited significantly reduced inflammatory infiltrates, ulceration and crypt damage (Fig. 2C) when compared to the DSS-treated controls.
We next examined proinflammatory cytokine expression by real-time RT-PCR analysis of TNF-α and IL1-β genes, which are good biomarkers for inflammation in IBD. DSS-treated mice exhibited significant increases in TNF-α (DSS: 4.6 fold vs. Control: 1.0; P < 0.001) expression when compared to control mice. Imiquimod (DSS + Imiquimod) treatment significantly reduced the TNF-α expression (Imiquimod; 2.4 fold vs. DSS; 4.6 P < 0.016) when compared with DSS-treated mice. Similarly, DSS treatments led to significant increases in IL1-β mRNA expression, while Imiquimod treatments (DSS + Imiquimod) significantly decreased the expression (Imiquimod: 3.8 vs. DSS: 1.3 fold; P < 0.03) (Fig 2D). Protein array analysis (data not shown) confirmed corresponding decreases in TNF-α and IL1-β protein expression. Similarly, antimicrobial peptides gene expression was increased with Imiquimod treatment compared to DSS treated or control mice (supplementary Fig.1). Taken together, these results suggest that oral administration of Imiquimod significantly ameliorates acute colitis in DSS-treated mice.
Topical Administration of Imiquimod also Induces Type I IFN in the Colon
Systemic induction of type I IFN results in flu-like symptoms. We next examined if topical treatment with Imiquimod could also induce colonic mucosal type I IFN expression. Balb/c mice were administered Imiquimod (30 mg/kg) by enema. Control mice received vehicle-enema only and groups were then sacrificed at 2, 6 & 18 hours. Total RNA isolated from colon was subjected to real-time RT-PCR analysis to examine the expression of type I IFN. Imiquimod treated mice exhibited a significant expression of IFN-α2 (16.2 fold P < 0.05) at 18 hours time point compared to control mice. However, no changes were noted with IFN-β expression at any time points. Spleen samples analyzed for type I IFN gene expression did not show any increase at any of the time points examined. This confirmed that systemic interferon response not induced by topical Imiquimod administration (Fig. 3). These results demonstrate that topical administration of Imiquimod has the ability to induce the expression of type I IFN in the colon at later time points.
Figure 3.
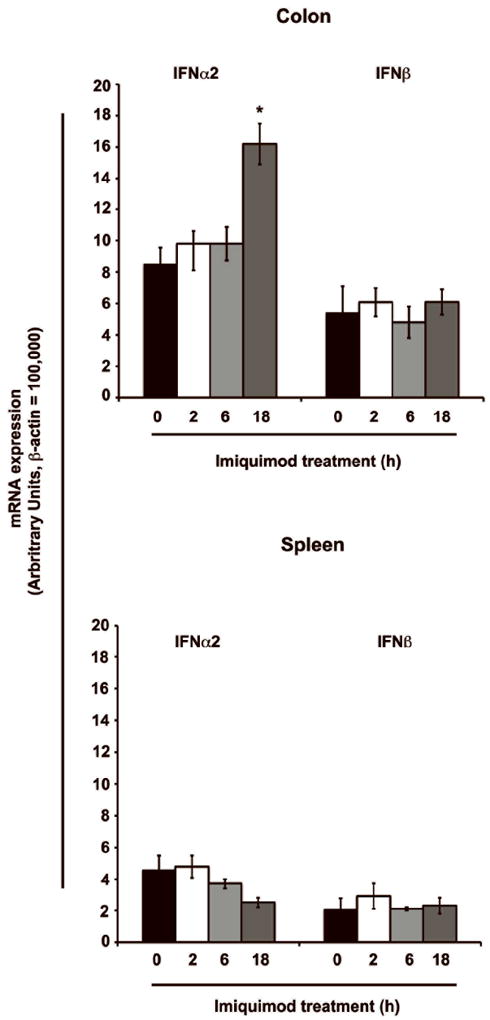
Topical administration of Imiquimod induces the expression of type I IFN in the colon. To determine the topical therapeutic effect, a similar dose of Imiquimod (30 mg/kg body weight) was administered rectally to Balb/c mice. Following real-time RT-PCR analysis, Imiquimod treatment resulted in a significant increase in IFN-α2 expression at 18 hours in the colon. No changes were noted with IFN-β expression. Increases in splenic type I IFN were not observed, arguing against systemic response. N = 5; results represent the data from two individual experiments and expressed as the mean ± SEM (* P < .05).
Topical administration of Imiquimod Improves DSS-induced Colitis
In DSS-treated mice, Imiquimod was administered topically by enema for 7 days. In contrast to oral administration, topical Imiquimod did not increase the spleen size, which confirmed its minimal systemic absorption. Colon length measured at necroscopy demonstrated a significantly less colonic shortening when compared to DSS-treated controls. MPO assay level was significantly reduced in the distal colon of Imiquimod-treated mice (Fig. 4A).
Figure 4.
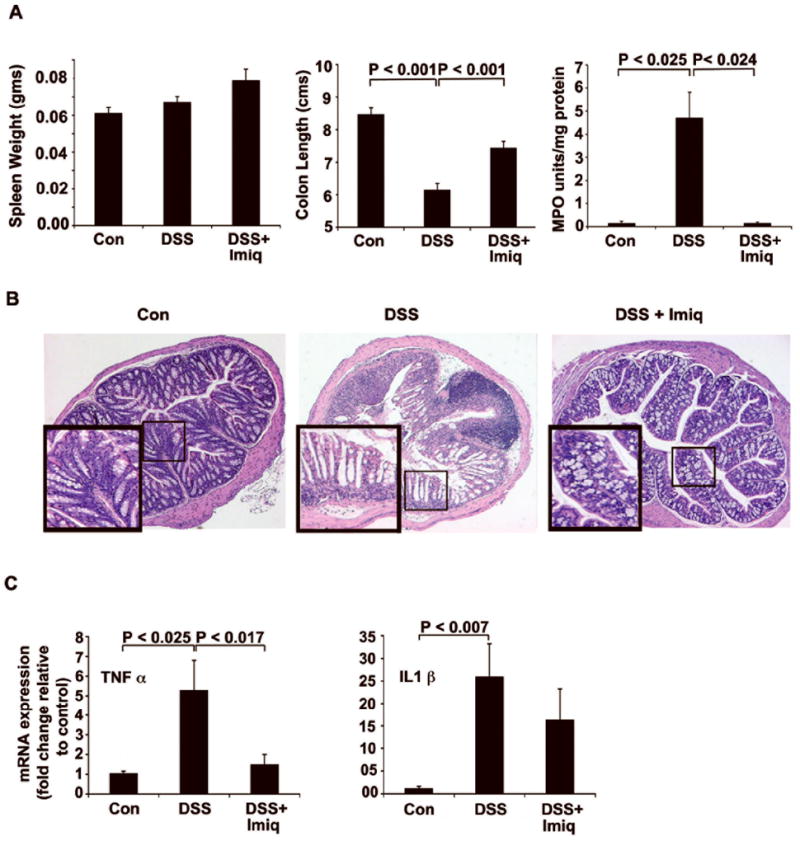
Topical administration of Imiquimod ameliorates DSS-induced colitis. (A) No significant increase in spleen size was noted following Imiquimod administration by enema. Imiquimod treatment reduced colonic shortening when compared to animals not treated with Imiquimod. Imiquimod treatments significantly reduced MPO activity in the distal colon. (B) Histological analysis of H&E-stained distal colon sections showed an Imiquimod-mediated decrease in the number of inflammatory infiltrates and maintenance of normal colonic morphology. (C) Imiquimod treatments led to significant decreases in proinflammatory cytokines TNF-α and IL1-β in the colon as assessed by real-time RT-PCR. Results suggest that topical administration of Imiquimod reduces DSS-induced colitis. N= 7; results are representative of three individual experiments.
Following histopathological analyses of distal colon sections by H&E staining, DSS-treated mice exhibited epithelial loss, ulceration, inflammatory infiltrates and total crypt damage. Imiquimod treatment reduced this damage and preserved normal crypt morphology (Fig. 4B). Following real-time RT-PCR analyses, mRNA expression of TNF-α (1.5 vs. 5.3; P < 0.017) and IL1-β (16.3 vs. 26) was decreased following Imiquimod treatment (Fig. 4C). Imiquimod treatment did not confer marked improvement in the DAI of DSS-treated mice. These results were expected as therapeutic effects were observed only in the colonic mucosa by the topical enema.
Imiquimod induces AMPs in CD11c+ cells and Prevents Extracellular Salmonella Survival
Dendritic cells reside in close proximity to the epithelia in the intestine and play a major role as sentinels of bacterial infection. Following microarray analysis, we observed Imiquimod-mediated increases in the expression of AMPs in the colon (Table I). To follow this finding, we next examined whether Imiquimod treatment induced AMPs in dendritic cells.
Imiquimod (10 μg/ml) treatments to CD11c+ cells led to a significant increases in the expression of AMPs including Defensins a1 (3.1 fold), defensins-related cryptdin inluding 4 (1.8 fold); 6 (2.1 fold) & 20 (2.2 fold), Cathelicidin (3.1 fold) and s1008 a (2.5 fold) (Fig. 5A). To study the effects of AMPs, media supernatants collected from CD11c+ cells inoculated with Salmonella typhimurium. Surviving colony forming units of Salmonella were significantly reduced (56%) with Imiquimod treatments when compared to vehicle-treated controls. Additional negative control included fresh serum free media (SFM) containing Imiquimod, which did not exhibit its own antimicrobial activity (Fig. 5B). These results suggest that Imiquimod treatment to CD11c+ cells results in the release of antimicrobial factors that results in effective extracellular killing of Salmonella.
Figure 5.
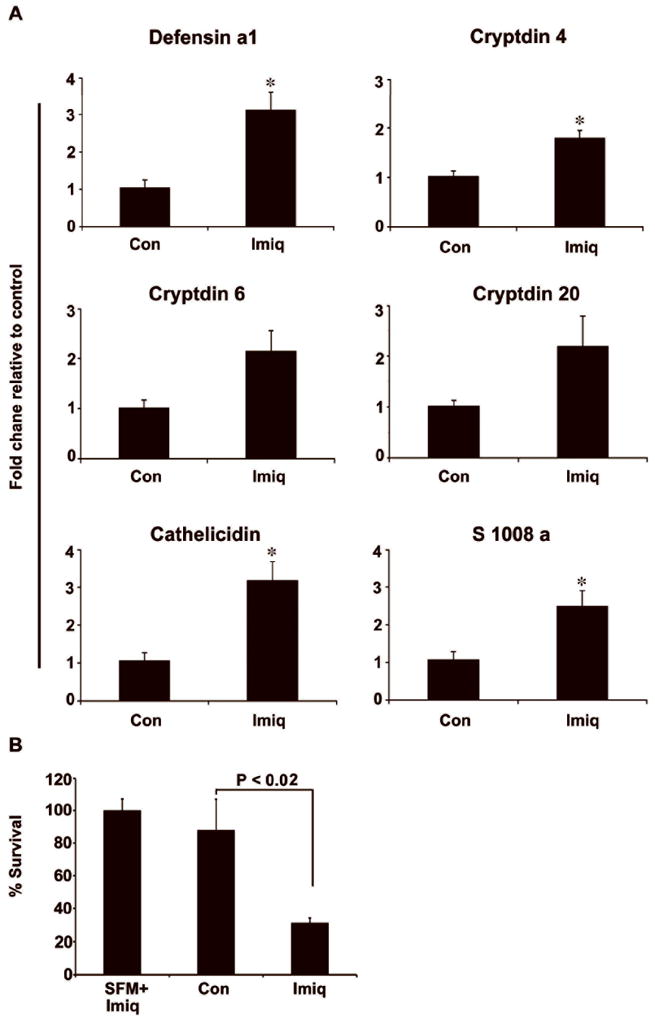
Imiquimod increases expression of AMPs in dendritic cells and reduces extracellular survival of Salmonella. CD11c+ cells isolated from GM-CSF-treated mice were stimulated with Imiquimod (10 μg/ml) for 24 hours. (A) Stimulated CD11c+ cells exhibited significant increases in expression of AMPs, including Defensins a1, Defensins related cryptdins (4, 6 & 20), Cathelicidin and s1008 a. (B) Media supernatant collected from the cultures of Imiquimod-treated CD11c+ cells were inoculated with Salmonella typhimurium for 1 hour. Serially diluted media were plated on agar plates for colony counts. Significantly reduced number of surviving Salmonella was observed with Imiquimod treatment. Results are representative of three individual experiments (* P < .05).
Imiquimod Induces AMPs and Protects Intestinal Epithelial Cells from Salmonella invasion
Intestinal epithelial cells express functional TLRs and are known to produce AMPs. 24 Seven day post-confluent HT29 cells were pretreated with Imiquimod (10 μg/ml) for 24 hours. To show that Imiquimod-mediated effects were due to TLR7 activation, we used chloroquine (100 μm), a known inhibitor of endosomal-TLR signaling. It acts by preventing endosomal acidification blocking TLR7 signaling. Intestinal epithelial cells treated with Imiquimod exhibited a significant increase in epithelial AMPs expression especially Defensins 3 (30.2 fold) and Defensins 2 (27.9 fold) and these effects were significantly reduced by the treatments of chloroquine. These results were further confirmed by performing ELISA to show respective changes in their protein levels (Figure 6A). A significant increase in human IFN-α2 mRNA expression (6.9 fold) was also observed (data not shown). These data show that Imiquimod significantly induces the epithelial expression of AMPs by TLR7 receptor activation.
Figure 6.
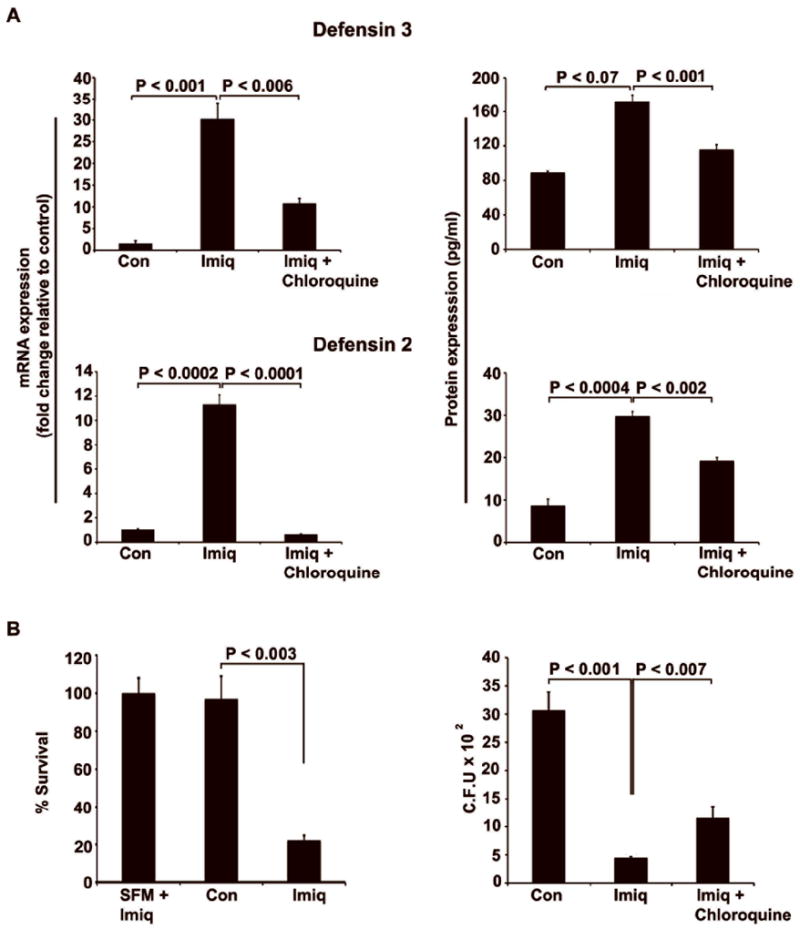
Imiquimod induces AMPs and reduces the cells susceptibility to Salmonella invasion. HT29 cells were treated with Imiquimod (10 μg/ml) for 24 hours. (A) Imiquimod treatment significantly induced mRNA expression of AMPs, including Defensins 3 and 2. These effects were significantly reduced when chloroquine was added to the culture media prior to Imiquimod treatment. Results were further confirmed by performing ELISA (B) Media supernatant from Imiquimod-treated cells significantly reduced surviving colony forming units (Left panel). Gentamicin protection assay performed in HT29 cells treated with Imiquimod led to significant decreases in colony forming units of viable intracellular Salmonella (Right panel). These effects were significantly reversed with chloroquine treatment. Results are representative of three individual experiments
To study the effects of AMPs, media supernatants collected from the HT29 cell culture were inoculated with Salmonella typhimurium and then plated on Macconkey agar plates. Following counting, surviving colony forming units were significantly reduced (75%) with Imiquimod treated group when (Figure 6B Left panel) compared to controls. We next performed a gentamicin protection assay to assess this increased AMP expression would be expected to increase epithelial resistance to Salmonella infection. Cells treated with Imiquimod exhibited a significant decrease in viable intracellular colony forming units when compared to controls (310 vs. 41). As expected, this effect was significantly reversed following chloroquine treatment (41 vs.116; Fig 6B Right panel). Similar results were observed in HCT116 cells with Imiquimod treatments (supplementary Fig 2). These results illustrate that Imiquimod-mediated protection to epithelial cells involves the expression of type I IFN and AMPs.
Discussion
Although the etiology of IBD is not clearly understood, it is thought to be related to an abnormal immune response in the GI tract. Studies have shown that hereditary and environmental factors interact with the immune system to induce chronic intestinal inflammation by dysregulating the mucosal immune response to commensal bacteria, microbial products and microbial antigens.2, 4 Defective epithelial immunity in the GI tract may allow bacteria or their products to trigger an immune reaction that results in prolonged inflammation. Mutation of NOD2/CARD15 and TLRs plays a role in innate immune dysfunction and may lead to the development of IBD.25-27 We previously reported a stimulation of innate immunity following treatment of granulocyte macrophage colony-stimulating factor (GM-CSF) in both human with Crohn’s disease and models of experimental colitis.28, 29 A type I IFN-dependant mechanism is supported in models of experimental colitis.29 In the present study, we found that oral administration of Imiquimod induced type I IFN expression in the small and large intestine of normal mice. Imiquimod-mediated increases in type I IFN expression occurred rapidly after the treatment. This result was in accordance with earlier studies, where Imiquimod-mediated increases in serum concentration of IFN-α and other cytokines were noted within 1-4 hours of treatments.8, 30
Plasmacytoid DCs (pDCs) are the main producers of type I IFN and strongly express TLR7 & 9.31 TLR9-mediated type I IFN production is also stimulated by bacterial products or synthetically derived DNA motifs, specifically cytosine-phosphate-guanosine (CpG) motifs. Administration of CpG immunostimulatory DNA has been shown to be effective in the treatment of experimental colitis in animal models by production of type I IFN. 5, 32-34 Stimulation of TLR7 has potent effects on PDC populations and potentially could mimic the effects of CpG or GM-CSF in colitis. In the present study, oral Imiquimod treatment led to significant increases in DC populations and type I IFN production that could be responsible for reduction in inflammation of DSS treated mice.
Therapeutic studies targeting proinflammatory cytokines like TNF-α and IL1-β have been extensively investigated in CD patients.35, 36 Imiquimod treatment in humans induces pro-inflammatory cytokines such as IFN-α, TNF-α, IL6 and other cytokines.19 Studies have shown that TLR4 and TLR8 agonists increases TNF-α, IL1-β, IL6 levels, which are associated with increased liver damage in HCV patients. Elevated levels of IL6 & TNF-α are also associated with a lower response to IFN-α therapy. 37, 38 A study comparing different TLR ligands in human peripheral blood mononuclear cells showed that TLR7 agonists did not induce proinflammatory cytokines significantly and recommended TLR7 as an attractive target for the development of new hepatitis C virus (HCV) therapies.39 The use of TLR activators as immunotherapeutic agents has been associated with side effects related to the release and systemic dispersion of proinflammatory cytokines. Clinical studies concerning the treatment of asymptomatic human immunodeficiency virus and refractory cancer have shown that Imiquimod treatments are effective, but limited by significant systemic side effects.23, 40 To address these side effects, in some studies TLR7 agonists were delivered as conjugates of macromolecules to confine the immunostimulatory activity to the local mucosal environment.41, 42
Since Imiquimod is approved as a topical applicator for the treatment of anogenital and perianal warts, we determined whether topical application of Imiquimod would revert colitis. Colonic administration of Imiquimod in normal mice significantly induced type I IFN expression. However, increases in type I IFN expression were observed in 18 hours treatment groups only, which likely are due to the limited permeability of the colon. Topical administration of Imiquimod in DSS-treated mice resulted in improvements in distal segments of colon only. Reduced colon shortening, improved crypt morphology and decreased level of proinflammatory cytokines were noted.
As IBD is thought to relate to an inappropriate response to gut bacteria, we examined Imiquimod effects on the epithelial and dendritic cells response to bacteria. We observed that Imiquimod-mediated increases in AMPs in dendritic cells and intestinal epithelial cells. These AMPs had the potential to reduce the survival of Salmonella. AMPs are expressed in the entire length of the GI tract, including the epithelium and paneth cells, and act as first-line extracellular defense in the intestine.43 The importance of AMPs was recently highlighted in mice with constitutive downregulated cryptdins associated with increased risk to intestinal inflammation.44 This study identifies the importance of type I IFN-mediated increases in AMP’s expression in setting of the antimicrobial activity in inflamed mucosa and maintaining a balance interaction between healthy mucosa and intraluminal pathogens.
In summary, Imiquimod has a therapeutic effect on experimental mouse models of DSS-induced acute colitis. The functional role of immune cells, especially DCs, establishes the importance of type I IFN and AMPs in mucosal innate defense. The present study identifies the TLR7 ligand, Imiquimod as a potent immunoregulator in the mucosa that holds promise for the treatments of IBD.
Supplementary Material
Acknowledgments
Grant support: NIH grants (DK 60106, AI 069390) and the Washington University DDRCC (P30 DK52574).
Abbreviations
- TLR7
toll like receptor-7
- IFN
interferons
- DSS
dextran sulfate sodium
- DC
dendritic cells
- Imiq
Imiquimod
- AMPs
antimicrobial peptides
References
- 1.Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:417–429. doi: 10.1056/NEJMra020831. [DOI] [PubMed] [Google Scholar]
- 2.Hanauer SB. Inflammatory bowel disease: epidemiology, pathogenesis, and therapeutic opportunities. Inflamm Bowel Dis. 2006;12(Suppl 1):S3–9. doi: 10.1097/01.mib.0000195385.19268.68. [DOI] [PubMed] [Google Scholar]
- 3.Present DH. 6-Mercaptopurine and other immunosuppressive agents in the treatment of Crohn’s disease and ulcerative colitis. Gastroenterol Clin North Am. 1989;18:57–71. [PubMed] [Google Scholar]
- 4.Blumberg RS. Inflammation in the intestinal tract: pathogenesis and treatment. Dig Dis. 2009;27:455–464. doi: 10.1159/000235851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Obermeier F, Dunger N, Strauch UG, et al. CpG motifs of bacterial DNA essentially contribute to the perpetuation of chronic intestinal inflammation. Gastroenterology. 2005;129:913–927. doi: 10.1053/j.gastro.2005.06.061. [DOI] [PubMed] [Google Scholar]
- 6.Obermeier F, Hofmann C, Falk W. Inflammatory bowel diseases: when natural friends turn into enemies-the importance of CpG motifs of bacterial DNA in intestinal homeostasis and chronic intestinal inflammation. Int J Inflam. 2010:641910. doi: 10.4061/2010/641910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Katakura K, Lee J, Rachmilewitz D, et al. Toll-like receptor 9-induced type I IFN protects mice from experimental colitis. J Clin Invest. 2005;115:695–702. doi: 10.1172/JCI22996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bottrel RL, Yang YL, Levy DE, et al. The immune response modifier imiquimod requires STAT-1 for induction of interferon, interferon-stimulated genes, and interleukin-6. Antimicrob Agents Chemother. 1999;43:856–861. doi: 10.1128/aac.43.4.856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lore K, Betts MR, Brenchley JM, et al. Toll-like receptor ligands modulate dendritic cells to augment cytomegalovirus- and HIV-1-specific T cell responses. J Immunol. 2003;171:4320–4328. doi: 10.4049/jimmunol.171.8.4320. [DOI] [PubMed] [Google Scholar]
- 10.Diebold SS, Kaisho T, Hemmi H, et al. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 11.Prins RM, Craft N, Bruhn KW, et al. The TLR-7 agonist, imiquimod, enhances dendritic cell survival and promotes tumor antigen-specific T cell priming: relation to central nervous system antitumor immunity. J Immunol. 2006;176:157–164. doi: 10.4049/jimmunol.176.1.157. [DOI] [PubMed] [Google Scholar]
- 12.Palamara F, Meindl S, Holcmann M, et al. Identification and characterization of pDC-like cells in normal mouse skin and melanomas treated with imiquimod. J Immunol. 2004;173:3051–3061. doi: 10.4049/jimmunol.173.5.3051. [DOI] [PubMed] [Google Scholar]
- 13.Wolf IH, Cerroni L, Kodama K, et al. Treatment of lentigo maligna (melanoma in situ) with the immune response modifier imiquimod. Arch Dermatol. 2005;141:510–514. doi: 10.1001/archderm.141.4.510. [DOI] [PubMed] [Google Scholar]
- 14.Gupta AK, Cherman AM, Tyring SK. Viral and nonviral uses of imiquimod: a review. J Cutan Med Surg. 2004;8:338–352. doi: 10.1007/s10227-005-0023-5. [DOI] [PubMed] [Google Scholar]
- 15.Gupta AK, Browne M, Bluhm R. Imiquimod: a review. J Cutan Med Surg. 2002;6:554–560. doi: 10.1007/s10227-001-0134-6. [DOI] [PubMed] [Google Scholar]
- 16.Edwards AD, Diebold SS, Slack EM, et al. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol. 2003;33:827–833. doi: 10.1002/eji.200323797. [DOI] [PubMed] [Google Scholar]
- 17.Hemmi H, Kaisho T, Takeuchi O, et al. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 18.Doxsee CL, Riter TR, Reiter MJ, et al. The immune response modifier and Toll-like receptor 7 agonist S-27609 selectively induces IL-12 and TNF-alpha production in CD11c+CD11b+CD8- dendritic cells. J Immunol. 2003;171:1156–1163. doi: 10.4049/jimmunol.171.3.1156. [DOI] [PubMed] [Google Scholar]
- 19.Miller RL, Gerster JF, Owens ML, et al. Imiquimod applied topically: a novel immune response modifier and new class of drug. Int J Immunopharmacol. 1999;21:1–14. doi: 10.1016/s0192-0561(98)00068-x. [DOI] [PubMed] [Google Scholar]
- 20.Loher F, Schmall K, Freytag P, et al. The specific type-4 phosphodiesterase inhibitor mesopram alleviates experimental colitis in mice. J Pharmacol Exp Ther. 2003;305:549–556. doi: 10.1124/jpet.102.039529. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Rey E, Delgado M. Therapeutic treatment of experimental colitis with regulatory dendritic cells generated with vasoactive intestinal peptide. Gastroenterology. 2006;131:1799–1811. doi: 10.1053/j.gastro.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 22.Sainathan SK, Tu L, Bishnupuri KS, et al. PEGylated murine Granulocyte-macrophage colony-stimulating factor: production, purification, and characterization. Protein Expr Purif. 2005;44:94–103. doi: 10.1016/j.pep.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 23.Savage P, Horton V, Moore J, et al. A phase I clinical trial of imiquimod, an oral interferon inducer, administered daily. Br J Cancer. 1996;74:1482–1486. doi: 10.1038/bjc.1996.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uehara A, Fujimoto Y, Fukase K, et al. Various human epithelial cells express functional Toll-like receptors, NOD1 and NOD2 to produce anti-microbial peptides, but not proinflammatory cytokines. Mol Immunol. 2007;44:3100–3111. doi: 10.1016/j.molimm.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 25.Figueroa C, Peralta A, Herrera L, et al. NOD2/CARD15 and Toll-like 4 receptor gene polymorphism in Chilean patients with inflammatory bowel disease. Eur Cytokine Netw. 2006;17:125–130. [PubMed] [Google Scholar]
- 26.Franchimont D, Vermeire S, El Housni H, et al. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut. 2004;53:987–992. doi: 10.1136/gut.2003.030205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Heel DA, Ghosh S, Hunt KA, et al. Synergy between TLR9 and NOD2 innate immune responses is lost in genetic Crohn’s disease. Gut. 2005;54:1553–1557. doi: 10.1136/gut.2005.065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Korzenik JR, Dieckgraefe BK, Valentine JF, et al. Sargramostim for active Crohn’s disease. N Engl J Med. 2005;352:2193–2201. doi: 10.1056/NEJMoa041109. [DOI] [PubMed] [Google Scholar]
- 29.Sainathan SK, Hanna EM, Gong Q, et al. Granulocyte macrophage colony-stimulating factor ameliorates DSS-induced experimental colitis. Inflamm Bowel Dis. 2008;14:88–99. doi: 10.1002/ibd.20279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reiter MJ, Testerman TL, Miller RL, et al. Cytokine induction in mice by the immunomodulator imiquimod. J Leukoc Biol. 1994;55:234–240. doi: 10.1002/jlb.55.2.234. [DOI] [PubMed] [Google Scholar]
- 31.Fitzgerald-Bocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. 2008;19:3–19. doi: 10.1016/j.cytogfr.2007.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rachmilewitz D, Karmeli F, Takabayashi K, et al. Immunostimulatory DNA ameliorates experimental and spontaneous murine colitis. Gastroenterology. 2002;122:1428–1441. doi: 10.1053/gast.2002.32994. [DOI] [PubMed] [Google Scholar]
- 33.Rachmilewitz D, Katakura K, Karmeli F, et al. Toll-like receptor 9 signaling mediates the anti-inflammatory effects of probiotics in murine experimental colitis. Gastroenterology. 2004;126:520–528. doi: 10.1053/j.gastro.2003.11.019. [DOI] [PubMed] [Google Scholar]
- 34.Obermeier F, Dunger N, Strauch UG, et al. Contrasting activity of cytosin-guanosin dinucleotide oligonucleotides in mice with experimental colitis. Clin Exp Immunol. 2003;134:217–224. doi: 10.1046/j.1365-2249.2003.02288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stack WA, Mann SD, Roy AJ, et al. Randomised controlled trial of CDP571 antibody to tumour necrosis factor-alpha in Crohn’s disease. Lancet. 1997;349:521–524. doi: 10.1016/s0140-6736(97)80083-9. [DOI] [PubMed] [Google Scholar]
- 36.Ferretti M, Casini-Raggi V, Pizarro TT, et al. Neutralization of endogenous IL-1 receptor antagonist exacerbates and prolongs inflammation in rabbit immune colitis. J Clin Invest. 1994;94:449–453. doi: 10.1172/JCI117345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Falasca K, Ucciferri C, Dalessandro M, et al. Cytokine patterns correlate with liver damage in patients with chronic hepatitis B and C. Ann Clin Lab Sci. 2006;36:144–150. [PubMed] [Google Scholar]
- 38.Farinati F, Cardin R, Bortolami M, et al. Oxidative damage, pro-inflammatory cytokines, TGF-alpha and c-myc in chronic HCV-related hepatitis and cirrhosis. World J Gastroenterol. 2006;12:2065–2069. doi: 10.3748/wjg.v12.i13.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Thomas A, Laxton C, Rodman J, et al. Investigating Toll-like receptor agonists for potential to treat hepatitis C virus infection. Antimicrob Agents Chemother. 2007;51:2969–2978. doi: 10.1128/AAC.00268-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goldstein D, Hertzog P, Tomkinson E, et al. Administration of imiquimod, an interferon inducer, in asymptomatic human immunodeficiency virus-infected persons to determine safety and biologic response modification. J Infect Dis. 1998;178:858–861. doi: 10.1086/515343. [DOI] [PubMed] [Google Scholar]
- 41.Wu CC, Hayashi T, Takabayashi K, et al. Immunotherapeutic activity of a conjugate of a Toll-like receptor 7 ligand. Proc Natl Acad Sci U S A. 2007;104:3990–3995. doi: 10.1073/pnas.0611624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan M, Hayashi T, Kuy CS, et al. Synthesis and immunological characterization of toll-like receptor 7 agonistic conjugates. Bioconjug Chem. 2009;20:1194–1200. doi: 10.1021/bc900054q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eckmann L. Defence molecules in intestinal innate immunity against bacterial infections. Curr Opin Gastroenterol. 2005;21:147–151. doi: 10.1097/01.mog.0000153311.97832.8c. [DOI] [PubMed] [Google Scholar]
- 44.Steinbrecher KA, Harmel-Laws E, Sitcheran R, et al. Loss of epithelial RelA results in deregulated intestinal proliferative/apoptotic homeostasis and susceptibility to inflammation. J Immunol. 2008;180:2588–2599. doi: 10.4049/jimmunol.180.4.2588. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.