

Abstract
A limitation to many polymer-based drug delivery systems is the ability to customize a particular polymer composition for tailoring drug release kinetics to a specific clinical application. In this study, we investigated the structure-function effects of conjugating various hydrophobic biocompatible side chains to poly(glycerol-co-caprolactone) copolymers with the goal of achieving prolonged and controlled release of a chemotherapeutic agent. The choice of side chain significantly affected the resulting polymer properties including thermal transitions, relative crystallinity (ΔHf), and hydrophobicity. Drug-loaded films cast from solutions of polymer and 10-hydroxycamptothecin demonstrated prolonged release from four to over seven weeks depending upon side chain structure without initial burst release behavior. Use of the stearic acid-conjugated poly(glycerol-co-caprolactone) films afforded substantial anti-cancer activity in vitro for at least 50 days when exposed to fresh cultures of A549 human lung cancer cells over 24-hour intervals, correlating well with the measured drug release kinetics.
Keywords: Hydrophobic polymer, controlled release, drug delivery, tunable release, film, chemotherapy
1. Introduction
A growing niche in polymeric-mediated drug delivery is focused on the localized delivery of anti-cancer agents to tumor cells.1-14 It is difficult to achieve both therapeutic dose and prolonged duration of chemotherapy at the disease site using conventional intravenous delivery for many first-line chemotherapeutic drugs due to poor aqueous solubility. Even with use of excipients to promote solubilization, the efficacy of intravenous chemotherapy treatments is significantly hindered by insufficient accumulation of drug at the tumor site as a consequence of dose-limiting toxicities and/or poor bioavailability at the location of disease. Localized treatment via controlled release polymer matrices (films, hydrogels, etc.) boasts a number of potential benefits over systemic delivery including: 1) the maintenance of therapeutic levels of drug through gradual and prolonged release at the site of disease; 2) ease of incorporation of hydrophobic drugs; 3) stabilization of water-sensitive drugs in their active form prior to release; 4) avoidance of bolus release kinetics felt to be detrimental during wound healing; 5) elimination of periodic and costly intravenous drug administration, and 6) reduced side effects and systemic toxicity.8,9 The number of polymer-based delivery systems that satisfy all of these requirements has been limited in both scope and efficacy in part due to challenges with achieving optimal drug release kinetics, biocompatibility, manufacturability, and/or implant degradation kinetics. As a result, effective control of local tumor is challenging with a polymer device alone and, clinically, a device may be used along with radiation therapy (e.g., Gliadel Wafer). In addition, optimal release kinetics depend on the tissue type, location, and pathology of the disease. Thus, the development of polymeric matrices with tunable and prolonged release of chemotherapeutics is critical towards optimizing local polymer implants for specific cancer applications.
One approach towards achieving controlled drug release kinetics is by utilization of polymers featuring modifiable functional side chains. These polymers have been based on naturally-occurring molecules including chitosan15,16, glycerol17-24, and various amino acids25-28, as well as synthetic polymers such as poly(lactic acid)29,30, poly(ε-caprolactone)31-33, or copolymers thereof. Functionalization of these side chains has traditionally been used for the covalent attachment of therapeutic moieties such as anti-inflammatory34, chemotherapeutic35, and anti-fungal drug molecules36. The subsequent drug release is dictated by an environmental trigger (i.e. temperature, pH) or by a cleavage of the linkage via exposure to esterases/proteases or hydrolysis. Since many clinical applications require available drug concentrations to remain high over an extended therapeutic window to maximize pharmacologic efficacy, there is a recognized need for drug delivery depots with non-burst prolonged release characteristics. For example, in cancer treatment, tumor cell susceptibility is frequently cell-cycle dependant since many chemotherapy drugs specifically target actively dividing cells. It is therefore imperative that the duration of drug exposure extend over multiple cell cycles, to ensure that no tumor cells escape the therapeutic window of treatment. This is the case for two ubiquitous chemotherapy agents, paclitaxel and camptothecin, whose mechanisms of action involve microtubule polymerization and DNA topoisomerase inhibition, respectively37,38. With the goal of extending chemotherapy exposure, we are exploring the variation in side chain composition of functionalizable polymers as this presents the opportunity to modify key structurally-influenced properties such as thermal transitions, crystallinity, and hydrophobicity which affect the release kinetics of an entrapped drug without altering the chemical structure of the drug.
We have previously demonstrated sustained chemotherapy release from novel stearic acid-functionalized poly(glycerol-co-caprolactone) films prevents local tumor recurrence in an in vivo murine model of lung cancer recurrence, as compared to an equivalent dose of drug given as a bolus injection locally or intraperitoneally.13 Functionalization of our polymer with a lipophilic side chain significantly prolonged and tempered the drug release kinetics from solvent-cast polymer films as compared to the unmodified polymer39. This substantial effect resulting from the conjugation of a hydrophobic side chain prompted us to explore in more detail the effects of side chain selection on important polymer properties that may affect controlled release kinetics. Herein, we report the synthesis and characterization of a series of poly(glycerol-co-caprolactone) copolymer analogues that have been functionalized with an array of biocompatible hydrophobic side chains, their corresponding release kinetics of a model hydrophobic chemotherapeutic agent from drug-loaded films, and in vitro evaluation of anti-proliferative efficacy against a human non-small cell lung cancer cell line over an extended time period.
2. Materials and Methods
2.1. General Procedures
All solvents were dried and freshly distilled prior to use. All other reagents were used as received, without further purification. Lauric acid, myristic acid, palmitic acid and stearic acid were purchased from Sigma Aldrich. Poly(glycerol-co-caprolactone) was prepared as previously described18. All reactions were performed under nitrogen atmosphere unless otherwise noted. 1H NMR spectra were recorded on a Varian INOVA spectrometer (400 MHz). Chemical shifts were referenced to residual solvent peaks (CHCl3 peak at 7.26 ppm). Thermal data was collected using a TA Instruments Q100 differential scanning calorimeter (ramp 10 °C/min to 80 °C, cool 5 °C/min to −70 °C, ramp 10 °C/min to 80 °C); thermal transitions were measured on the second heating cycle. Static contact angles were recorded using a Kruss DSA100 Contact angle goniometer and determined using a polynomial fitting function. DCM = dichloromethane, Chol = 5-androsten-3β-ol-17β-carboxylic acid, PGC = poly(glycerol-co-ε-caprolactone), PCL = poly(ε-caprolactone), PBS = phosphate buffered solution.
2.2. Functionalization of poly(glycerol-co-ε-caprolactone)
Functionalized analogues were synthesized via the attachment of hydrophobic side chains to the free pendant hydroxyl groups on the glycerol units. Specifically, alkyl chains of varying length (C12, C14, C16, C18) were reacted to the copolymer backbone. A representative synthetic procedure is as follows. Poly(glycerol-co-ε-caprolactone) (500 mg, 0.875 mmol), lauric acid (193 mg, 0.962 mmol), and dicyclocarbodiimide (216 mg, 1.05 mmol) were dissolved in distilled dichloromethane. The solution was stirred at room temperature for 18 h. The dicyclohexylurea was filtered and the solvent evaporated. The product was precipitated in cold methanol, the solvent decanted, and the polymer dried under reduced pressure to yield a white solid (90 % yield). Addition of the lauric acid side chain was determined by the presence of the methylene group at the end of the alkyl chain, with a corresponding peak in the 1H NMR spectrum at 0.82. (m, 3H, CH3).
2.3. Preparation of drug-loaded films
Drug-loaded films were prepared as previously reported39. Films were cast from polymer solutions onto glass cover-slips using a microsyringe. First, 10-hydroxycamptothecin (HCPT) was added to CH2Cl2 (0.1 % w/v) in a glass vial and homogenized for 60 min in a sonication bath to break apart aggregates. Polymer (5% w/v) was then dissolved into the solution by vortexing for 1 min. The solution was slowly added to the glass surface and left to evaporate for 24 h, followed by further drying under reduced pressure for another 24 h. The film casting method was repeatable and resulted in very little deviation in thicknesses within and between samples. All the solid samples were the same thickness (40 ± 1 μm). Thicker or thinner samples were discarded.
2.4. In vitro drug release studies
The release kinetics from HCPT-loaded films were assessed in PBS buffer at 37 °C. Specifically, films comprised of 5 mg polymer and 100 μg HCPT were cast onto glass cover-slips and submerged in 150 mL of PBS. At specific time points, an aliquot of release media was removed and the concentration of HCPT was measured. An HP 1090 HPLC system was used with a fluorescence detector (λex = 380 nm, λem = 540 nm), with a Phenomenex Prodigy 5 ODS reverse-phase column (150 × 4.6 mm, 5 μm). The mobile phase was composed of 25% acetonitrile and 75% ammonium acetate buffer adjusted to a pH of 6.4 and delivered at 0.8 mL/min. Calibration curves were constructed for both lactone (rt = 3.7 min) and carboxylate (rt = 2.8 min) forms with sensitivities of 0.5 ng/mL and 2 ng/mL respectively. Total drug release was calculated by adding the contributions of both drug forms at a given time point. After the completion of release, the HCPT remaining in each film was quantified. Films were dissolved in CH2Cl2 to release encapsulated drug, the solvent evaporated, and PBS (150 mL) was added under rigorous stirring. The fluorescence spectrum was recorded and remaining drug measured.
2.5. Anti-proliferative studies with a human cancer cell line
Long-term in vitro efficacy was evaluated using a human non-small cell lung cancer cell line (A549). Cells were cultured with Dulbecco's Modified Eagle Medium containing 10% fetal bovine serum and 1% penicillin/streptomycin (100 U/100 lg/mL). Cells were plated at a density of 30,000 cells/well in 12-well Transwell plates (Corning Inc.) and incubated overnight. Films were placed on individual tissue culture treated polyester membrane inserts (0.4 μm pore size) and then exposed to cultured cells for 24 hours. Following co-incubation, the films were transferred to holding wells for four days containing fresh phosphate buffered saline to maintain sink conditions, before their next 24 hour cell exposure. Treated cells were incubated for an additional 4 days and cell viability was quantified using a standard MTS cell proliferation assay. To assess continued drug release and cytotoxicity, this procedure was repeated every 5 days in the setting of new media and freshly plated tumor cells.
3. Results
Synthesis and Characterization
The polymer was synthesized with glycerol units within the structure since glycerol-based polymers have been successfully used for several biomedical applications as well as the structure itself can provide a site for further functionalization (e.g., 1° OH or 2° OH).18,19,21,24,40-44 Specifically, the pendant secondary-hydroxyl groups of poly(glycerol-co-caprolactone) were modified with hydrophobic side chains in order to evaluate the structure-property relationship of drug release kinetics from poly(glycerol-co-caprolactone) polymer matrices (Figure 1). It was previously demonstrated that if the hydroxyl of the glycerol unit in the polymer is not free but coupled to a group (e.g., benzyl alcohol), the poly(glycerol-co-caprolactone) copolymer backbone does not appreciably degrade over at least six months upon exposure to phosphate buffered solution at 37°C18. Thus, the drug release kinetics from functionalized polymer cast films were expected to be diffusion controlled. Characterization of the various film analogues was performed to identify trends affecting the release kinetics of a drug from the polymer matrix, including thermal transitions, relative crystallinity, and contact angle.
Figure 1.

Synthetic scheme and structures for hydrophobic poly(glycerol-co-ε-caprolactone) analogue copolymers. a. BnBr, KOH, THF, 0°C, b. 1M HCl, MeOH, 80°C, c. EtOCOCl, TEA, THF, 0°C, d. ε-caprolactone, Sn(Oct)2, 140°C, e. Pd(OH)2/C, H2, f. R-H, DCM/DMF, DCC.
Poly(glycerol-co-caprolactone) was synthesized as described previously and shown in Figure 118. Briefly, 5-benzyloxy-1,3-dioxan-2-one was copolymerized with ε-caprolactone by a ring-opening polymerization using tin(II) 2-ethylhexanoate as the catalyst. The resulting copolymer was comprised of 20 mol % glycerol subunits; this ratio of glycerol to caprolactone has been demonstrated to yield the best combination of thermal/mechanical properties while maximizing the number of available hydroxyl groups for further modification. The benzylprotected secondary hydroxyl groups were removed using Pd catalyzed hydrogenolysis. Functionalized analogues of poly(glycerol-co-caprolactone) (or PGC copolymers) were prepared via the attachment of hydrophobic side chains, including alkyl chains of various length (lauric, C12; myristic, C14; palmitic, C16; or stearic acid, C18) to the copolymer backbone (Fig. 1). These lipophilic groups were selected because they are natural metabolites and were hypothesized to increase the hydrophobicity of the resultant copolymers, thus increasing the barrier to diffusion into and out of the polymer matrix upon exposure to an aqueous environment.
The extent of pendant hydroxyl conjugation of poly(glycerol-co-caprolactone) with the various side chains was verified by proton NMR by comparing integrations between the added side chain structure and the caprolactone units (Fig. 2). Additionally, the corresponding proton peak to the α-carbon of the caproic acid unit is distinctly shifted downfield when the hydroxyl group on the glycerol unit is not reacted, giving a convenient secondary method of evaluating extent of modification. All of the structures conjugated to PGC-OH resulted in complete conversion.
Figure 2.
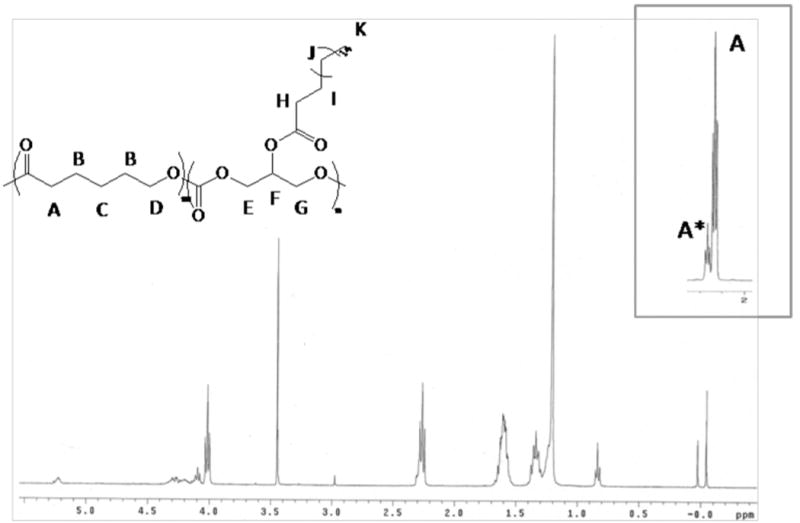
1H NMR of a representative functionalized copolymer, poly(glycerol stearate-co-ε-caprolactone). The extent of side chain modification is observed by monitoring the shift and disappearance of A* as the pendant hydroxy groups of the copolymer are conjugated.
Changes in thermal properties were observed relative to the choice of side chain structure for the polymers (Fig. 3). Analogues with linear side chains had higher heat of fusions (ΔHf), a measure of relative crystallinity between similar polymers than the unmodified polymer. The addition of larger side chains resulted in lowered melting transitions such as for the stearic acid derivative of PGC, PGC-C18 (31 °C). Polymers modified with lauric, myristic, or palmitic acid had significantly higher melting points (40 – 41 °C) compared to PGC-OH (23 °C). One possible explanation for the lower melting transition for the C18 over the C12 analog is that the longer alkyl side chain are more predisposed towards integrating in to the semi-crystalline domains of the copolymer due to their increased accessibility, hence increasing the number of defects in those regions and lowering the melting point of the polymer. It should be emphasized that PGC-OH and PGC-C18, had melting temperatures below 37 °C, thus would lack crystallinity in vivo and are better described as ‘polymer melts’.
Figure 3.

Melting temperature (Tm) and heat of fusion (ΔHf) for the copolymer analogues. All glass transition temperatures were below -40°C (data not shown).
The hydrophobicity of solvent cast polymer films was assessed by static contact angle measurements (Fig. 4). Increasing the chain length for the linear fatty acid analogues increased the contact angle from 82° for the unmodified PGC-OH to 98° with the 12 carbon side chain PGC-C12, and up to 124° for the 18 carbon side chain PGC-C18. For comparison, DuPont's Teflon-AF™ has a reported contact angle of 104°.
Figure 4.
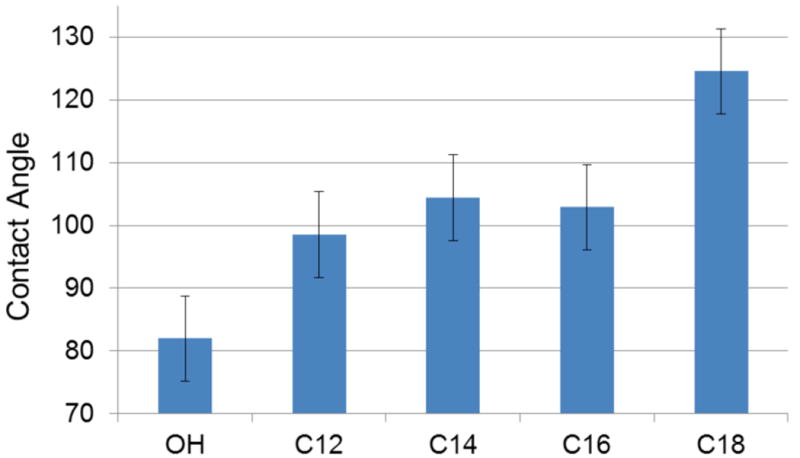
Static contact angle measurements from polymer cast films (n=3).
Release kinetics of 10-hydroxycamptothecin from polymer films
Unmodified poly(glycerol-co-caprolactone) films (PGC-OH) demonstrated unfavorable HCPT release kinetics in vitro for our interests, characterized by a burst release over the first 48 hours amounting to 46% of the total drug released (Fig. 5). Furthermore, by the end of release at four weeks the PGC-OH films still retained 49% of their initial loading. In addition, the unmodified polymer fragmented or flaked apart over the first week of release and did not retain its original form. Conjugation with hydrophobic moieties was expected to temper and prolong the release kinetics via inhibition of aqueous penetration and result in a more stable cast film. Films formed from the four linear fatty acid chains including lauric (PGC-C12), myristic (PGC-14), palmitic (PGC-16), and stearic (PGC-C18) showed decreasing trends of release rate and final cumulative release with increasing alkyl chain length. These chains were anticipated to increase hydrophobicity while minimizing the disruptive effects of bulky steric hindrance on polymer chain packing. All of these films maintained their shape and eluted HCPT for a more prolonged duration than the unmodified polymer, with continued release for at least 7 weeks. The PGC-C12 films released 68% of their total loading, at rate of about 2.2%/day through day 21 diminishing thereafter, while the PGC-C18 films eluted only 41% of their initial loading over the same time 21 day interval with a release rate of only 1.05% per day. All of the analogue films maintained their original form throughout the course of the experiment.
Figure 5.
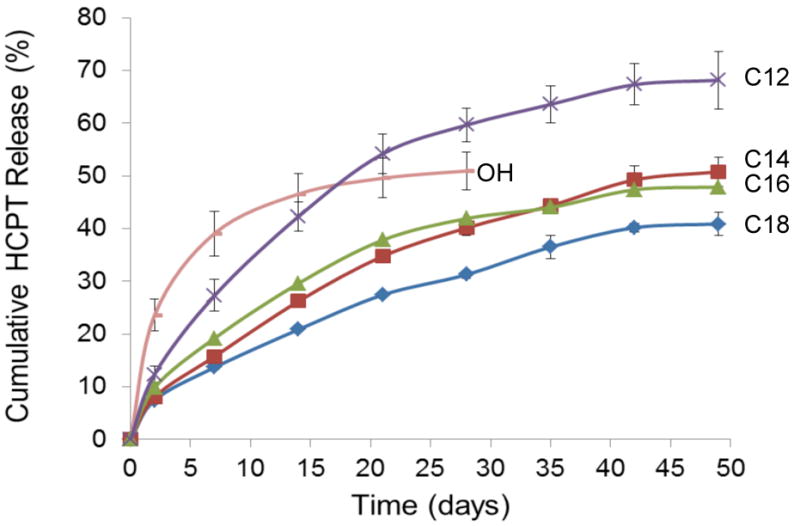
Cumulative release of 10-hydroxycamptothecin from drug-loaded films in PBS at 37°C (n = 3).
Long-term anti-proliferative efficacy of HCPT-loaded films
After characterizing the various polymer analogues, the anticancer activity of HCPT-loaded PGC-OH and PGC-C18 films (2% w/w) was evaluated in vitro against cultured human non-small cell lung cancer cells (A549) over successive 5-day intervals for a total of 50 days (Fig. 6). The stearic acid-modified copolymer (PGC-C18) was chosen for its ease of synthesis, prolonged release kinetics, and lack of a significant initial burst. The films were co-incubated with cells for 24 hour exposures, and then transported to holding wells under sink conditions between each cell exposure. Freshly cultured cells were utilized for each cell exposure. In this manner, the effective longevity of the films' ability to inhibit cancer cell proliferation was determined. The initial HCPT loading was chosen to ensure that the concentration of hydroxycamptothecin would exceed the IC50 of the cells by the end of each 5-day exposure interval.
Figure 6.
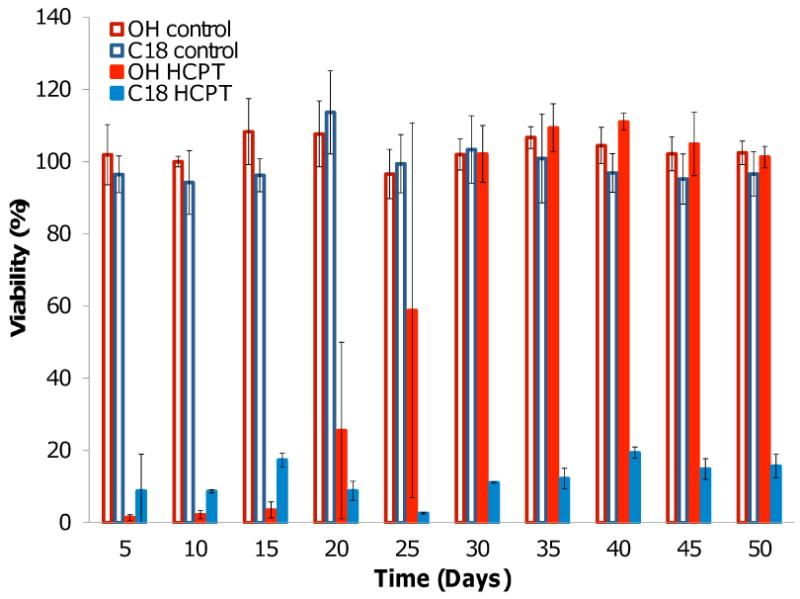
Anti-proliferative efficacy of HCPT-loaded and unloaded PCG-OH and PCG-C18 films exposed to A549 human non-small cell lung cancer cells over 24 hour intervals (n=3).
A direct correlation was observed between the measured release kinetics and duration of anti-proliferative efficacy for the HCPT-loaded PGC-OH and PGC-C18 films. The potency of the HCPT-loaded PGC-OH diminished following 15 days of cumulative release until becoming ineffective by the 30 day time point. This was to be expected, as those films were not shown to elute significant amounts of drug after 30 days of release. Conversely, the PGC-C18 films exhibited significant cytotoxicity for each exposure during the 50 day experiment, demonstrating that the films are capable of releasing a sustained dose of therapeutic drug for at least 7 weeks and that HCPT retains its anti-proliferative activity while embedded in the polymer and following release from the film. Unloaded PGC-OH and PGC-C18 films were not cytotoxic for the duration of the experiment.
4. Discussion
The prolonged release of chemotherapy at the site of tumor cells is desired in order to maintain a localized therapeutic concentration of drug over multiple cell cycles to maximize tumor cytotoxicity. 10-Hydroxycamptothecin was selected as a representative chemotherapeutic agent because of its demonstrated strong anti-tumor activity in vitro and in vivo. Unfortunately, its practical use has been limited by poor aqueous solubility, instability in aqueous solution, and dose-limiting systemic toxicities observed during clinical trials45. The entrapment of HCPT in the copolymer matrices was expected to circumvent the solubility issue, reduce aqueous exposure to the drug, and potentially diminish or eliminate systemic side effects in vivo by locally delivering the therapeutic agent directly to the site of tumor.
While several trends were evident from the release data in Figure 5, it is clear that release is affected by a combination of properties. It is known that the presence of crystallinity can decrease release rates by increasing the packing and density of the polymer. For example, Pitt et. al. showed that increasing percent crystallinity of poly(ε-caprolactone) polymers from 45% up to 59% by various annealing methods led to as much as a 3-fold decrease in the drug release rate of Norgestrel from poly(ε-caprolactone) films46. They proposed that increasing crystallinity decreases polymer permeability emphasizing diffusion as the rate-limiting process of drug release. For the current study, however, the relative percent crystallinity was not predictive of drug release kinetics. The polymers conjugated with the lauric (PGC-C12), myristic (PGC-C14), and palmitic acid (PGC-C16) side chains had the highest relative crystallinity and the films cast from these analogues all released drug continuously for about 50 days. PGC-C18 had the slowest overall release rate and the lowest total amount of drug release over the 50 days. With a melting temperature below 37°C, this data suggests crystallinity was not the most important factor in predicting release for these polymers.
Slower release rates have been attributed to hydrophobicity by diminishing aqueous diffusion into the matrix and by increasing polymer-drug interactions when the encapsulated agent is hydrophobic. Hydrophobicity was a good predictor of relative release rates in this study; films with higher contact angles generally had slower release rates. Several groups have investigated fatty acid alkyl chains for the side group functionalization of macromolecules to optimize diffusion/degradation properties resulting in the observation of several trends. Increased alkyl chain length functionalization and percent of substitution to chitosan matrices led to significantly slower degradation, erosion, and release rates.15,16 Acyl modification of a polymer has been shown to increase drug encapsulation efficiency for drug-loaded nanoparticles, indicating enhanced polymer-drug interaction, but lower encapsulation efficiencies for high levels of substitution suggested that free volume also affects loading.19 In addition to hydrophobicity surface measurements, it is worth noting the partition coefficients of the various side chain molecules in understanding the microenvironment the drug resides within the polymer. The partition coefficients normalized to their testing environment (pH = 7.4, 0.16M phosphate buffer; P.C./10pH-pK) for laurate, myristate, palmitate, and oleate are reported as 4.70, 86.3, 489, and 2340, respectively47. Thus the chemical environment present in PGC-C18 may favor the partitioning of hydrophobic drug more so than PGC-C12, for example. These studies, along with the results reported here, demonstrate that substantial changes in drug release profile can be obtained by tuning the hydrophobicity.
Traditionally, there are a limited number of options for modifying the drug release kinetics of a particular polymer system, including varying the monomer ratio within a copolymer or by introducing an additive into the polymer matrix for a given system. The use of functionalizable polymers such as poly(glycerol-co-caprolactone) provides another variable for affecting release rates without directly modifying the polymer backbone composition. We have demonstrated in a previous study that the poly(glycerol-co-caprolactone) hydroxyl groups can also be conjugated with short amine, carboxyl, and hydroxyl-bearing linkages for further reaction with various functionalities leaving the number of possible side chain additions, and polymer properties to suit a wide variety of applications18. In the current study, the various analogues were prepared from the same batch of unmodified poly(glycerol-co-caprolactone), resulting in initial release rates ranging from 2.5 - 11.8 % release over the first two days with continued release from 28 days to more than 7 weeks depending on the conjugated side chain structure. Such increase in release duration directly translated into prolonged in vitro anti-cancer efficacy.
4. Conclusion
A series of poly(glycerol-co-caprolactone) copolymers was synthesized with backbones conjugated to various hydrophobic biocompatible side chains for evaluation as polymer matrices for controlled release of a chemotherapeutic agent. The choice of side chain significantly affected the resulting polymer properties including thermal transitions, relative crystallinity (ΔHf), and hydrophobicity. Drug-loaded films cast from solutions of polymer and 10-hydroxycamptothecin demonstrated prolonged release from four to over seven weeks depending upon side chain structure without initial burst release behavior. The stearic acid-conjugated poly(glycerol-co-caprolactone) films (PGC-C18), chosen as a representative copolymer for its ease of synthesis and slow release kinetics, resulted in substantial anti-cancer activity in vitro for at least 50 days when exposed for 24 hours to fresh cultures of A549 human lung cancer cells. By expanding the repertoire of polymer structures available for controlled release applications, clinical treatment of recurrent or established tumors can be customized (i.e., we can pay more consideration to the nuances of each patient's disease state). For example, one can envision a set of polymeric drug delivery implants exhibiting slow, moderate, or fast release kinetics depending on the location, extent, type, and genetic predisposition of an individual's disease. Through judicious selection of polymer side chain structure, the drug release rate and duration can be better optimized to maximize drug delivery directly to tumor sites as the next step in personalized cancer care.
Acknowledgments
This work was supported in part by BU, BWH, CIMIT, NIH R01CA149561, and the Boston University's Nanomedicine Program and Cross-Disciplinary Training in Nanotechnology for Cancer, NIH R25 CA153955.
References
- 1.Wolinsky JB, Colson YL, Grinstaff MW. J Control Release. 2012 doi: 10.1016/j.jconrel.2011.11.031. on line. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Souza R, Zahedi P, Allen CJ, Piquette-Miller M. Drug Delivery. 2010;17:365–375. doi: 10.3109/10717541003762854. [DOI] [PubMed] [Google Scholar]
- 3.Kim S, Kim JH, Jeon O, Kwon IC, Park K. Eur J Pharm Biopharm. 2009;71:420–430. doi: 10.1016/j.ejpb.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu P, Grainger DW. Biomaterials. 2006;27:2450–2467. doi: 10.1016/j.biomaterials.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 5.Guse C, Koennings S, Blunk T, Siepmann J, Goepferich A. Int J Pharmaceutics. 2006;314:161–169. doi: 10.1016/j.ijpharm.2005.12.051. [DOI] [PubMed] [Google Scholar]
- 6.Langer R, Tirrell DA. Nature. 2004;428:487–92. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 7.Saltzman WM, Fung LK. Adv Drug Deliv Rev. 1997;26:209–230. doi: 10.1016/s0169-409x(97)00036-7. [DOI] [PubMed] [Google Scholar]
- 8.Langer R. Science. 1990;249:1527–1533. doi: 10.1126/science.2218494. [DOI] [PubMed] [Google Scholar]
- 9.Langer R, Peppas NA. Biomaterials. 1981;2:201–214. doi: 10.1016/0142-9612(81)90059-4. [DOI] [PubMed] [Google Scholar]
- 10.Manabe T, Okino H, Maeyama R, Mizumoto K, Tanaka M, Matsuda T. J Biomed Mater Res B Appl Biomater. 2005;73:203–7. doi: 10.1002/jbm.b.30186. [DOI] [PubMed] [Google Scholar]
- 11.Azab AK, Kleinstern J, Doviner V, Orkin B, Srebnik M, Nissan A, Rubinstein A. J Control Release. 2007;123:116–22. doi: 10.1016/j.jconrel.2007.07.014. [DOI] [PubMed] [Google Scholar]
- 12.Brem H, Mahaley MS, Jr, Vick NA, Black KL, Schold SC, Jr, Burger PC, Friedman AH, Ciric IS, Eller TW, Cozzens JW, Kenealy JN. J Neurosurg. 1991;74:441–6. doi: 10.3171/jns.1991.74.3.0441. [DOI] [PubMed] [Google Scholar]
- 13.Liu R, Wolinsky JB, Walpole J, Southard E, Chirieac LR, Grinstaff MW, Colson YL. Ann Surg Oncol. 2010;17:1203–1213. doi: 10.1245/s10434-009-0856-z. [DOI] [PubMed] [Google Scholar]
- 14.Liu R, Wolinsky JB, Catalano PJ, Chirieac RL, Wagner AJ, Grinstaff MW, Colson YL, Raut CP. Ann Surg Oncol. 2011 doi: 10.1245/s10434-011-1871-4. [DOI] [PubMed] [Google Scholar]
- 15.Martin L, Wilson CG, Koosha F, Tetley L, Gray AI, Senel S, Uchegbu IF. J Control Release. 2002;80:87–100. doi: 10.1016/s0168-3659(02)00005-6. [DOI] [PubMed] [Google Scholar]
- 16.Le Tien C, Lacroix M, Ispas-Szabo P, Mateescu MA. J Control Release. 2003;93:1–13. doi: 10.1016/s0168-3659(03)00327-4. [DOI] [PubMed] [Google Scholar]
- 17.Ray WC, G MW. Macromolecules. 2003;36:3557–3562. [Google Scholar]
- 18.Wolinsky JB, Ray WC, III, Colson YL, Grinstaff MW. Macromolecules. 2007;40:7065–7068. [Google Scholar]
- 19.Kallinteri P, Higgins S, Hutcheon GA, St Pourcain CB, Garnett MC. Biomacromolecules. 2005;6:1885–94. doi: 10.1021/bm049200j. [DOI] [PubMed] [Google Scholar]
- 20.Carnahan MA, Grinstaff MW. J Am Chem Soc. 2001;123:2905–2906. doi: 10.1021/ja005726+. [DOI] [PubMed] [Google Scholar]
- 21.Weiser JR, Zawaneh PN, Putnam D. Biomacromolecules. 2011;12:977–986. doi: 10.1021/bm101342p. [DOI] [PubMed] [Google Scholar]
- 22.Wilms D, Stiriba SE, Frey H. Acc Chem Res. 2010;43:129–141. doi: 10.1021/ar900158p. [DOI] [PubMed] [Google Scholar]
- 23.Calderón M, Quadir MA, Sharma SK, Haag R. Adv Mater. 2010;22:190–218. doi: 10.1002/adma.200902144. [DOI] [PubMed] [Google Scholar]
- 24.Steinhilber D, Seiffert S, Heyman JA, Paulus F, Weitz DA, Haag R. Biomaterials. 2011;32:1311–1316. doi: 10.1016/j.biomaterials.2010.10.010. [DOI] [PubMed] [Google Scholar]
- 25.Barrera D, A Z, E L, P T, L R. J Am Chem Soc. 1993;115:11010–11011. [Google Scholar]
- 26.Zhou QX, Kohn J. Macromolecules. 1990;23:3399–3406. [Google Scholar]
- 27.Kumar R, Gao W, Gross RA. Macromolecules. 2002;35:6835–6844. [Google Scholar]
- 28.Shen Y, Chen X, Gross RA. Macromolecules. 1999;32:3891–3897. [Google Scholar]
- 29.Wang LA, Ha XH, Yuan Z. Polymer. 2006;47:6978–6985. [Google Scholar]
- 30.He B, Poon YF, Feng J, Chan-Park MB. J Biomed Mater Res A. 2008 [Google Scholar]
- 31.Tian D, Dubois P, Grandfils C, Jérôme R. Macromolecules. 1997;30:406–409. [Google Scholar]
- 32.Mahmud A, Xiong XB, Lavasanifar A. Macromolecules. 2006;39:9419–9428. [Google Scholar]
- 33.Trollsas M, Lee V, Mecerreyes D, Lowenhielm P, Moller M, Miller R, Hedrick J. Abstracts of Papers of the American Chemical Society. 2000;219:U371–U371. [Google Scholar]
- 34.Liu XM, Quan LD, Tian J, Laquer FC, Ciborowski P, Wang D. Biomacromolecules. 2010;11:2621–2628. doi: 10.1021/bm100578c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai PS, Lou PJ, Peng CL, Pai CL, Yen WN, Huang MY, Young TH, Shieh MJ. J Control Release. 2007;122:39–46. doi: 10.1016/j.jconrel.2007.06.012. [DOI] [PubMed] [Google Scholar]
- 36.Sedlak M, Pravda M, Kubicova L, Mikulcikova P, Ventura K. Bioorg Med Chem Lett. 2007;17:2554–7. doi: 10.1016/j.bmcl.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 37.Liu LF, Desai SD, Li TK, Mao Y, Sun M, Sim SP. Ann N Y Acad Sci. 2000;922:1–10. doi: 10.1111/j.1749-6632.2000.tb07020.x. [DOI] [PubMed] [Google Scholar]
- 38.Foa R, Norton L, Seidman AD. Int J Clin Lab Res. 1994;24:6–14. doi: 10.1007/BF02592403. [DOI] [PubMed] [Google Scholar]
- 39.Wolinsky JB, Liu R, Walpole J, Chirieac LR, Colson YL, Grinstaff MW. J Control Release. 2010;144:280–7. doi: 10.1016/j.jconrel.2010.02.022. [DOI] [PubMed] [Google Scholar]
- 40.Weinhart M, Gröger D, Enders S, Dernedde J, R H. Biomacromolecules. 2011;12:2502–2511. doi: 10.1021/bm200250f. [DOI] [PubMed] [Google Scholar]
- 41.Zawaneh PN, Singh SP, Padera RF, Henderson PW, Spector JA, Putnam D. Proc Natl Acad Sci U S A. 2010;107:11014–11019. doi: 10.1073/pnas.0811529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Calderón M, Welker P, Licha K, Fichtner I, Graeser R, Haag R, Kratz F. J Control Release. 2011;151:295–301. doi: 10.1016/j.jconrel.2011.01.017. [DOI] [PubMed] [Google Scholar]
- 43.Zelikin AN, Zawaneh PN, Putnam D. Biomacromolecules. 2006;7:3239–44. doi: 10.1021/bm060544e. [DOI] [PubMed] [Google Scholar]
- 44.Stiriba SE, Frey H, Haag R. Angew Chem Int Ed Engl. 2002;41:1329–1334. doi: 10.1002/1521-3773(20020415)41:8<1329::aid-anie1329>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 45.Ping YH, Lee HC, Lee JY, Wu PH, Ho LK, Chi CW, Lu MF, Wang JJ. Oncol Rep. 2006;15:1273–9. [PubMed] [Google Scholar]
- 46.Pitt CG, Gratzl MM, Jeffcoat AR, Zweidinger R, Schindler A. J Pharm Sci. 1979;68:1534–8. doi: 10.1002/jps.2600681219. [DOI] [PubMed] [Google Scholar]
- 47.Simpson RB, Ashbrook JD, Santos EC, Spector AA. J Lipid Res. 1974;15:415–422. [PubMed] [Google Scholar]