

Abstract
Loss of function mutations and deletions encompassing the PHF6 gene are present in about 20% of T-cell acute lymphoblastic leukemias. Here we report the identification of recurrent mutations in PHF6 in 10/353 adult acute myeloid leukemias (AML). Genetic lesions in PHF6 found in AML are frameshift and nonsense mutations distributed through the gene or point mutations involving the second PHD-like domain of the protein. As in the case of T-ALL, where PHF6 alterations are found almost exclusively in males, mutations in PHF6 were 7 times more prevalent in males than in females with AML. Overall these results identify PHF6 as a tumor suppressor mutated in AML and extend the role of this X-linked tumor suppressor gene in the pathogenesis of hematologic tumors.
Keywords: PHF6, mutations, AML
Introduction
Acute myeloid leukemia (AML) consists of a heterogeneous group of aggressive neoplasms that is characterized by clinical and genetic heterogeneity and shows an increasing incidence with age(1). Insights in the molecular genetic basis of AML initially came from the characterization of recurrent chromosomal rearrangements, including t(8;21), t(15;17), inv(16), and different 11q23 translocations (2). Such clonal chromosome aberrations are detectable in the leukemic blasts of approximately 55% of adults with AML and have been recognized as important prognostic factors. Moreover, the characterization of genes located in the breakpoints of these rearrangements identified critical fusion oncogenes involved in the pathogenesis of AML including RUNX1-MTG8/AML1-ETO, PML-RARA, CBFB/SMMHC and MLL-AF9. Subsequently, intense sequencing efforts of specific candidate genes, including NPM1, FLT3, CEBPA, MLL, NRAS, WT1, RUNX1, NF1 and TET2, further broadened the spectrum of genetic lesions towards a wide variety of somatic mutations implicated in AML pathogenesis (2–5). Finally, sequencing of complete AML genomes (6, 7) revealed the presence of new somatically acquired mutations and led to the identification of recurrent mutations in the IDH1 and IDH2 genes (8, 9).
Recently, we identified the plant homeodomain finger 6 (PHF6) gene as a new tumor suppressor in T-cell acute lymphoblastic leukemia (T-ALL)(10). PHF6 deletions and inactivating mutations are found in about 20% of T-ALL samples and are present almost exclusively in male leukemia cases(10). Notably, PHF6 mutations were not identified in precursor B-lineage ALL samples suggesting that loss of PHF6 might be restricted to lymphoid tumors of the T-cell lineage(10). In the past, detailed molecular characterization of T-ALL and AML revealed a number of common genetic lesions shared by these hematological tumors including the CALM-AF10(11, 12) and SET-NUP214(13, 14) gene fusions, MLL translocations(15) and somatically acquired mutations in RAS(16, 17), WT1(18, 19), FLT3(20–22) and NF1(23). Given these similarities, we hypothesized that mutational loss of PHF6 might also be implicated in the pathogenesis of specific subtypes of AML. To address this question, we sequenced all coding exons of PHF6 in a cohort of 353 AML patients. In addition, we used real-time quantitative DNA PCR to assess the presence of genomic PHF6 deletions in 41 cases. The results of this analysis show the presence of recurrent loss of function mutations in PHF6 in AML and characterize the spectrum of associated genetic alterations cooperating with PHF6 loss in myeloid malignancies.
Methods
Patient samples
Leukemic DNA and cryopreserved lymphoblast samples were provided by collaborating institutions in the US [Eastern Cooperative Oncology Group (ECOG) and Memorial Sloan-Kettering Cancer Center (MSKCC)], Spain [Hospital Central de Asturias, Oviedo] and Belgium [Department of Pediatric Hemato-Oncology, Leuven University Hospital, Leuven]. All samples were collected with informed consent and under the supervision of local IRB.
Sequence analysis
PHF6 mutations were analyzed by PCR amplification of PHF6 exons 2–10 followed by direct bidirectional DNA sequencing as previously described(10). Sequence analysis of IDH1, IDH2, TET2, ASXL1, FLT3, NPM1, CEBPA, WT1, KRAS and NRAS was performed as previously described(25).
Sorting of hematopoietic stem cell (HSC), myeloid progenitor and lymphoid populations
Murine bone marrow, thymus and spleen cells were sorted using a Dako Cytomation Mo-Flo Fluorescence Activated Cell Sorter. Antibody staining was performed as previously described(26). The antibodies used for sorting included c-kit (2B8), Sca-1 (D7), Mac-1 (M1/70), Gr-1 (RB6-8C5), NK1.1 (PK136), TER-119, CD3 (145-2C11), CD4 (L3T4), CD8α (53-6.7), CD19 (1D3), IgM (II/41), IL7Rα (A7R34), CD25 (PC61), TCRβ (H57-597), CD34 (RAM34), FcgammaRII/III (2.4G2), CD150 (9D1) and were purchased from BD-Pharmingen or e-Bioscience. The bone marrow lineage cocktail included antibodies against Mac-1, Gr-1, NK1.1, TER-119, CD3 and CD19. Sorted hematopoietic stem cell populations included total LSK (lin−/sca-1+/c-kit+), CD150− LSK and CD150+ LSK. Myeloid progenitor populations included common myeloid progenitors (CMP, lin−/sca-1−/c-kit+/CD34+/FcgammaRII/IIIlow), granulocyte-macrophage progenitors (GMP, lin−/sca-1−/c-kit+/CD34+/FcgammaRII/IIIhigh) and megakaryocyte-erythroid progenitors (MEP, lin−/sca-1−/c-kit+/CD34+/FcgammaRII/III−). Lymphocyte populations included bone marrow pro B (IgM−/CD19+/cKit+/CD25−) and pre B cells (IgM−/CD19+/cKit−/CD25+), mature splenic B cells (CD19+/IgM+), thymic double negative 1 T cells (DN1, CD4−/CD8−/cKit+/CD25−), double negative 2 T cells (DN2, CD4−/CD8−/cKit+/CD25+), double negative 3 T cells (DN3, CD4−/CD8−/cKit−/CD25+), double negative 4 T cells (DN4, CD4−/CD8−/cKit−/CD25low), intermediate single positive (ISP, CD4−/CD8+/TCRβ−) and double positive T cells (DP, CD4+/CD8+) and finally splenic peripheral mature single positive CD4 T cells (SP-CD4+, CD4+/CD8−) and single positive CD8 T cells (SP-CD8+, CD4−/CD8+).
Quantitative real time PCR
RNA preparation of sorted cell population was achieved using the RNeasy plus mini kit (QIAGEN) according to manufacturer’s protocol. cDNA was generated with the ThermoScript RT-PCR system (Invitrogen) and analyzed by quantitative real-time PCR using the SYBR Green RT-PCR Core Reagents kit (Applied Biosystems) and the 7300 Real-Time PCR System (Applied Biosystems). Phf6 expression levels were calculated using Gapdh as reference. PCR primers sequences are available upon request.
Real-time quantification of DNA copy number
Real-time quantitative DNA PCR analysis was performed to screen AML cases for the presence of genomic PHF6 deletions using the FastStart Universal SYBR Green Master Mix (Roche) and the 7300 Real-Time PCR System (Applied Biosystems) as previously described(10) using TIE2 as control gene. Data were analyzed using the comparative ddCT method (Applied Biosystems).
Statistical analysis
The Fisher’s exact test was used to compare the frequency of PHF6 mutations between male and female AML patients.
Results
PHF6 mutations in adult AML
PHF6 was recently identified as a novel X-linked tumor suppressor gene recurrently mutated and deleted in pediatric and adult T-ALL(10). To evaluate if PHF6 inactivation might also contribute to the pathogenesis of AML, we sequenced all coding exons of PHF6 and used real-time quantitative DNA PCR to assess the presence of genomic PHF6 deletions in AML samples. DNA sequencing analysis of PHF6 in AML revealed the presence of PHF6 mutations in 10/353 (3%) AMLs analyzed. Most PHF6 mutations present in AML were characteristically loss of function alleles with 3 nonsense and 4 frameshift truncating mutations (Figure 1a,b). In addition, we identified 3 missense mutations located in the N-terminal region (A40G) and the second PHD2 domain (H302Y and H329L) of PHF6 (Figure 1a,b). DNA copy number analysis of the PHF6 locus failed to detect any genomic PHF6 deletions in 41 AML (22 male and 19 female) cases analyzed.
Figure 1. PHF6 mutations in AML.

(A) Schematic representation of the PHF6 protein depicting the location of 4 nuclear localization signals and 2 imperfect PHD zinc finger domains. Overview of PHF6 mutations identified in primary AML samples. Filled circles represent nonsense and frameshift mutations, whereas missense mutations are depicted as open circles. The circle filled in gray indicates a frameshift PHF6 mutation identified in a female AML sample. (B) Representative DNA sequencing chromatograms of AML DNA samples showing mutations in exons 7, 9 and 10 of PHF6.
Cooperative genetic lesions in PHF6 mutated adult AML
PHF6 mutated AML cases in this series, corresponded to FAB subtype’s M0, M1 and M2, or presented as a secondary AML (Table 1). At the genetic level, AML is a heterogeneous disease characterized by the accumulation of acquired somatic genetic lesions that cooperate in the transformation of myeloid progenitor cells. In order to identify genetic defects that might cooperate with PHF6 inactivation in the pathogenesis of AML, we sequenced IDH1, IDH2, TET2, ASXL1, FLT3, NPM1, CEBPA, WT1, KRAS and NRAS, in PHF6 mutated AML samples. This analysis revealed mutations affecting IDH2, ASXL1, FLT3, CEBPα and NRAS as additional genetic events that may cooperate with PHF6 inactivation in the pathogenesis of AML (Table 1).
Table 1.
Characteristics of 10 primary AML samples showing PHF6 inactivation
| ID | Sex | Age (yr) | FAB | Cytogenetics |
PHF6 lesion
|
Cooperative gene mutations
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Type of alteration | Predicted protein | IDH1 | IDH2 | TET2 | ASXL1 | FLT3 | NPM1 | CEBPA | WT1 | KRAS | NRAS | |||||
| 1 | M | 60 | M0 | normal | Missense | H329L | WT | WT | WT | WT | TKD | WT | WT | WT | WT | WT |
| 2 | M | 45 | M1 | normal | Nonsense | R274X | WT | WT | WT | WT | WT | WT | WT | WT | WT | WT |
| 3 | M | 72 | 2nd | tri(8) | Frameshift | R335fs | WT | WT | WT | A637fs | WT | WT | WT | NA | NA | NA |
| 4 | M | 66 | M2 | tri(8) | Nonsense | R342X | WT | R140Q | WT | WT | WT | WT | WT | NA | NA | NA |
| 5 | M | 65 | 2nd | normal | Missense | H302Y | WT | WT | WT | WT | WT | WT | WT | NA | NA | NA |
| 6 | M | 41 | M2 | tri(8) | Frameshift | C20fs | WT | WT | WT | WT | ITD | WT | WT | NA | NA | NA |
| 7 | M | 74 | 2nd | del(5q),-7,-20 | Missense | A40G | WT | WT | WT | WT | ITD | WT | H195fs | NA | NA | NA |
| 8 | M | NA | M0 | NA | Frameshift | P200fs | WT | WT | WT | E1192X | WT | WT | WT | WT | WT | WT |
| 9 | F | NA | M0* | NA | Frameshift | N171fs | WT | WT | WT | WT | WT | WT | WT | WT | WT | G12V |
| 10 | M | NA | M0 | NA | Nonsense | R319X | WT | R172K | WT | E558X | WT | WT | P39H | WT | WT | WT |
2nd, secondary AML; TKD, tyrosine kinase domain mutation; ITD, internal tandem duplication
Biphenotypic T-myeloid leukemia
Phf6 expression in murine HSC, myeloid progenitor and lymphoid populations
PHF6 is highly conserved among vertebrates(28) and shows ubiquitous expression in a wide variety of human tissues(10, 28). The presence of recurrent mutations in PHF6 in AML suggests a possible role of this tumor suppressor gene in the control of myeloid development. In order to evaluate Phf6 expression in hematopoietic stem cells (HSCs) and myeloid progenitors, we performed quantitative RT-PCR analysis of sorted mouse myeloid progenitor and lymphoid cell populations. These analyses revealed ubiquitous but slightly lower expression levels of Phf6 transcripts in HSC and myeloid cell progenitor populations as compared to different subsets of lymphoid cells (Figure 2). Within the myeloid progenitor populations, we noticed higher Phf6 levels in LSK progenitors compared to CMP and GMP populations (Figure 2). The murine thymocyte populations at different stages of development showed a similar pattern of variable Phf6 expression as previously identified in human T-cell subsets(10). Finally, in the murine B-cell populations, we noticed a marked upregulation of Phf6 transcripts in pre-B cells compared to both pro-B and mature B cells.
Figure 2. Phf6 expression in HSC and myeloid progenitor populations.
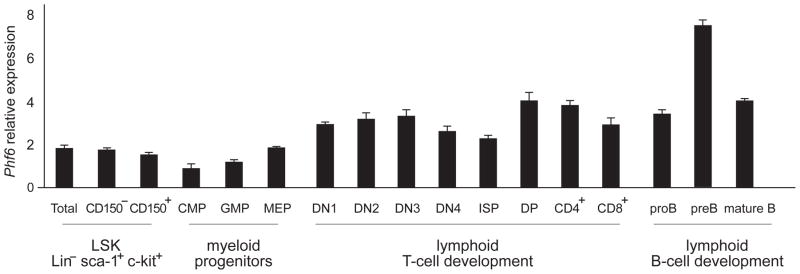
Quantitative RT-PCR analysis of murine Phf6 expression normalized to Gapdh in HSC, myeloid progenitor and lymphoid populations. Error bars indicate s.d.
PHF6 mutations are characteristically present in male patients with in AML
One of the most notorious features of PHF6 mutations in T-ALL is that they are almost exclusively found in male patients with this disease (10), which may explain in part the 3:1 higher prevalence of T-ALL in males than in females. Notably, although to a less extent than in T-ALL, AML is also more frequently found in males with a male to female ratio of 1.3 to 1. Analysis of the gender distribution in PHF6-mutated AML patients demonstrated that genetic alterations in PHF6 are 7 times more frequent in male (9/195; 4.6%) than in female (1/158; 0.6%) AML patient samples (P<0.05, Figure 3).
Figure 3. Gender distribution of PHF6 mutations in AML.
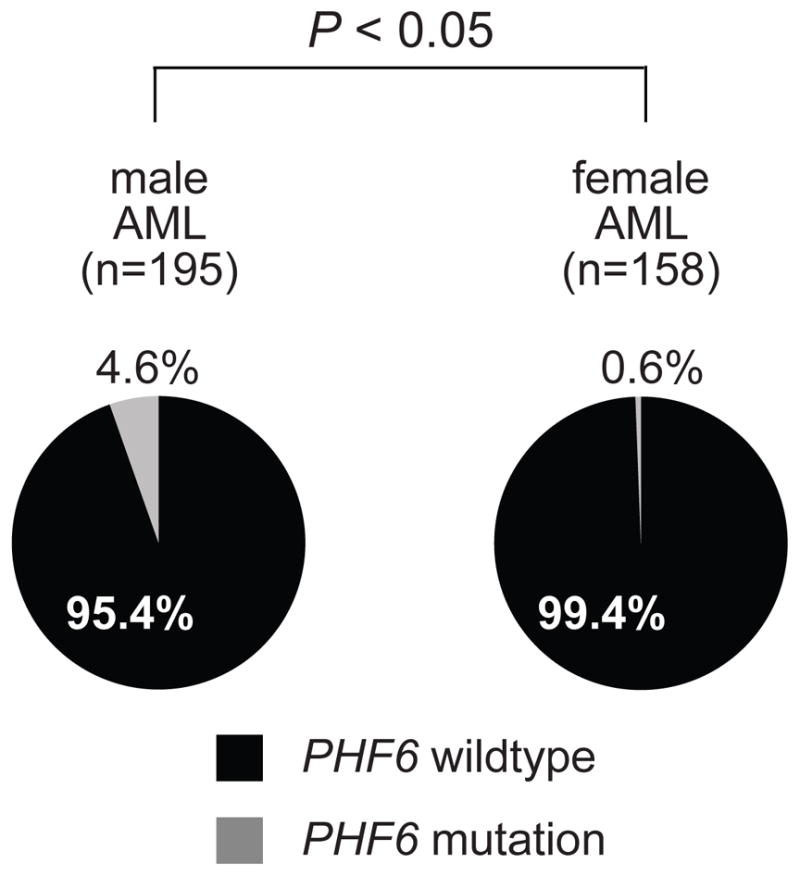
Differential distribution of PHF6 mutations in AML samples from male and female patients.
Discussion
The PHF6 tumor suppressor gene encodes a plant homeodomain (PHD) protein containing 4 nuclear localization signals and 2 imperfect PHD zinc finger domains(27) with a proposed role in transcriptional regulation and/or chromatin remodeling(27, 28). Inactivating mutations in PHF6 cause the Börjeson-Forssman-Lehman syndrome(29) (BFLS), a relatively uncommon type of X-linked mental retardation that mainly affects males, and shows milder clinical features in affected carrier females(30). A recent report described a male BFLS patient that developed T-ALL, suggesting that BFLS represents a cancer predisposition syndrome(31).
In this study, we evaluated if mutational loss of PHF6 might also be implicated in the pathogenesis of adult AML and identified PHF6 mutations in ~3% (10/353) of adult AML samples analyzed. PHF6 mutated primary AML cases were predominantly immature leukemias (FAB subtypes M0–M2), however, they showed definite AML immunophenotypes. Only in one case, retrospective analysis of one of the PHF6 mutated AML M0 samples, showed weak cytoplasmic CD3 positivity, together with 8% myeloperoxidase positive blasts and CD15/CD33 expression. Moreover, the presence of additional cooperative mutations affecting prototypical AML associated oncogenes and tumor suppressor genes such as IDH2, ASXL1, FLT3, CEBPα and NRAS (Table 1), in PHF6 mutated AML cases, further confirms the true myeloid nature of these samples.
Nonsense and frame-shift PHF6 mutations accounted for 70% (7/10) of all PHF6 mutations identified in our series and were distributed throughout the complete PHF6 gene (Figure 1a,b). Missense mutations accounted for the remaining 30% (3/10) of PHF6 lesions and mainly involved the second plant homeodomain (PHD)-like zinc finger domain of the PHF6 protein (Figure 1a,b). This includes an amino acid substitution (A40G) in the N terminus region of PHF6, a variant that was unique among 546 hematologic tumors analyzed in our lab. However, no remission material was available to test the somatic origin of this change. Thus, this particular variant may correspond to a novel point mutation disrupting the tumor suppressor function of PHF6 or alternatively correspond to a previously unreported polymorphism. Notably, the other two missense mutations found in AML involved residues R319 and H329 located in the second PHD-like domain of PHF6 (Figure 1a,b, Table 1), which have been previously found mutated in T-ALL(10), further strengthening the idea that the second PHD-like domain may mediate critical tumor suppressor functions of PHF6(10).
Overall, our results identify PHF6 as an X-linked tumor suppressor gene that is mutated in a fraction of both de novo as well as secondary adult AMLs. PHF6 mutations occur at a lower frequency in AML compared with T-ALL, but target mainly male patients in both hematological malignancies. The prognostic impact of PHF6 mutations in AML will need to be assessed in larger cohorts of patients collected on multi center clinical trials. In addition, these results suggest the possibility that PHF6 mutations might occur in male patients with other myeloid malignancies, such as myelodysplasia or myeloproliferative disorders which should be the addressed in future studies.
Acknowledgments
This study was supported by the Fund for Scientific Research (FWO) Flanders (postdoctoral grants to P.V.V. and project grants G.0198.08 and G.0869.10N to F.S.); the GOA-UGent (grant no. 12051203); the IWT-Vlaanderen (SBO grant no. 060848); the Children Cancer Fund Ghent (F.S.); the Belgian Program of Interuniversity Poles of Attraction; the Belgian Foundation Against Cancer; the ECOG and MSKCC tumor banks; a Physical Sciences-Oncology Center grant from the NCI (R.L.L.); the National Institutes of Health (R01CA120196 and R01CA129382 to A.F.); the Rally Across America Foundation (A.F); the Swim Across America Foundation (A.F.) and the Golfers Against Cancer Foundation (A.F.). R.L.L. is the Geoffrey Beene Junior Chair and an Early Career Award Recipient of the Howard Hughes Medical Institute. A.F. is a Leukemia & Lymphoma Society Scholar.
We appreciate the assistance of Adriana Heguy with DNA resequencing.
Footnotes
Author contributions
P.V.V. performed mutation analysis of PHF6 and wrote the manuscript. J.P., O.A. and R.L. performed mutation analysis of PHF6 on the ECOG patient series (E1900). C.L. and I.A. performed the isolation of murine hematopoietic stem cell (HSC) and myeloid progenitor populations. E.P. and A.M. provided samples and correlative data from ECOG. M.B., C.N. and A.P provided samples and correlative data from Hospital Central de Asturias. P.V., J.C. and F.S. provided samples and correlative data from University Hospital Leuven. R.L. and C.H. provided samples and correlative data from MSKCC. A.F. designed the studies, directed research and wrote the manuscript.
Disclosure of conflicts of interest
No conflicts of interest to disclose.
References
- 1.Renneville A, Roumier C, Biggio V, Nibourel O, Boissel N, Fenaux P, et al. Cooperating gene mutations in acute myeloid leukemia: a review of the literature. Leukemia. 2008 May;22(5):915–931. doi: 10.1038/leu.2008.19. [DOI] [PubMed] [Google Scholar]
- 2.Dohner K, Dohner H. Molecular characterization of acute myeloid leukemia. Haematologica. 2008 Jul;93(7):976–982. doi: 10.3324/haematol.13345. [DOI] [PubMed] [Google Scholar]
- 3.Langemeijer SM, Kuiper RP, Berends M, Knops R, Aslanyan MG, Massop M, et al. Acquired mutations in TET2 are common in myelodysplastic syndromes. Nat Genet. 2009 Jul;41(7):838–842. doi: 10.1038/ng.391. [DOI] [PubMed] [Google Scholar]
- 4.Jankowska AM, Szpurka H, Tiu RV, Makishima H, Afable M, Huh J, et al. Loss of heterozygosity 4q24 and TET2 mutations associated with myelodysplastic/myeloproliferative neoplasms. Blood. 2009 Jun 18;113(25):6403–6410. doi: 10.1182/blood-2009-02-205690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Delhommeau F, Dupont S, Della Valle V, James C, Trannoy S, Masse A, et al. Mutation in TET2 in myeloid cancers. N Engl J Med. 2009 May 28;360(22):2289–2301. doi: 10.1056/NEJMoa0810069. [DOI] [PubMed] [Google Scholar]
- 6.Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008 Nov 6;456(7218):66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mardis ER, Ding L, Dooling DJ, Larson DE, McLellan MD, Chen K, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009 Sep 10;361(11):1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marcucci G, Maharry K, Wu YZ, Radmacher MD, Mrozek K, Margeson D, et al. IDH1 and IDH2 gene mutations identify novel molecular subsets within de novo cytogenetically normal acute myeloid leukemia: a Cancer and Leukemia Group B study. J Clin Oncol. May 10;28(14):2348–2355. doi: 10.1200/JCO.2009.27.3730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thol F, Damm F, Wagner K, Gohring G, Schlegelberger B, Hoelzer D, et al. Prognostic impact of IDH2 mutations in cytogenetically normal acute myeloid leukemia. Blood. Apr 26; doi: 10.1182/blood-2010-03-272146. [DOI] [PubMed] [Google Scholar]
- 10.Van Vlierberghe P, Palomero T, Khiabanian H, Van der Meulen J, Castillo M, Van Roy N, et al. PHF6 mutations in T-cell acute lymphoblastic leukemia. Nat Genet. 2010 Apr;42( 4):338–342. doi: 10.1038/ng.542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asnafi V, Radford-Weiss I, Dastugue N, Bayle C, Leboeuf D, Charrin C, et al. CALM-AF10 is a common fusion transcript in T-ALL and is specific to the TCRgammadelta lineage. Blood. 2003 Aug 1;102(3):1000–1006. doi: 10.1182/blood-2002-09-2913. [DOI] [PubMed] [Google Scholar]
- 12.Bohlander SK, Muschinsky V, Schrader K, Siebert R, Schlegelberger B, Harder L, et al. Molecular analysis of the CALM/AF10 fusion: identical rearrangements in acute myeloid leukemia, acute lymphoblastic leukemia and malignant lymphoma patients. Leukemia. 2000 Jan;14(1):93–99. doi: 10.1038/sj.leu.2401614. [DOI] [PubMed] [Google Scholar]
- 13.Quentmeier H, Schneider B, Rohrs S, Romani J, Zaborski M, Macleod RA, et al. SET-NUP214 fusion in acute myeloid leukemia- and T-cell acute lymphoblastic leukemia-derived cell lines. J Hematol Oncol. 2009;2:3. doi: 10.1186/1756-8722-2-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Vlierberghe P, van Grotel M, Tchinda J, Lee C, Beverloo HB, van der Spek PJ, et al. The recurrent SET-NUP214 fusion as a new HOXA activation mechanism in pediatric T-cell acute lymphoblastic leukemia. Blood. 2008 May 1;111(9):4668–4680. doi: 10.1182/blood-2007-09-111872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ferrando AA, Armstrong SA, Neuberg DS, Sallan SE, Silverman LB, Korsmeyer SJ, et al. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: dominance of HOX dysregulation. Blood. 2003 Jul 1;102(1):262–268. doi: 10.1182/blood-2002-10-3221. [DOI] [PubMed] [Google Scholar]
- 16.Bos JL, Toksoz D, Marshall CJ, Verlaan-de Vries M, Veeneman GH, van der Eb AJ, et al. Amino-acid substitutions at codon 13 of the N-ras oncogene in human acute myeloid leukaemia. Nature. 1985 Jun-Jul;315(6022):726–730. doi: 10.1038/315726a0. [DOI] [PubMed] [Google Scholar]
- 17.Kawamura M, Ohnishi H, Guo SX, Sheng XM, Minegishi M, Hanada R, et al. Alterations of the p53, p21, p16, p15 and RAS genes in childhood T-cell acute lymphoblastic leukemia. Leuk Res. 1999 Feb;23(2):115–126. doi: 10.1016/s0145-2126(98)00146-5. [DOI] [PubMed] [Google Scholar]
- 18.Hollink IH, van den Heuvel-Eibrink MM, Zimmermann M, Balgobind BV, Arentsen-Peters ST, Alders M, et al. Clinical relevance of Wilms tumor 1 gene mutations in childhood acute myeloid leukemia. Blood. 2009 Jun 4;113(23):5951–5960. doi: 10.1182/blood-2008-09-177949. [DOI] [PubMed] [Google Scholar]
- 19.Tosello V, Mansour MR, Barnes K, Paganin M, Sulis ML, Jenkinson S, et al. WT1 mutations in T-ALL. Blood. 2009 Jul 30;114(5):1038–1045. doi: 10.1182/blood-2008-12-192039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horiike S, Yokota S, Nakao M, Iwai T, Sasai Y, Kaneko H, et al. Tandem duplications of the FLT3 receptor gene are associated with leukemic transformation of myelodysplasia. Leukemia. 1997 Sep;11(9):1442–1446. doi: 10.1038/sj.leu.2400770. [DOI] [PubMed] [Google Scholar]
- 21.Paietta E, Ferrando AA, Neuberg D, Bennett JM, Racevskis J, Lazarus H, et al. Activating FLT3 mutations in CD117/KIT(+) T-cell acute lymphoblastic leukemias. Blood. 2004 Jul 15;104(2):558–560. doi: 10.1182/blood-2004-01-0168. [DOI] [PubMed] [Google Scholar]
- 22.Van Vlierberghe P, Meijerink JP, Stam RW, van der Smissen W, van Wering ER, Beverloo HB, et al. Activating FLT3 mutations in CD4+/CD8− pediatric T-cell acute lymphoblastic leukemias. Blood. 2005 Dec 15;106(13):4414–4415. doi: 10.1182/blood-2005-06-2267. [DOI] [PubMed] [Google Scholar]
- 23.Balgobind BV, Van Vlierberghe P, van den Ouweland AM, Beverloo HB, Terlouw-Kromosoeto JN, van Wering ER, et al. Leukemia-associated NF1 inactivation in patients with pediatric T-ALL and AML lacking evidence for neurofibromatosis. Blood. 2008 Apr 15;111(8):4322–4328. doi: 10.1182/blood-2007-06-095075. [DOI] [PubMed] [Google Scholar]
- 24.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009 Jul-Aug;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 25.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, et al. The Common Feature of Leukemia-Associated IDH1 and IDH2 Mutations Is a Neomorphic Enzyme Activity Converting alpha-Ketoglutarate to 2-Hydroxyglutarate. Cancer Cell. Mar 16;17(3):225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Aifantis I, Feinberg J, Fehling HJ, Di Santo JP, von Boehmer H. Early T cell receptor beta gene expression is regulated by the pre-T cell receptor-CD3 complex. J Exp Med. 1999 Jul 5;190(1):141–144. doi: 10.1084/jem.190.1.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lower KM, Turner G, Kerr BA, Mathews KD, Shaw MA, Gedeon AK, et al. Mutations in PHF6 are associated with Borjeson-Forssman-Lehmann syndrome. Nat Genet. 2002 Dec;32(4):661–665. doi: 10.1038/ng1040. [DOI] [PubMed] [Google Scholar]
- 28.Voss AK, Gamble R, Collin C, Shoubridge C, Corbett M, Gecz J, et al. Protein and gene expression analysis of Phf6, the gene mutated in the Borjeson-Forssman-Lehmann Syndrome of intellectual disability and obesity. Gene Expr Patterns. 2007 Oct;7(8):858–871. doi: 10.1016/j.modgep.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Borjeson M, Forssman H, Lehmann O. An X-linked, recessively inherited syndrome characterized by grave mental deficiency, epilepsy, and endocrine disorder. Acta Med Scand. 1962 Jan;171:13–21. doi: 10.1111/j.0954-6820.1962.tb04162.x. [DOI] [PubMed] [Google Scholar]
- 30.Turner G, Lower KM, White SM, Delatycki M, Lampe AK, Wright M, et al. The clinical picture of the Borjeson-Forssman-Lehmann syndrome in males and heterozygous females with PHF6 mutations. Clin Genet. 2004 Mar;65(3):226–232. doi: 10.1111/j.0009-9163.2004.00215.x. [DOI] [PubMed] [Google Scholar]
- 31.Chao M, Todd MA, Kontny U, Neas K, Sullivan MJ, Hunter AG, Picketts DJ, Kratz CP. T-cell acute lymphoblastic leukemia in association with Börjeson-Forssman-Lehmann syndrome due to a mutation in PHF6. Pediatric Blood & Cancer. 2010 doi: 10.1002/pbc.22574. [DOI] [PMC free article] [PubMed] [Google Scholar]