

Abstract
Background
Francisellosis, caused by the bacterium Francisella noatunensis subsp. noatunensis, remains a serious threat to Atlantic cod (Gadhus morhua) farming in Norway and potentially in other countries. As outbreak strains appear clonal in population structure, access to highly discriminatory typing tools is critical for understanding the epidemiology of francisellosis infections in aquaculture. In this study, a simplified multiple-locus variable-number of tandem repeat analysis (MLVA) targeting five highly polymorphic variable number of tandem repeat (VNTR) loci in a single multiplex PCR was developed to rapidly discriminate between outbreak strains.
Results
The assay resulted in identification of at least 13 different allelic profiles or subpopulations among 91 F. noatunensis isolates from farmed cod in Norway. The VNTR loci appear relatively stable, with isolates originating from individual outbreaks showing identical MLVA profiles following repeated passage. MLVA displayed greater discriminatory power than pulse-field gel electrophoresis (PFGE). Both MLVA and PFGE show good epidemiological concordance by their abilities to separate outbreak strains from epidemiologically unrelated isolates.
Conclusions
The MLVA method presented here is robust, easy to perform and provides a good alternative to other typing systems for F. noatunensis subsp. noatunensis and epidemiological study of francisellosis in cod.
Keywords: VNTR, Capillary electrophoresis, Francisella noatunensis, Fish
Background
Francisella noatunensis is an emergent fish pathogen of major concern. It causes francisellosis, a systemic bacterial disease characterized by the presence of multi-organ granuloma with high morbidity and varying associated mortality levels [1]. The disease affects several important cultured fish species in fresh, brackish and marine water environments. Francisellosis in farmed Atlantic cod Gadhus morhua L. was first reported in 2004/2005, when an outbreak caused by F. noatunensis subsp. noatunensis was discovered in Norway [2,3]. Since then, many cases of F. noatunensis infection have been diagnosed in cod from numerous grow-out facilities spanning most of the mid-and south-western Norwegian coastline. Outbreaks caused by different strains of F. noatunensis have also been linked to other important cultured fish species around the globe [1]. As no vaccine is yet available and antibiotic treatment largely ineffective, understanding the infection dynamics and spread of the disease may be important in regard to management of francisellosis in aquaculture.
Genetically, F. noatunensis subsp. noatunensis is a monomorphic pathogen, showing very little intra-species variation [4,5]. As a result, few genetic tools with the necessary resolution to track and link individual disease outbreaks are available. An increasingly applied molecular typing tool for discrimination of bacterial species with stable clonal population structures is multiple locus variable-number tandem repeat analysis (MLVA). The method is based upon PCR amplification of variable tandem repeats (VNTRs), which are short polymorphic DNA sequences located at several loci in many microbial genomes [6]. Isolates belonging to single bacterial species generally maintain the same sequence elements but show variation in the number of repeat units through the activity of a strand-slippage mechanism introduced by DNA polymerase during replication [7-9]. The different variants are most commonly resolved by standard agarose gel electrophoresis or capillary electrophoresis on a DNA sequencer [10].
To date, there is limited information on MLVA application relating to pathogenic fish Francisella species. In a recently published study, DNA sequencing was used to determine variations in the number of repeats at seven VNTR loci [11]. Although the study highlighted the value of using MLVA as a typing system for F. noatunensis subsp. noatunensis, DNA sequencing is costly and labour intensive. Typing of a single isolate may require seven different PCRs and 14 sequencing reactions. The primary aim of the present study was, therefore, to improve the efficiency of MLVA as a typing scheme for discrimination of F. noatunensis isolates, through development of a single tube multiplex PCR amplification followed by automated fragment analysis using capillary electrophoresis. The epidemiological utility of the developed assay was compared with Pulsed-field gel electrophoresis (PFGE), which has been the gold standard method for epidemiological investigation of disease outbreaks in many pathogenic bacteria.
Results
MLVA
The MLVA assay developed in this study is based on capillary electrophoresis of the five most polymorphic loci identified within available fish-pathogenic Francisella genomes. Four of these loci, fnVNTR-2, fnVNTR-3, fnVNTR-4 and fnVNTR-5 were independently identified as Fnn-VNTR2, Fnn-VNTR3, Fnn-VNTR1 and Fnn-VNTR4, respectively, in a previous study by Brevik et al. [11]. The performance of the present assay was optimised in a single multiplex by determining the appropriate concentrations of primers, PCR amplification and capillary run conditions. Primers chosen for each marker yielded PCR products compatible with the internal size standard which generated peaks from 50 to 625 bp. DNA extracted by boiling was found adequate for further analysis, thus a separate DNA purification step was considered unnecessary. Individual PCR amplicons were easily differentiated based on colour, size and peak height in the electropherograms (Figure 1). Most markers appeared as single sharp peaks, while some showed split peaks, which is a common artefact in capillary electrophoresis, due to non-templated (incomplete) 3′A nucleotide additions (Applied Biosystems). Sizing of individual fragments was reproducible across replicated capillary electrophoretic runs. Sequencing of selected isolates (including all variants identified at each locus) confirmed that the size determined by fragment analysis was due to varying numbers of repeats at each targeted loci. However, calculation of the number of repeats in locus fnVNTR-1 based on automated fragment sizing was inaccurate. Capillary electrophoresis consistently estimated fragment sizes larger than direct sequencing of the different alleles, regardless of the dye (FAM or VIC) used for labeling. Nevertheless, the size differences were consistent across the entire size range for this locus.
Figure 1.
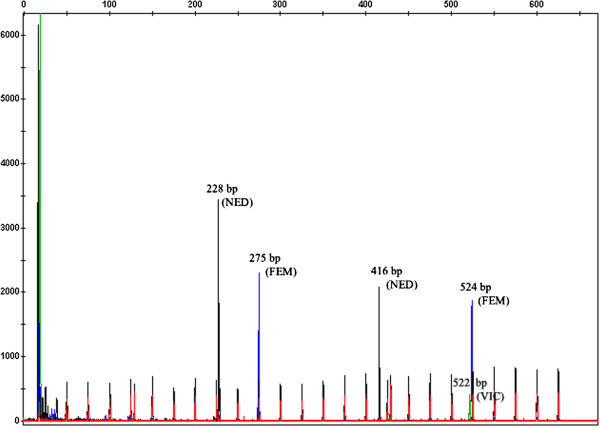
Electropherograms showing PCR fragments of all five VNTR loci co-amplified in a single PCR reaction. The fragments were resolved by size and dye colour using capillary electrophoresis.
In vitro stability of the VNTR loci was evaluated after multiple subcultures of four F. noatunensis subsp. noatunensis isolates displaying genetically distinct MLVA profiles. The isolates were passaged at 5 day intervals for nearly 30 weeks on CHAB incubated at 22°C. Screening of single colonies picked from passage 10, 20, 30 and 40 revealed the same MLVA profile as the original isolates (data not shown).
MLVA performed on 91 F. noatunensis subsp. noatunensis isolates from Atlantic cod, yielded PCR products for all VNTR loci. The number of individual alleles ranged between four (fnVNTR-4) and nine (fnVNTR-1) (Table 1). FnVNTR-1 carried the largest number of repeats (20–33 repeats), while fnVNTR-4 revealed only a limited number of repeat units (4–7 repeats). Differences in diversity indices (DI) were also observed for the 5 VNTR loci. The highest DI was associated with fnVNTR-1 (76.5%) and the lowest with fnVNTR-2 (57.7%). The discriminatory power for the combined set of VNTR markers was 83.3%, indicating the high probability of the MLVA assay separating two isolates from different diagnostic cases.
Table 1.
Characteristics of VNTR loci used in MLVA for F. noatunensis subsp. noatunensis isolates
| Marker locus | Alternate designation a | GeneBank accession no. | Repeat sequence | Number of repeats | Fragment size range (bp) | No. of alleles | DI (%) | Coding functions |
|---|---|---|---|---|---|---|---|---|
| fnVNTR-1 |
NA |
CP000937 |
TCTTTATTG |
20-33 |
427-549 |
9 |
76.5 |
TPR repeat protein |
| fnVNTR-2 |
Fnn-VNTR2 |
GU385768 |
AGTTATT |
8-14 |
275-440 |
4 |
57.7 |
ThiJ/PfpI family protein |
| fnVNTR-3 |
Fnn-VNTR3 |
GU385769 |
TAGAT |
7-11 |
213-234 |
5 |
67.6 |
UDP-N-acetylglucosamine 2-epimerase |
| fnVNTR-4 |
Fnn-VNTR1 |
GU385767 |
TTAAGGTA |
4-7 |
507-532 |
5 |
65.5 |
NA |
| fnVNTR-5 | Fnn-VNTR4 | GU385770 | CCACAA | 9-31 | 404-539 | 7 | 65.5 | DNA-directed DNA polymerase |
aFrom Brevik et al. [11].
NA, not applicable/available.
The population linkage disequilibrium was calculated to be significant by LIAN analysis ( = 0.2604, Ppara = 2.21 × 10-29), suggesting a low rate of recombination between the alleles. In total, 13 different MLVA types were found among the 91 Norwegian cod isolates (Additional file 1: Figure S1 in supplementary material). These MLVA profiles grouped into three major clonal clusters (designated I, II and III) using minimum spanning tree analysis to display the relationship between the various MLVA types (Figure 2). Cluster I was the largest group containing 8 different MLVA types (types 1, 2, 3, 4, 5, 6, 7 and 8) with MLVA type 1 being the dominant clone. The isolates in this cluster had no specific geographical or temporal associations (Figure 3). Cluster II consisted of 3 different MLVA types (type 9, 12 and 13), with the majority belonging to MLVA type 9 (Figure 2). These isolates were collected from a confined geographical area and included the type strain NCIMB 14265T. Cluster III comprised of two closely related MLVA types (types 10 and 11), differing only in one allele. All isolates in this cluster were collected from a limited geographical area in the period of 2006–2008. A large degree of genetic heterogeneity was evident among F. noatunensis subsp. noatunensis isolates from Chile, Ireland and Norway e.g. the Chilean salmon isolate shared only two VNTR loci with the other F. noatunensis subsp. noatunensis isolates from cod in Norway (fnVNTR3 and fnVNTR5) and Ireland (fnVNTR2 and fnVNTR3). The few isolates of F. noatunensis subsp. orientalis and F. philomiragia included in this study were distinctly separated from the F. noatunensis subsp. noatunensis isolates.
Figure 2.
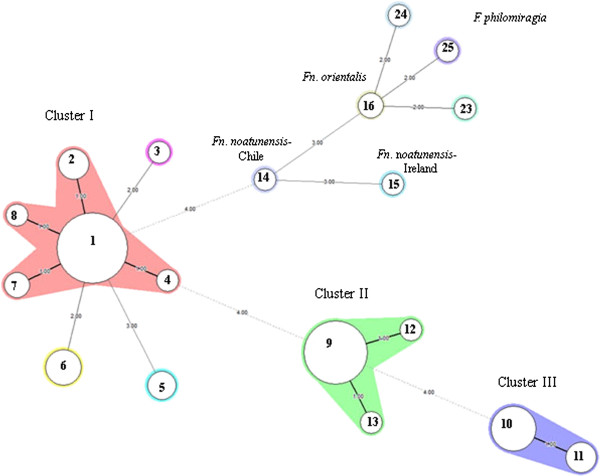
Minimum spanning tree (MST) of MLVA showing the genetic relatedness among the isolates included in this study. A categorical coefficient and the priority rule using the highest number of single-locus changes was used for generation of the minimum spanning tree. Each circle in the tree represents a different MLVA type (MVT), with the different MVTs indicated by the number in the circle. MVTs differing by a single VNTR locus are represented by thick short lines, while double-locus variants are shown by thin longer lines and dotted lines for those differing by more than two loci. The number of loci that differ between two MVTs is indicated on the lines linking them together. Clusters were defined as MVTs having a maximum of three differing loci and a minimum cluster size of two. The Norwegian isolates grouped into three main clusters (I, II and III). Cluster I is shaded pink, Cluster II in green and cluster III in violet.
Figure 3.

Map of Norway showing the geographical distribution of F. noatunensis subsp. noatunensis MLVA clonal groups generated by minimum spanning tree (MST) analysis. Cluster I is shaded pink, cluster II in green and cluster III in violet.
PFGE analysis
Overall, PFGE separated the 47 Norwegian cod isolates studied into five PFGE types with a lower discriminatory index (DI = 63.7%; CI 95% = 0.569-0.704) than MLVA typing. The isolates grouped into two main PFGE clusters designated PFGE I and II (Figure 4). The distribution pattern of PFGE types was generally consistent with the clustering obtained for the MLVA typing. In most cases, isolates belonging to the same MLVA cluster belonged to the same PFGE type. Exceptions included isolate 5341 (MLVA cluster I) and NCIMB14265T (MLVA cluster II) that differed in PFGE type from other isolates in their clusters, displaying a single band difference. The non-Norwegian isolates displayed unique PFGE types, which separate distinctly from each other (see Additional file 1: Figure S1 in supplementary material). The concordance between PFGE and MLVA, as determined by the adjusted Rand index, was moderate (50%) for the isolates typed by both methods. The calculated Wallace coefficients, which measures directional congruence between the typing methods were 80.8% and 37.1% for MLVA and PFGE, respectively. This suggests MLVA as more predictive in assigning isolates to the same PFGE group, than vice versa.
Figure 4.
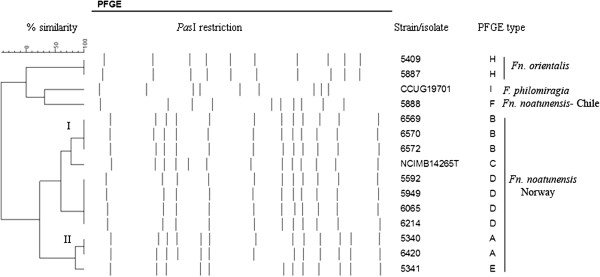
Dendrogram showing the genetic relatedness among selected isolates based on PFGE analysis. The dendrogram was constructed using the Dice coefficient correlation and WARD algorithm. The Norwegian isolates grouped into two clusters (I and II).
Discussion
In this study we validated a novel MLVA assay for the fish pathogen F. noatunensis subsp. noatunensis using capillary electrophoresis. The presented assay relies on multiplex PCR amplification of five VNTR loci visualised in a single electrophoretic run, allowing typing of many or few isolates simultaneously. The MLVA protocol is robust, easy to perform and generates unambiguous numerical profiles for individual strain identification.
Not surprisingly, independent searches within highly similar genomes performed during the same time periods identified several common informative VNTR loci. Brevik et al. [11] published an MLVA assay for F. noatunensis, in which sequencing was used to identify polymorphisms in seven VNTR loci. Four of these loci were also identified and included in the present study. The fnVNTR-1 marker was, however, unique to the present study. The high degree of polymorphism identified in each VNTR loci suggests that the optimal markers were selected by both MLVA schemes. For all loci, with the exception of fnVNTR-1, accurate size estimation of PCR products was achieved by capillary electrophoresis. Sizing by capillary, although reproducible for this locus, failed to correspond to the actual fragment size identified by sequencing. This discrepancy could be due to the nature of the gel matrix, or to slightly biased flanking sequences or differences in mobility patterns of specific repeat units as previously mentioned by other investigators [12,13]. Nonetheless, this did not interfere with the overall results, as alleles were correctly assigned with or without adjustment of the number of repeats deduced by capillary electrophoresis.
The specificity of the present assay was demonstrated by the amplification of all 5 VNTRs in all Norwegian isolates. As the primers were designed to exclusively detect F. noatunensis subsp. noatunensis isolates from cod, the lack of amplification from some loci in other bacterial subspecies/species may be due to either sequence variability in the primer regions or the absence of the locus altogether.
Generally, informative VNTR loci require a good balance between variability and stability. Although the studied loci appeared relatively stable over several generations in vitro, a single repeat change was previously reported for fnVNTR-4 in F. noatunensis subsp. noatunensis GM2212 following successive culturing at 20°C [11]. Nevertheless, the fact that isolates from different outbreaks in the same locality (exemplified by MVT 10), displayed identical MLVA patterns over a period of 3 years, indicates that the loci are of sufficient stability to be used as VNTR markers for typing of F. noatunensis subsp. noatunensis.
Our MLVA assay provided high resolution typing and clustered the Norwegian F. noatunensis subsp. noatunensis isolates into three major clonal groups, consistent with previous MLVA findings [11]. However, in the previous study, they indicated low diversity among 17 F. noatunensis subsp. noatunensis isolates collected from outbreaks in cultured cod populations. A total of four different allelic profiles were identified compared with 13 MLVA profiles detected from the 91 outbreak isolates included in the present study. Although polymorphisms in fnVNTR-1 may have contributed to the greater discriminatory ability of the present assay, it is more likely related to the greater number of isolates included in the present analysis. It was also reported that isolates from wild cod are more diverse than those from farmed cod [11]. That the least polymorphic locus identified in the present study (fnVNTR-2) was among the most diverse loci identified in isolates from wild cod [11], suggests that the discriminatory power of the present assay for F. noatunensis subsp. noatunensis may be underestimated.
At the time of sampling, the epidemiological situation was complex with movement of juvenile fish common over long distances along the Norwegian coast. Thus, the information generated during the present study is difficult to interpret in terms of ‘natural’ or anthropological spread of infection. Our data shows, however, that several clones were present within the Norwegian cod-farming industry over a short period of time. Interpretation of these results in relation to microevolution will require knowledge of the mutation rates and understanding of how VNTRs in F. noatunensis subsp. noatunensis change with time. The situation could also be explained by the existence of many clones of which we have only identified a few. MVT1 may represent a highly successful spreading clone, as this genotype was detected at high frequency each sampling year. As with other theories relating to bacterial clonal expansion, the widespread dissemination of MVT1 could have been caused by human activities [11,14]. While any significance of the demonstrated diversity within noatunensis to pathogenesis remains unclear, the current study helps define and differentiate epizootic F. noatunensis clones causing francisellosis in Norwegian cod populations.
Our data indicated better resolution for MLVA compared to PFGE. The two methods, however, assess different genetic events in different parts of the chromosome. While mutation, recombination or replication error may affect the number and frequency of tandem repeats, variations in PFGE profiles are caused by mutations in restriction enzyme sites. Generally, isolates belonging to the same cluster or PFGE type were identified with the same MLVA type. It should be noted, however, that isolates used for PFGE analysis were chosen based on differences in MLVA profile, which might have introduced selection bias. We observed two isolates, including the type strain NCIMB14265T that were inseparable by MLVA, but were resolved by PFGE. The PFGE profiles of these isolates differed by only one band, consistent with a single genetic event such as a point mutation, insertion or deletion [15]. NCIMB14265T has been cultured repeatedly over time, which could explain the minor genetic changes.
Conclusions
We found the described MLVA scheme to meet all the performance criteria proposed for a good typing method [16]. It shows good stability, provided 100% typeability of the isolates, is reproducible and has a high discriminatory power. These performance qualities do not differ significantly from the previous F. noatunensis MLVA assay [11], however, the main advantage of our MLVA is the high throughput that is facilitated by using a single multiplex PCR and capillary electrophoresis. The epidemiological concordance was demonstrated by the ability of MLVA typing to correctly cluster isolates within an outbreak and separate these from epidemiologically unrelated isolates. Thus, the assay has a great potential as a high-resolution molecular typing tool for the study of outbreaks of cod francisellosis. By using allele string codes based on repeat copy numbers for strain identification, the assay can easily be standardised to facilitate exchange of data among laboratories. Compared to PFGE, it lacks subjectivity, is less time-consuming and more discriminatory. However, as MLVA and PFGE target different parts of the chromosome, combined analyses of these two methods may result in a more discriminatory approach to understanding the processes of transmission of this fish pathogen in aquaculture.
Methods
Bacterial strains and culture conditions
Ninety-one isolates of F. noatunensis subsp. noatunensis, including the type strain NCIMB14265T, from disease outbreaks in Norwegian cod farms during the period 2005 to 2011, were studied. For comparative purposes, single isolates derived originally from Atlantic salmon Salmo salar L. (F. noatunensis subsp. noatunensis PQ 1106) in Chile, three line-grunt Parapristipoma trilineatum Thunberg (F. noatunensis subsp. orientalis Ehime-1) in Japan, and from a captive wild Atlantic cod (F. noatunensis subsp. noatunensis) in Ireland were included in the analysis. The collection also included three reference strains of F. philomiragia (CCUG 12603, CCUG 13404 and CCUG 19701). Details of source of isolation and country of origin of the strains can be found in supplementary material Additional file 1: Figure S1. Stock cultures of bacterial strains were stored in cryo-broth with 20% (v/v) glycerol at -80°C. Individual colonies of each strain were revived by streaking on Cysteine Heart Agar with Blood (CHAB) and incubated at 22°C for 7 days. F. philomiragia was incubated under the same conditions for 3 days. The Irish isolate was kindly provided by Dr Neil Ruane, Marine Institute, Oranmore, Ireland. The Chilean PQ 1106 isolate and the two F. noatunensis subsp. orientalis strains were kindly provided by PHARMAQ AS, Norway.
Identification and selection of VNTR loci
Tandem repeats (TRs) were identified in the published complete genome sequence of F. philomiragia subsp. philomiragia (ATCC 25017) and three draft genome sequences of F. noatunensis subsp. noatunensis (NCIMB14265T, GM 2212T, PQ 1106) [17], using Tandem Repeats Finder (http://tandem.bu.edu/trf/trf.html) [18]. Initially, variability of 25 potential VNTR loci were tested in temporally and geographically discrete F. noatunensis subsp. noatunensis isolates using conventional PCR and gel-based analysis. Primers targeting flanking regions of each locus were designed using Primer3 software [19]. DNA was prepared by boiling bacterial cells in TZ lysis buffer [20] and used directly in PCR after brief centrifugation at 10,000 × g for 3 minutes. Alternatively, genomic DNA was purified using the QIAamp DNA mini kit (Qiagen) following the manufacturer’s instructions. PCR amplification reactions were carried out with a PTC- 100 Programmable Thermal Controller (MJ Research Inc. Watertown, Massachusetts, USA). Each 25-μl reaction mixture contained 1 × PCR buffer, 0.4 μM of each primer (Invitrogen), 0.2 mM dNTP mix, 1.5 mM MgCl2, 2U Taq DNA polymerase (GE Healthcare) and 2 μl of DNA as template. The thermal cycling conditions were as follows: Initial denaturation cycle at 95°C for 5 min, followed by 35 cycles of amplification at 95°C for 30 s, 53°C for 30 s and 72°C for 1 min and a final extension at 72°C for 4 min. A portion of the amplified PCR products (10 μl) were size-fractionated by electrophoresis on 3% agarose gel in 1 × TBE buffer at constant voltage of 70 V (3–4 V/cm) for 4 h and visualized after staining with ethidium bromide. As control, DNA from NCIMB14265T was included in each experiment. Loci resulting in polymorphic banding patterns were considered suitable for further analysis.
MLVA typing
Five VNTR loci were selected for MLVA and multiplexed in a single PCR reaction. Forward primers were labelled at the 5′ end with either VIC, NED or 6-carboxyfluorescein (6-FAM) fluorescent reporter dyes (Applied Biosystems). Reverse primers were unlabeled (Invitrogen). All primer sequences are shown in Table 2. PCRs were performed using the Qiagen multiplex PCR kit (Qiagen) in a 25 μl reaction following the manufacturer’s instructions. The optimum primer concentrations are shown in Table 2. The thermal cycling conditions were as follows: 15 min initial denaturation at 95°C, 30 cycles of amplification (30 s at 94°C, 90 s at 57°C and 90 s at 72°C), with a final extension of 30 min at 60°C. Arbitrarily chosen PCR products were verified by gel electrophoresis prior to fragment analysis using capillary electrophoresis. The PCR products were diluted 1:67 in deionized water and 1 μL of the diluted sample was added to 12 μL formamide and 1 μL of Geneflo-625 TAMRA internal size standard (CHIMERx, Madison, WI, USA). All samples were denaturated for 2 min at 94°C and cooled to room temperature before being subjected to capillary electrophoresis on a 3130xl Genetic Analyzer (Applied-Biosystems). Samples were run for 35 min at 60°C with an injection voltage of 15 kV for 5 s and a running voltage of 15 kV using POP-7 polymer (Applied-Biosystems). Each VNTR locus was identified by colour and size using GeneMapper software® 3.7 (Applied-Biosystems). Allele variation was identified by differences in fragment size. Fragment sizes were converted into repeat numbers based on the formula: number of repeats (bp) = fragment size (bp) − flanking regions (bp)/repeat size (bp). Flanking regions were deduced from the sequenced PCR products of several strains. Each isolate was assigned an MLVA type (MVT) based on a 5-digit allele string in the order fnVNTR-1, fnVNTR-2, fnVNTR-3, fnVNTR-4 and fnVNTR-5, reflecting the number of repeats at each locus. Absent PCR-products were designated an allele number of ‘0’. The allele strings were entered into BioNumerics software v6.1 (Applied Maths) as character values for clustering analysis.
Table 2.
Primers used for MLVA in this study
|
Marker |
Primer sequence
a
|
Primer concentration |
|
|---|---|---|---|
| Forward | Reverse | ||
| fnVNTR-1 |
V-CAGCTAGAAGCTTATTCGCCTCTT |
CAGGGTAATGCCTTAACGCATAT |
0.2 μM |
| fnVNTR-2 |
F-GAGTATTCCCTGCACCTACAATGAT |
TGTCATGCCTTTTTCTCTAGAGGAT |
0.2 μM |
| fnVNTR-3 |
N-CAAACTCTTTCACAAGAGGAAGCAT |
TCTGAACTCTGCTCTTTTCCCTCTA |
0.2 μM |
| fnVNTR-4 |
F-TTGTGCAAACACATCGATAGGAGAT |
AACTGCATCATCAGCATCTCTTCTA |
0.2 μM |
| fnVNTR-4b |
GTGCTTTTGCCTGTACCACCTT |
GCATCATTTGATACACCGTCCA |
NA |
| fnVNTR-5 |
N-CAATCACTCATCAACCACTAGCCAT |
TGCTGACTAATGGCTGACTGTAGTT |
0.3 μM |
| fnVNTR-5b | CTCATCAACCACTAGCCATCACAT | AAAGCCTGGGCTATCTAAATGCT | NA |
aFluorescent dyes are abbreviated as follows: F, FAM (6-carboxyfluorescein); N, NED; V, VIC. bsequencing primers; NA, not applicable.
Verification by DNA sequencing
To verify the accuracy of sizing determined by capillary electrophoresis, PCR fragments representing all variants of each tandem repeat were sequenced. For fnVNTR-4 and fnVNTR-5 new sequencing primers were designed (Table 2), while for the other loci the primers used to amplify the VNTRs were also used for sequencing. PCR products were purified using Nucleospin® Extract II (Macherey-Nagel, Germany) following the manufacturer’s instructions. Two μL of purified PCR product was added to 4 μL DYEnamic™ Dye Terminator Cycle Sequencing Kit (Amersham Biosciences), 1 μL sequencing primer (5 μM) and 3 μl sterile dH2O. Sequencing products were purified using the DYE Terminator Removal Kit (ABgene®) and sequenced on MegaBACE 1000 sequencing instrument (Amersham Biosciences). Consensus sequences were determined using Vector NTI (Invitrogen).
Stability of VNTR loci after in-vitro passage
The stability of each VNTR locus was determined in-vitro after a number of laboratory passages (40 passages at 5 days intervals) using four F. noatunensis subsp. noatunensis isolates showing distinct MLVA profiles. For each passage, single colonies were streaked on CHAB agar plates. Bacterial growth conditions, preparation of DNA, VNTR PCR amplification and MLVA were carried out on single colonies from passage 10, 20, 30 and 40 as described above.
PFGE typing
Prior to PFGE analysis, in silico searches for rare-cutting restriction enzymes were made from the available in-house Francisella genomes [17]. In all, five candidate endonucleases (PasI, SacII, ApaI, NaeI) were evaluated. Of these enzymes, PasI (Fermentas) was the most discriminatory on the preliminary panel of five F. noatunensis isolates tested (data not shown). PFGE using PasI was then performed on a subset of 54 isolates including members from each representative MLVA group, using the PulseNet protocol for subtyping of Francisella tularensis (CDC, Atlanta, GA, USA).
Bacterial cells were embedded in 1.2% agarose plugs (SeaKem Gold agarose; FMC Bioproduct, Rockland, ME), lysed, washed, and genomic DNA digested with 30 U PasI enzyme for at least 3 h at 37°C. Salmonella enterica serotype Braenderup (H9812) was used as a reference standard, and restricted with 50 U XbaI (Roche Diagnostics, Indianapolis, IN, USA) for 3 hours at 37°C. The digests were resolved by electrophoresis on the CHEF-DRIII apparatus (Bio-Rad Laboratories, Hercules, CA) with 1% agarose gel in 0.5× Tris-borate –EDTA (TBE) buffer and running conditions set at voltage, 6 V/cm; initial switch time, 5.0 s; final switch time 30 s; runtime 18 h at 14°C. Gels were stained with ethidium bromide (1 mg/mL) and images of individual DNA fingerprints were captured using the Bio-Rad Gel Doc system (Bio-Rad Laboratories).
Data analysis
Cluster analysis of MLVA and PFGE profiles was performed in BioNumerics 6.1 (Applied Maths, Saint-Martens-Latem, Belgium). Dendrograms were created using the categorical similarity coefficient or the Dice correlation coefficient coupled with WARD algorithm. A minimum spanning tree (MST) was created to illustrate the distribution and inter-relationships of the MLVA genotypes within the F. noatunensis subsp. noatunensis population (Figure 2). The discriminatory capacity of the MLVA assay was evaluated using the Simpson’s index of diversity [21], which was calculated for both individual and combined VNTR markers on a data subset containing single representatives of each genotype identified in individual diagnostic cases. The degree of linkage disequilibrium, as an indicator of the statistical independence of each of the five loci was tested using the LIAN Linkage analysis 3.5 Software as described by Haubold & Hudson [22] on a data subset representing single representatives of each genotype.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DS was a major contributor to project design, performed some of the experiments and responsible for writing of the manuscript. WX identified the VNTR loci in the draft genome, performed some of the experiments and contributed to writing of the manuscript. TNM performed and optimized the MLVA assay. LP, MK, SA identified the VNTR and restriction enzyme markers and contributed to completion of the manuscript. MF was involved in the interpretation of the data and contributed to the completion of the manuscript. CDJ conceived and coordinated the study, and contributed to completion of the manuscript. All authors read and approved the final manuscript.
Supplementary Material
Description of the Francisella isolates and strains used in the present study. The dendrogram was generated based on their MLVA genetic relatedness using categorical coefficient and Ward algorithm. nd = no data.
Contributor Information
Samuel Duodu, Email: samuel.duodu@vetinst.no.
Xihe Wan, Email: wanxihe@hotmail.com.
Nora Martinussen Tandstad, Email: nora_tandstad@hotmail.com.
Pär Larsson, Email: par.larsson@foi.se.
Kerstin Myrtennäs, Email: kerstin.myrtennas@foi.se.
Andreas Sjödin, Email: andreas.sjodin@foi.se.
Mats Forsman, Email: mats.forsman@foi.se.
Duncan J Colquhoun, Email: duncan.colquhoun@vetinst.no.
Acknowledgements
This work was supported financially by the Norwegian Research Council (Grant no. 185362), the Swedish Civil Contingencies Agency (Grant no. 08-H189) and the Swedish Ministry of Defence (Project A4042) to whom we express our gratitude. We thank Hilde Welde for technical assistance.
References
- Colquhoun DJ, Duodu S. Francisella infections in farmed and wild aquatic organisms. Vet Res. 2011;9:47. doi: 10.1186/1297-9716-42-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylund A, Ottem KF, Watanabe K, Karlsbakk E, Krossøy B. Francisella sp. (Family Francisellaceae) causing mortality in Norwegian cod (Gadus morhua) farming. Arch Microbiol. 2006;9:383–392. doi: 10.1007/s00203-006-0109-5. [DOI] [PubMed] [Google Scholar]
- Olsen AB, Mikalsen J, Rode M, Alfjorden A, Hoel E, Straum-Lie K, Haldorsen R, Colquhoun DJ. A novel systemic granulomatous inflammatory disease in farmed Atlantic cod Gadus morhua L., associated with a bacterium belonging to the genus Francisella. J Fish Dis. 2006;9:307–311. doi: 10.1111/j.1365-2761.2006.00714.x. [DOI] [PubMed] [Google Scholar]
- Mikalsen J, Olsen AB, Tengs T, Colquhoun DJ. Francisella philomiragia subsp. noatunensis subsp. nov., isolated from farmed Atlantic cod (Gadus morhua L.) Int J Syst Evol Microbiol. 2007;9:1960–1965. doi: 10.1099/ijs.0.64765-0. [DOI] [PubMed] [Google Scholar]
- Ottem KF, Nylund A, Karlsbakk E, Friis-Møller A, Kamaishi T. Elevation of Francisella philomiragia subsp. noatunensis Mikalsen et al. (2007) to Francisella noatunensis comb. nov. [syn. Francisella piscicida Ottem et al. (2008) syn. nov.] and characterization of Francisella noatunensis subsp. orientalis subsp. nov., two important fish pathogens. J Appl Microbiol. 2009;9:1231–1243. doi: 10.1111/j.1365-2672.2008.04092.x. [DOI] [PubMed] [Google Scholar]
- van Belkum A, Scherer S, Van Alphen L, Verbrugh H. Short-sequence repeats in prokaryotic genomes. Microbiol Mol Biol Rev. 1998;9:275–293. doi: 10.1128/mmbr.62.2.275-293.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belkum A. The role of short sequence repeats in epidemiologic typing. Curr Opin Microbiol. 1999;9:306–311. doi: 10.1016/S1369-5274(99)80053-8. [DOI] [PubMed] [Google Scholar]
- Farlow J, Smith KL, Wong J, Abrams M, Lytle M, Keim P. Francisella tularensis strain typing using multiple-locus, variable-number tandem repeat analysis. J Clin Microbiol. 2001;9:3186–3192. doi: 10.1128/JCM.39.9.3186-3192.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzgar D, Thomas E, Davis C, Field D, Wills C. The microsatellites of Escherichia coli: rapidly evolving repetitive DNAs in a non-pathogenic prokaryote. Mol Microbiol. 2001;9:183–190. doi: 10.1046/j.1365-2958.2001.02245.x. [DOI] [PubMed] [Google Scholar]
- Jenkins AO, Venter EH, Hutamo K, Godfroid J. Comparison of the capillary and agarose electrophoresis based multiple locus VNTR (variable number of tandem repeats) analysis (MLVA) on Mycobacterium bovis isolates. Vet Microbiol. 2010;9(1–2):172–176. doi: 10.1016/j.vetmic.2010.02.036. [DOI] [PubMed] [Google Scholar]
- Brevik ØJ, Ottem KF, Nylund A. Multiple-locus, variable number of tandem repeat analysis (MLVA) of the fish-pathogen Francisella noatunensis. BMC Vet Res. 2011;9:5. doi: 10.1186/1746-6148-7-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lista F, Faggioni G, Valjevac S, Ciammaruconi A, Vaissaire J, le Doujet C, Gorgé O, De Santis R, Carattoli A, Ciervo A, Fasanella A, Orsini F, D'Amelio R, Pourcel C, Cassone A, Vergnaud G. Genotyping of Bacillus anthracis strains based on automated capillary 25-loci multiple locus variable-number tandem repeats analysis. BMC Microbiol. 2006;9:33. doi: 10.1186/1471-2180-6-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualotto AC, Denning DW, Anderson MJ. A cautionary tale: lack of consistency in allele sizes between two laboratories for a published multilocus microsatellite typing system. J Clin Microbiol. 2007;9:522–528. doi: 10.1128/JCM.02136-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keim P, Price LB, Klevytska AM, Smith KL, Schupp JM, Okinaka R, Jackson PJ, Hugh-Jones ME. Multiple-locus variable-number tandem repeat analysis reveals genetic relationships within Bacillus anthracis. J Bacteriol. 2000;9:2928–2936. doi: 10.1128/JB.182.10.2928-2936.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenover FC, Arbeit RD, Goering RV, Mickelsen PA, Murray BE, Persing DH, Swaminathan B. Interpreting chromosomal DNA restriction patterns produced by pulsed-field gel electrophoresis: criteria for bacterial strain typing. J Clin Microbiol. 1995;9:2233–2239. doi: 10.1128/jcm.33.9.2233-2239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Belkum A, Tassios PT, Dijkshoorn L, Haeggman S, Cookson B, Fry NK, Fussing V, Green J, Feil E, Gerner-Smidt P, Brisse S, Struelens M. Guidelines for the validation and application of typing methods for use in bacterial epidemiology. Clin Microbiol. 2007;9:1–46. doi: 10.1111/j.1469-0691.2007.01786.x. Infect. [DOI] [PubMed] [Google Scholar]
- Sjodin A, Svensson K, Ohrman C, Ahlinder J, Lindgren P, Duodu S, Johansson A, Colquhoun DJ, Larsson P, Forsman M. Genome characterisation of the genus Francisella reveals insight into similar evolutionary paths in pathogens of mammals and fish. BMC Genomics. 2012;9:268. doi: 10.1186/1471-2164-13-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson G. Tandem repeats finder:a program to analyze DNA sequences. Nucleic Acids Res. 1999;9:573–580. doi: 10.1093/nar/27.2.573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen S, Skaletsky HJ. In: Bioinformatics Methods and Protocols: Methods in Molecular Biology. Krawetz S, Misener S, editor. Totowa, NJ: Humana Press; 2000. Primer3 on the WWW for general users and for biologist programmers; pp. 365–386. [DOI] [PubMed] [Google Scholar]
- Abolmaaty A, Vu C, Oliver J, Levin RE. Development of a new lysis solution for releasing genomic DNA from bacterial cells for DNA amplification by polymerase chain reaction. Microbios. 2000;9:181–189. [PubMed] [Google Scholar]
- Simpson EH. Measurement of diversity. Nature. 1949;9:688. doi: 10.1038/163688a0. [DOI] [Google Scholar]
- Haubold B, Hudson RR. LIAN 3.0: detecting linkage disequilibrium in multilocus data. Bioinformatics. 2000;9:847–848. doi: 10.1093/bioinformatics/16.9.847. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of the Francisella isolates and strains used in the present study. The dendrogram was generated based on their MLVA genetic relatedness using categorical coefficient and Ward algorithm. nd = no data.