

The chemical diversity encoded by natural microbial communities has been significantly underexplored due to limitations associated with the inability to culture the majority of environmental bacteria[1] and the silencing of biosynthetic pathways under laboratory conditions.[2] Soils are predicted to contain thousands of unique bacterial species, which potentially harbor tens of thousands of functionally unexplored natural product biosynthetic gene clusters.[3] With the development of metagenomic cloning methods, it is now possible to use DNA extracted directly from soil (environmental DNA, eDNA) to construct libraries that capture the enormous biosynthetic diversity present in soil environments.[4] These libraries provide a means of functionally examining unexplored soil biosynthetic gene clusters and are therefore, appealing resources for sequence guided natural product discovery programs.[5] Based on the magnitude of the biosynthetic diversity captured in saturating soil eDNA libraries, we believed that it would be possible to use these libraries to identify novel members of clinically relevant natural product families with potentially improved biological activities. To this end, we used a natural product sequence-tag driven approach to guide the discovery of an anthracycline-based aromatic polyketide that shows improved in vitro antiproliferative activity compared to the natural product anthracyclines that are currently in clinical use. In heterologous expression experiments the eDNA-derived arm cluster was found to encode for arimetamycin A, an anthracycline that is more potent than clinically used natural anthracyclines and retains activity against multidrug-resistant (MDR) cancer cells.
Aromatic polyketides comprise a large class of structurally and functionally diverse bacterial natural products.[6] The biosyntheses of these metabolites, although differing in detail, all originate with the production of a polyacetate precursor by a conserved minimal polyketide synthase (min-PKS) that is composed of three proteins [β-ketoacyl synthase alpha (KSα), β-ketoacyl synthase beta (KSβ), acyl carrier protein (ACP)] (Figure 1).[6] KSα and KSβ genes clade into groups that correlate strongly with the specific aromatic polyketide structural classes that are encoded by the gene clusters in which they reside.[7] Extrapolating from this observation, we hypothesized that it should be possible to systematically screen large eDNA libraries for biosynthetic gene clusters capable of encoding novel members of clinically relevant aromatic polyketide structural classes using only PCR amplified min-PKS gene fragments as sequence tags. To test the utility of this approach, cosmid DNA isolated from four previously archived and arrayed soil eDNA mega-libraries (AB,, AZ and TX) was screened by PCR using min-PKS specific degenerate primers (Figure 1).[8] PCR amplicons generated during the screening of individual sub-libraries were sequenced and phylogenetically compared to the corresponding KSβ fragments from biosynthetic gene clusters known to encode for clinically relevant polyketide families. eDNA-derived KSβ sequences that fell into clades associated with the biosynthesis of a variety of clinically useful antibiotics and anticancer structural classes were identified in this analysis, suggesting that gene clusters capable of expanding the structural diversity seen within these molecule families were potentially present in our archived libraries (Figure 1).
Figure 1.

Sequence guided discovery of clinically relevant polyketides from arrayed soil metagenomic libraries. DNA extracted directly from soil is used to construct saturating cosmid mega-libraries (>10,000,000 clones), which are arrayed as pools of unique 5,000 membered sub-libraries. Aromatic polyketide min-PKS gene cassettes are amplified in PCR reactions with degenerate primers using DNA from each unique sub-library within the California (AB), Utah (UT), Arizona (AZ) and Texas (TX) mega-libraries as template. The phylogenetic grouping of eDNA sequences with KSβ sequences from gene clusters that encode clinically relevant natural products is used to guide the recovery of clones containing gene clusters of interest from specific sub-libraries. Heterologous expression of bioinformatically unique eDNA-derived gene clusters can be used to expand the structural diversity seen within known families of natural products and to potentially identify metabolites with improved activities.
A group of KSβ sequence tags of particular interest to us was one that fell into the clades with KSβ genes used in the biosynthesis of anthracycline-type molecules (Figure 1). The anthracyclines doxorubicin and daunorubicin, derived from the cultured bacterium Streptomyces peucetius, are both potent anticancer agents.[9] They are used extensively in the treatment of diverse cancers including soft tissue sarcomas, hematological malignancies and a variety of carcinomas. The clinical use of these drugs is hampered, however, by the development of resistance and cumulative cardiotoxicity issues.[9a, 9b] Despite extensive synthetic efforts,[10] natural compounds remain the most widely used compounds from this structural class. The results of min-PKS screening suggested that our arrayed soil eDNA libraries contained multiple biosynthetic gene clusters that could potentially encode additional natural anthracyclines.
Cosmid clones associated with three KSβ sequence tags (AZ129, AZ515 and TX19) that clade with known anthracycline KSβs were recovered from the eDNA libraries, sequenced, annotated and then compared to known anthracycline biosynthetic gene clusters (Figure 2). Two of these clones (AZ129 and TX19) were found to contain gene clusters distinct from any previously described anthracycline gene clusters. The gene cluster captured on the third clone (AZ515) is identical in both gene content and organization to the nogalamycin gene cluster.[11] Sequencing of the TX19 clone revealed two min-PKSs, one containing a KSβ gene that clades with type I (21-carbon core) anthracyclines (e.g. doxorubicin, cosmomycin and aclacinomycin) and a second containing a KSβ gene with type II (20-carbon core) anthracyclines (e.g. nogalamycin and steffimycin) (Figure 2). No previously sequenced anthracycline gene clusters are predicted to encode for the biosynthesis of both Type I and Type II anthracycline core structures. The gene cluster captured on clone AZ129 is most closely related to the biosynthetic gene cluster encoding for the S. steffisburgensis derived steffimycin family of anthracyclines.[12] However, it contains two additional glycosyltransferases as well as a set of genes predicted to encode for the biosynthesis of aminodeoxy sugars, which are not seen in known steffimycin structures (Figure 2).
Figure 2.
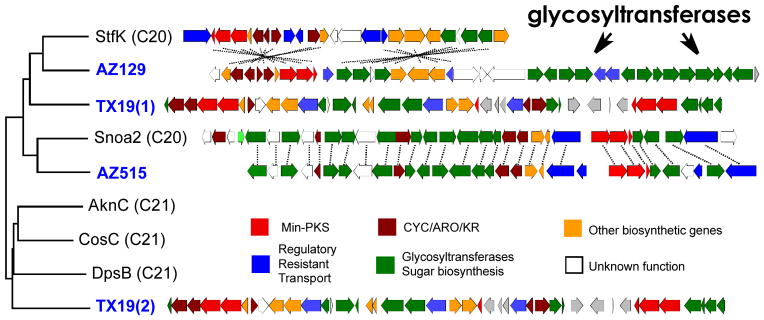
Anthracycline gene clusters. ClustalW phylogenetic tree of KSβ gene fragments from eDNA libraries (blue labels) and from culture-based studies (black labels). The gene clusters from clone AZ515 and clone AZ129 are compared to sequenced anthracycline gene cluster relatives (dashed lines connect functional homologs). The TX19 gene cluster does not closely resemble any anthracycline clusters deposited in public databases.
The steffimycins are a small subgroup of Type II anthracyclines (C20) that has gained only limited attention due to the poor antitumor activity of previously described steffimycin congeners.[13] Extensive structure activity relationship studies within the anthracycline family suggest that the sugar moiety plays a critical role in the observed antitumor activity of anthracyclines by interacting with the minor groove of DNA and topoisomerase II in a ternary complex.[14] In fact, these studies suggest that the ternary complex interactions mediated by the sugar moiety and the fourth ring of the anthracycline core are likely more critical for determining antitumor potency than the intercalation of DNA by the anthraquinone portion of the anthracycline core. The weak biological activity observed for known steffimycins is, at least in part, due to the fact that all steffimycins reported to date are functionalized with rhamnose-like sugar moieties, while most clinically relevant anthracyclines contain aminodeoxy sugar derivatives (e.g. L-daunosamine and L-rhodosamine).[13c] Owing to the importance of the sugar moiety for anthracycline bioactivity and the presence of the unprecedented set of aminodeoxy sugar biosynthesis genes found on clone AZ129, we elected to study this gene cluster using heterologous expression methods.
To permit heterologous expression studies in Streptomyces spp., the AZ129 cosmid was retrofitted with the 6.8 kb DraI fragment from pOJ436 which contains an origin of transfer, an apramycin resistance gene and the ΦC31 phage integration system. Retrofitted AZ129 was then transferred by intergenic conjugation into S. albus to generate S. albus/AZ129. In fermentation analyses this strain was found to produce the known steffimycin aglycone biosynthetic intermediate, 8-demethoxy-10-deoxysteffimycinone (Figure 4b, compound 4).[12] The production of an aglycone was not unexpected as AZ129, like the steffimycin cluster, is missing two key genes (dNDP-D-glucose synthase and a dNDP-D-glucose-4,6-dehydratase) that are required to initiate aminodeoxy sugar biosynthesis. These sugar biosynthesis genes appear in the primary metabolic background of some, but not all, Streptomyces spp.[15] The early genes required for deoxy sugar biosynthesis were therefore provided in trans (pIJRham) by cloning the rhamnose biosynthetic operon (oleL, oleS, oleE and oleU)[16] from oleandromycin biosynthesis under the control of the constitutive ermE* promoter within the conjugative integrative shuttle expression vector pIJ10257.[17] Conjugation of this construct into S. albus/AZ129 yielded S. albus/AZ129//pIJRham.
Figure 4.

Heterologous production and proposed biosynthesis of the arimetamycins based on gene fuction prediction by sequence homology and domain analysis. (a) The arm (arimetamycin) gene cluster spanning roughly 40 kb and containing 36 predicted ORFs (GenBank KF040454). (b) HPLC analysis of fermentation broth extracts showing arimetamycins A – C production by S. albus/AZ129/pIJRham. (c) Proposed biosynthetic scheme for the arimetamycins based on the gene prediction with the aid of Pfam domain analysis. See supplementary figure 3 for an expanded biosynthesis scheme and supplementary table 4 for a complete arm cluster gene table.
LCMS analysis of organic extracts obtained from cultures of S. albus/AZ129/pIJRham showed the presence of three major clone-specific metabolites with masses suggesting the successful addition of either one (MW 544 and 558) or two (MW 756) sugars onto the anthracycline aglycone core (Figure 4b). These three metabolites were isolated by C18 reversed-phase HPLC from large-scale ethyl acetate extracts and named arimetamycins A – C (1 –3: 2.5 mg/L for 1, 0.5 mg/L for 2 and 3 mg/L for 3). The structures of 1 – 3 were determined by HRESIMS, and 1D and 2D NMR analysis including 1H, 13C, COSY, TOCSY, HMQC, HMBC and ROESY NMR experiments (Supplementary discussions 1 and 2, Supplementary figures 1 and 2, Supplementary tables 1 and 2).
The arimetamycins feature a 20-carbon (Type II) anthracycline core (compound 4) with two distinct glycosylation patterns (Figure 3a). The dissacharide arimetamycin A (1) contains two rare N,N-dimethyl aminodeoxy sugar moieties, brasiliose and lemonose. To the best of our knowledge, lemonomycin[18] and nocathiacin[19] families are the only natural products reported to date that contain a lemonose sugar moiety. All previously reported steffimycin family anthracyclines are functionalized with a single rhamnose-based sugar moiety.[13c] The monoglycosylated arimetamycins B (2) and C (3) contain L-olivose-based sugars which differ from L-rhamnose by the absence of the C-2′ hydroxyl. Arimetamycin B (2) contains an actual L-olivose sugar while arimetamycin C (3) contains a methylated version of this sugar, 3′-OMe-L-olivose.
Figure 3.

Structure and activity of the arimetamycins. (a) Chemical structures of arimetamycins A – C (1 – 3). Only relative configuration is shown for the aglycone moiety. (b) Cytotoxicity assay against the four human carcinoma cell lines. Previously reported IC50 values for steffimycin against the MDAMB231 and HT-29 (WiDr) are 4,870 and 6,790 nM, respectively.[21]
Arimetamycins A – C (1 – 3) were evaluated for anticancer activity against representative colon, lung and breast cancer cell lines. The lung cancer line (H69AR) is multidrug resistant (MDR), showing high-level resistance to anthracyclines (50-fold) as well as many other anticancer agents including vincristine, etoposide and mitoxantrone (Figure 3b).[20] The disaccharide functionalized arimetamycin A (1) exhibits nanomolar IC50 against all cell lines including the H69AR MDR cell line. The monosaccharide functionalized arimetamycins B (2) and C (3) show only moderate activity in these cells (micromolar IC50), which is comparable to the activity reported for previously described anthracyclines in the steffimycin family.[21] IC50s obtained for arimetamycin A are consistently 2–10 fold lower than those observed for either doxorubicin or daunorubicin, and unlike doxorubicin or daunorubicin, arimetamycin A is highly active against both MDR and drug sensitive cell lines. P-glycoprotein expression-dependent MDR is the most clinically significant resistance mechanism affecting the utility of anthracyclines.[9a, 9b] The sugar moiety has been shown to play a key role in anthracycline bioactivity, in particular N-alkylation of the sugar has proved important.[22] Arimetamycin A provides a new N-alkylated lead structure for use in the potential development of threapies for MDR cancers.
The biosynthesis of the arimetamycins can be rationalized on the basis of the predicted gene functions for the genes found within the AZ129 clone (Figure 4a and supplementary figure 3). In our biosynthetic proposal scheme for the arimetamycins, the Type II (C20) anthryacycline 4 is predicted to be synthesized by enzymes encoded by nine genes (arm1-9) that show a high sequence identity (67 – 85%) to their counterparts from the steffimycin gene cluster.[12] In our biosynthetic proposal, intermediate 4 is predicted to enter into two distinct glycosylation pathways (Figure 4c) resulting in the formation of both the dissacharide containing 1 and the monosaccharide containing 2 and 3. The tentative assignment of glycosyl transfer specificities shown in our biosynthetic scheme is based on the high sequence identity observed between arm12 and the steffimycin glycocysotranferase (stfG) and the position of the arm25/32 glycosyl transfer genes in the sugar biosynthesis cassette. As with all discovery based on heterologous expression studies, the genetic background, in this case S. albus expressing rhamnose biosynthesis, may play an un-natural role in the generation of the specific compounds observed in these studies.
The structures of compounds 1 – 3 together with the overall organization of the arm cluster and the proposed branched biosynthetic scheme suggest that the genetic instructions for the biosynthesis and addition of the two aminodeoxy sugars seen in arimetamycin A were likely acquired at some point by an existing monosaccharide generating anthracycline biosynthetic gene cluster that is closely related to the known steffimycin gene cluster (Figure 4).[12] Mimicking this natural evolutionary phenomenon recombinantly by transferring the arm-derived aminodeoxy sugar biosynthesis cassette to other anthracycline producers could prove useful for improving the activity of other compounds in this family as well.
Resistance mechanisms related to many of those found in human health care settings, like MDR efflux pumps, are also seen in lower eukaryotes and bacteria.[23] The ultimate consequence of a natural microbial small molecule “arms race” may be a situation where resistance and molecules capable of circumventing resistance evolve hand in hand. In this scenario, the arm gene cluster, in particular the aminodeoxy sugar biosynthesis cassette (arm22-36) would arise in response to naturally occurring MDR-like resistance mutations. The systematic screening of eDNA libraries for analogs of clinically relevant families of natural products should prove useful for identifying additional compounds capable of overcoming clinically relevant resistance mechanisms.
The work presented here demonstrates that the pool of biosynthetic gene clusters captured in saturating eDNA soil libraries is large enough to permit the directed discovery of gene clusters that encode for metabolites with improved activity compared to known biomedically relevant natural products. The sequence-tag guided gene cluster discovery approach we outline enables the rapid profiling of chemical diversity encoded by gene clusters previously hidden in microbial communities and the subsequent prioritization of these gene clusters for heterologous expression efforts. The strong correlation observed between individual biosynthetic gene phylogeny and the chemical structures of natural products associated with these sequences is not unique to aromatic polyketide biosynthesis. In fact, this phenomenon is likely to be seen universally throughout natural product biosynthesis as a result of functionally related gene clusters having common evolutionary ancestors. The approach outlined here should therefore be generically useful for rapidly assessing nature’s ability to encode for metabolites capable of improving the efficacy of diverse families of biomedically relevant natural products.
Supplementary Material
Footnotes
This work was supported by NIH GM077516. S.F.B. is an HHMI Early Career Scientist. We thank Dr José A. Salas for the generous gift of pRham.
References
- 1.a) Torsvik V, Ovreas L. Curr Opin Microbiol. 2002;5:240–245. doi: 10.1016/s1369-5274(02)00324-7. [DOI] [PubMed] [Google Scholar]; b) Rappe MS, Giovannoni SJ. Annu Rev Microbiol. 2003;57:369–394. doi: 10.1146/annurev.micro.57.030502.090759. [DOI] [PubMed] [Google Scholar]
- 2.Scherlach K, Hertweck C. Org Biomol Chem. 2009;7:1753–1760. doi: 10.1039/b821578b. [DOI] [PubMed] [Google Scholar]
- 3.a) Handelsman J, Rondon MR, Brady SF, Clardy J, Goodman RM. Chem Biol. 1998;5:R245–249. doi: 10.1016/s1074-5521(98)90108-9. [DOI] [PubMed] [Google Scholar]; b) Harvey A. Drug Discov Today. 2000;5:294–300. doi: 10.1016/s1359-6446(00)01511-7. [DOI] [PubMed] [Google Scholar]; c) Daniel R. Curr Opin Biotechnol. 2004;15:199–204. doi: 10.1016/j.copbio.2004.04.005. [DOI] [PubMed] [Google Scholar]; d) Schmeisser C, Steele H, Streit WR. Appl Microbiol Biotechnol. 2007;75:955–962. doi: 10.1007/s00253-007-0945-5. [DOI] [PubMed] [Google Scholar]; e) Miao V, Davies J. In: Uncultivated Microorganisms. Epstein SS, editor. Vol. 10. Springer; Berlin Heidelberg: 2009. pp. 217–236. [Google Scholar]; f) Iqbal HA, Feng Z, Brady SF. Curr Opin Chem Biol. 2012;16:109–116. doi: 10.1016/j.cbpa.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a) Zhou J, Bruns MA, Tiedje JM. Appl Environ Microbiol. 1996;62:316–322. doi: 10.1128/aem.62.2.316-322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Courtois S, Cappellano CM, Ball M, Francou FX, Normand P, Helynck G, Martinez A, Kolvek SJ, Hopke J, Osburne MS, August PR, Nalin R, Guerineau M, Jeannin P, Simonet P, Pernodet JL. Appl Environ Microbiol. 2003;69:49–55. doi: 10.1128/AEM.69.1.49-55.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bertrand H, Poly F, Van VT, Lombard N, Nalin R, Vogel TM, Simonet P. J Microbiol Methods. 2005;62:1–11. doi: 10.1016/j.mimet.2005.01.003. [DOI] [PubMed] [Google Scholar]; d) Brady SF. Nat Protoc. 2007;2:1297–1305. doi: 10.1038/nprot.2007.195. [DOI] [PubMed] [Google Scholar]
- 5.a) Ginolhac A, Jarrin C, Gillet B, Robe P, Pujic P, Tuphile K, Bertrand H, Vogel TM, Perriere G, Simonet P, Nalin R. Appl Environ Microbiol. 2004;70:5522–5527. doi: 10.1128/AEM.70.9.5522-5527.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fisch KM, Gurgui C, Heycke N, van der Sar SA, Anderson SA, Webb VL, Taudien S, Platzer M, Rubio BK, Robinson SJ, Crews P, Piel J. Nat Chem Biol. 2009;5:494–501. doi: 10.1038/nchembio.176. [DOI] [PubMed] [Google Scholar]; c) King RW, Bauer JD, Brady SF. Angew Chem. 2009;121:6375–6379. doi: 10.1002/anie.200901209. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2009;48:6257–6261. doi: 10.1002/anie.200901209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Hertweck C, Luzhetskyy A, Rebets Y, Bechthold A. Nat Prod Rep. 2007;24:162–190. doi: 10.1039/b507395m. [DOI] [PubMed] [Google Scholar]; b) Hertweck C. Angew Chem. 2009;121:4782–4811. [Google Scholar]; Angew Chem Int Ed. 2009;48:4688–4716. doi: 10.1002/anie.200806121. [DOI] [PubMed] [Google Scholar]
- 7.a) Ridley CP, Lee HY, Khosla C. Proc Natl Acad Sci. 2008;105:4595–4600. doi: 10.1073/pnas.0710107105. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Reddy BV, Kallifidas D, Kim JH, Charlop-Powers Z, Feng Z, Brady SF. Appl Environ Microbiol. 2012;78:3744–3752. doi: 10.1128/AEM.00102-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seow KT, Meurer G, Gerlitz M, Wendt-Pienkowski E, Hutchinson CR, Davies J. J Bacteriol. 1997;179:7360–7368. doi: 10.1128/jb.179.23.7360-7368.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Pharmacol Rev. 2004;56:185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]; b) Monneret C. Eur J Med Chem. 2001;36:483–493. doi: 10.1016/s0223-5234(01)01244-2. [DOI] [PubMed] [Google Scholar]; c) Grimm A, Madduri K, Ali A, Hutchinson CR. Gene. 1994;151:1–10. doi: 10.1016/0378-1119(94)90625-4. [DOI] [PubMed] [Google Scholar]
- 10.a) Vasey P. Int J Oncol. 1993;2:677–681. doi: 10.3892/ijo.2.4.677. [DOI] [PubMed] [Google Scholar]; b) Weiss RB, Sarosy G, Clagett-Carr K, Russo M, Leyland-Jones B. Cancer Chemother Pharmacol. 1986;18:185–197. doi: 10.1007/BF00273384. [DOI] [PubMed] [Google Scholar]
- 11.Torkkell S, Kunnari T, Palmu K, Mantsala P, Hakala J, Ylihonko K. Mol Genet Genomics. 2001;266:276–288. doi: 10.1007/s004380100554. [DOI] [PubMed] [Google Scholar]
- 12.Gullon S, Olano C, Abdelfattah MS, Brana AF, Rohr J, Mendez C, Salas JA. Appl Environ Microbiol. 2006;72:4172–4183. doi: 10.1128/AEM.00734-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.a) Wiley PF, Elrod DW, Marshall VP. J Org Chem. 1978;43:3457–3461. [Google Scholar]; b) Matsuzawa Y, Oki T, Takeuchi T, Umezawa H. J Antibiot. 1981;34:1596–1607. doi: 10.7164/antibiotics.34.1596. [DOI] [PubMed] [Google Scholar]; c) Laatsch H, Fotso S. Top Curr Chem. 2008;282:3–74. [Google Scholar]
- 14.Zunino F, Pratesi G, Perego P. Biochem Pharmacol. 2001;61:933–938. doi: 10.1016/s0006-2952(01)00522-6. [DOI] [PubMed] [Google Scholar]
- 15.Rodriguez E, Menzella HG, Gramajo H. Methods Enzymol. 2009;459:339–365. doi: 10.1016/S0076-6879(09)04615-1. [DOI] [PubMed] [Google Scholar]
- 16.Rodriguez L, Oelkers C, Aguirrezabalaga I, Brana AF, Rohr J, Mendez C, Salas JA. J Mol Microbiol Biotechnol. 2000;2:271–276. [PubMed] [Google Scholar]
- 17.Hong HJ, Hutchings MI, Hill LM, Buttner MJ. J Biol Chem. 2005;280:13055–13061. doi: 10.1074/jbc.M413801200. [DOI] [PubMed] [Google Scholar]
- 18.He H, Shen B, Carter GT. Tetrahedron Lett. 2000;41:2067–2071. [Google Scholar]
- 19.Ding Y, Yu Y, Pan H, Guo H, Li Y, Liu W. Mol BioSyst. 2010;6:1180–1185. doi: 10.1039/c005121g. [DOI] [PubMed] [Google Scholar]
- 20.Mirski SE, Gerlach JH, Cole SP. Cancer Res. 1987;47:2594–2598. [PubMed] [Google Scholar]
- 21.Olano C, Abdelfattah MS, Gullon S, Brana AF, Rohr J, Mendez C, Salas JA. Chem Bio Chem. 2008;9:624–633. doi: 10.1002/cbic.200700610. [DOI] [PubMed] [Google Scholar]
- 22.a) Capranico G, Supino R, Binaschi M, Capolongo L, Grandi M, Suarato A, Zunino F. Mol Pharmacol. 1994;45:908–915. [PubMed] [Google Scholar]; b) Schaefer A, Westendorf J, Lingelbach K, Schmidt CA, Mihalache DL, Reymann A, Marquardt H. Cancer Chemother Pharmacol. 1993;31:301–307. doi: 10.1007/BF00685675. [DOI] [PubMed] [Google Scholar]; c) Fang L, Zhang G, Li C, Zheng X, Zhu L, Xiao JJ, Szakacs G, Nadas J, Chan KK, Wang PG, Sun D. J Med Chem. 2006;49:932–941. doi: 10.1021/jm050800q. [DOI] [PubMed] [Google Scholar]
- 23.Saier MH, Jr, Paulsen IT, Sliwinski MK, Pao SS, Skurray RA, Nikaido H. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 1998;12:265–274. doi: 10.1096/fasebj.12.3.265. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.