

Summary
Crizotinib, an inhibitor of anaplastic lymphoma kinase (ALK), has also recently shown efficacy in the treatment of lung cancers with ROS1 translocations. Resistance to crizotinib developed in a patient with metastatic lung adenocarcinoma harboring a CD74–ROS1 rearrangement who had initially shown a dramatic response to treatment. We performed a biopsy of a resistant tumor and identified an acquired mutation leading to a glycine-to-arginine substitution at codon 2032 in the ROS1 kinase domain. Although this mutation does not lie at the gatekeeper residue, it confers resistance to ROS1 kinase inhibition through steric interference with drug binding. The same resistance mutation was observed at all the meta-static sites that were examined at autopsy, suggesting that this mutation was an early event in the clonal evolution of resistance. (Funded by Pfizer and others; ClinicalTrials.gov number, NCT00585195.)
Since the pivotal discovery of imatinib as a safe and effective drug for the treatment of chronic myeloid leukemia,1 kinase inhibitors have emerged as powerful agents in the treatment of several cancers. The identification of activating mutations within the kinase domain of the epidermal growth factor receptor (EGFR)2,3 has led to the widespread use of kinase inhibitors in this genetically defined subset of lung cancers.4 More recently, chromosomal translocations that create fusion proteins involving the tyrosine kinase domains of ALK5 or ROS16 have been the subject of intense investigation in lung adenocarcinoma because of the availability of clinically active inhibitors of these kinases. Although kinase inhibitors can be highly effective, most cancers invariably acquire resistance to these targeted therapies, an effect that represents a major limitation to their long-term clinical benefit. By identifying the ways in which cancers evolve to evade kinase inhibitors, researchers have been able to develop new therapeutic strategies to overcome resistance in several cancers.7,8
The multitargeted tyrosine kinase inhibitor crizotinib is highly active in patients with lung cancer who harbor rearrangements in ALK9,10 or ROS1.11-13 Prompt identification of secondary ALK mutations that confer resistance to crizotinib14-16 has led to the clinical investigation of novel compounds that are able to overcome crizotinib resistance.17,18 Similarly, elucidating the molecular mechanisms of resistance in ROS1-rearranged cancers will be essential to the rapid development of more durable treatments for these patients.
A woman with metastatic lung adenocarcinoma harboring a ROS1 rearrangement initially showed a response to treatment with crizotinib, but the disease ultimately progressed. In her resistant tumors, we identified an acquired mutation affecting the ROS1 kinase domain that confers resistance to crizotinib.
Case Report
A 48-year-old woman with a distant history of light smoking presented with progressive dyspnea. Cytologic analysis of a malignant pleural effusion in the right lung was performed (Fig. 1A), and a diagnosis of metastatic lung adenocarcinoma was made. Genetic studies of the patient's cancer cells revealed no mutations in KRAS or EGFR and no ALK translocation (not shown). She was started on first-line chemotherapy with carboplatin and pemetrexed, but her condition deteriorated, as indicated by worsening discomfort in the right side of her chest, increased fluid output from a right pleural catheter (up to 1 liter per day), and progressive fatigue, weight loss, intermittent fevers, hypoxemia, and hypercalcemia. After three cycles of chemotherapy, repeat imaging of the chest confirmed marked progression of cancer throughout the right lung and pleura.
Figure 1. The CD74–ROS1 Translocation in the Patient's Lung Cancer before Treatment with Crizotinib.
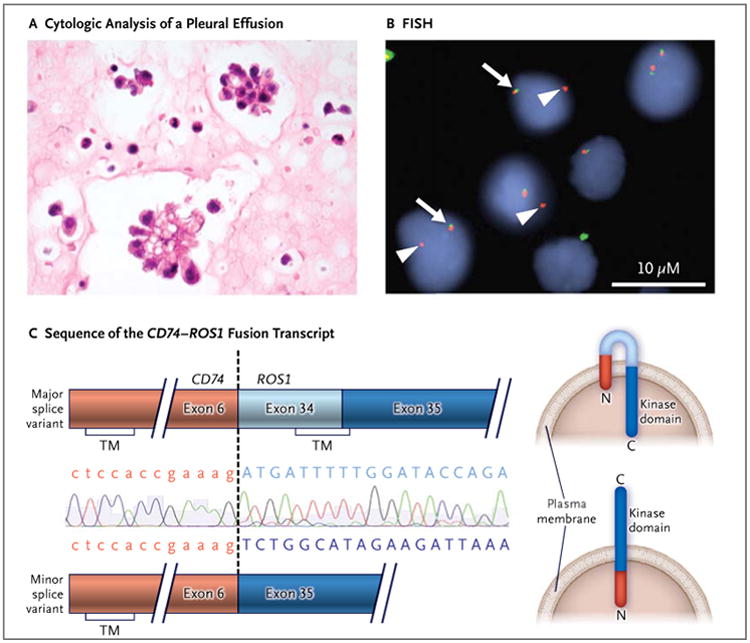
In Panel A, a cell block prepared from a malignant pleural effusion shows clusters of tumor cells in a micropapillary pattern (hematoxylin and eosin). In Panel B, a break-apart fluorescence in situ hybridization (FISH) assay11 with a 5′ ROS1 probe (green) and a 3′ ROS1 probe (red) shows the ROS1 rearrangement, as indicated by the presence of single isolated red 3′ ROS1 probes (arrowheads). The normal ROS1 locus is shown as unsplit red and green pairs of probes (arrows). The nuclei are stained with 4′,6-diamidino-2-phenylindole. In Panel C, Sanger sequencing of the product of reverse-transcriptase–polymerase chain reaction shows the fusion of CD74 exon 6 (red) to either ROS1 exon 34 (light blue) or exon 35 (dark blue). The predicted plasma-membrane orientations for each splice form are shown at right. The N-terminal (N) of CD74 is intracellular and contains a transmembrane (TM) domain. Only the major splice form (CD74 fused to ROS1 exon 34), which contains a second transmembrane domain, is expected to be pathogenic, as a result of the placement of the tyrosine kinase domain of the ROS1 C-terminal (C) in the intracellular compartment.
Additional molecular testing was performed. A ROS1 rearrangement within tumor cells was revealed on fluorescence in situ hybridization (FISH) (Fig. 1B). Reverse-transcriptase–polymerase-chain-reaction (RT-PCR) assays that were performed on total RNA extracted from tumor cells showed that this rearrangement leads to expression of a fusion transcript joining exon 6 of CD74 to either exon 34 (a major splice form) or exon 35 (a minor splice form) of ROS1 (Fig. 1C). Since CD74 is a type II integral membrane protein with a cytoplasmic N-terminal,19 we would expect that only the major splice variant that contains an additional transmembrane domain (encoded by ROS1 exon 34) would be oncogenic because of the positioning of the ROS1 tyrosine kinase domain in the intracellular compartment (Fig. 1C).
The patient was enrolled in a clinical trial investigating the safety and efficacy of crizotinib in cancers with ROS1 translocations (ClinicalTrials.gov number, NCT00585195). She began taking 250 mg of crizotinib twice daily, and within a week she noted a substantial reduction in her dyspnea and fatigue and a substantial increase in appetite. As compared with a computed tomographic (CT) scan obtained before she began treatment with crizotinib, a repeat CT scan of the chest obtained after 2 months of treatment with crizotinib revealed a dramatic response to treatment (Fig. 2A). However, 1 month later, the patient's respiratory symptoms worsened while she was still taking crizotinib, and imaging of the chest showed disease progression (Fig. 2A).
Figure 2. Identification of an Acquired ROS1 G2032R Mutation at the Time of Resistance to Crizotinib.

Axial CT scans of the chest (Panel A) show the patient's disease burden before treatment, after a response to crizotinib, and at the time of crizotinib resistance. Sanger sequencing of RT-PCR products (Panel B) before and after treatment with crizotinib shows the acquired c.6094G→A mutation, which encodes for p.Gly2032Arg. (These coding and amino acid sequences are numbered in accordance with National Center for Biotechnology Information [NCBI] reference sequences CCDS5116 and NP_002935.2, respectively.) In all six malignant sites examined at autopsy (Panel C), the c.6094G→A ROS1 mutation was detected by means of Sanger sequencing of RT-PCR products. Genomic DNA sequencing of ROS1 exon 38 in the patient's grossly and microscopically normal liver tissue shows the nonmutated ROS1 sequence; the CD74–ROS1 fusion transcript could not be detected on RT-PCR in the normal liver. In the summary of autopsy findings at right, the presence of the G2032R mutation is indicated by a plus sign, and the absence of the G2032R mutation by a minus sign.
Methods
Study Oversight
The patient and her family provided written informed consent for the genetic research studies, which were performed in accordance with protocols approved by the institutional review board at Massachusetts General Hospital. The first author wrote the manuscript in collaboration with the senior academic authors; no one other than the authors was involved in the writing of this report. All authors vouch for the accuracy and completeness of the report. The patient participated in a clinical trial funded by Pfizer.
Genetic Studies
A break-apart ROS1 FISH assay11 was used to identify ROS1 rearrangements and gene copy number in specimens of malignant tissue. Complementary DNA was reverse-transcribed from total RNA that was extracted from samples obtained from the patient and then amplified by means of PCR with primers to CD74 and ROS1. Sanger sequencing was performed on RT-PCR products. Genomic DNA extracted from patient samples was used both for deep sequencing of ROS1 exons 34 to 42 and for exon sequencing of a panel of 409 cancer-related genes. Detailed information on the isolation and sequencing of nucleic acid and descriptions of other laboratory techniques are provided in the Supplementary Appendix, available with the full text of this article at NEJM.org.
Analysis of the ROS1 G2032R Mutant
Expression plasmids were made containing the nonmutated or mutated CD74–ROS1 coding sequence and were used for transient transfection assays in 293T cells. For soft-agar assays, NIH-3T3 cells were infected with lentivirus encoding the nonmutated or mutated CD74–ROS1. The ROS1 kinase domain (residues 1934 to 2232) was expressed in Sf21 cells and purified for use in the in vitro enzymatic assays and for the purpose of determining the crystal structure of the nonmutant ROS1–crizotinib complex (structure deposited in the Worldwide Protein Data Bank, entry 3zbf).
Results
Genetic Analysis of Resistant Tumors
To identify the mechanisms of resistance to crizotinib, we obtained a biopsy specimen of a lesion in the right lung that was enlarging while the patient was receiving crizotinib and compared this specimen with one obtained before treatment with crizotinib began. The persistence of the ROS1 rearrangement in the resistant lesion was confirmed by means of FISH, which also showed no ROS1 gene amplification (Fig. S1 in the Supplementary Appendix). Total RNA was extracted from tumor tissue, and the sequencing of an RT-PCR product spanning the CD74–ROS1 fusion junction revealed a c.6094G→A, p.Gly2032Arg (G2032R), mutation that had not been detected in the pretreatment specimen (Fig. 2B, and Fig. S2 in the Supplementary Appendix).
After the patient died, an autopsy was performed. All the sites of disease that were examined had the G2032R mutation (Fig. 2C), suggesting that its occurrence was an early event in the clonal expansion of crizotinib-resistant tumor cells. Deep sequencing (>10,000 reads) of the 3′ portion of the ROS1 gene involved in the rearrangement (exons 34 to 42) was performed on all autopsy specimens; no somatic mutations other than G2032R were revealed within ROS1, and the G2032R mutation was not detected in the sample of malignant cells assessed before treatment with crizotinib (Fig. S2 in the Supplementary Appendix). Exon sequencing of a panel of 409 genes implicated in carcinogenesis independently confirmed the presence of the G2032R mutation in tumors that were resistant to treatment and also identified mutations in three other genes — TP53, ataxia–telangiectasia mutated (ATM), and NOTCH2 — in both the pretreatment and the crizotinib-resistant tumor specimens but not in normal tissue (Fig. S2 in the Supplementary Appendix). No mutations in EGFR or KRAS were detected on exon sequencing.
Molecular Basis of crizotinib Resistance
The glycine at position 2032 is conserved in all human ROS1 paralogs and in several other, more distantly related tyrosine and serine–threonine kinases (Fig. 3A). To determine whether an arginine substitution at this highly conserved residue conferred resistance to ROS1 inhibitors, we transfected 293T cells with an expression plasmid encoding either nonmutated or G2032R CD74–ROS1. The transfected cells were subsequently treated with increasing doses of tyrosine kinase inhibitors. Although crizotinib and TAE68414 (Fig. 3B) inhibited the phosphorylation of nonmutant CD74–ROS1 in a dose-dependent manner, with IC50 (half-maximal inhibitory concentration) values of approximately 30 nM and 50 nM, respectively (Fig. S3 in the Supplementary Appendix), they were ineffective against the G2032R mutant, for which IC50 values for both compounds were greater than 1000 nM. We did not observe activity of the selective ALK inhibitor CH542480215 against either the nonmutant or mutant forms of CD74–ROS1 (Fig. S4 in the Supplementary Appendix).
Figure 3. Mutation of Highly Conserved Glycine at Residue 2032 to Arginine and Resistance to Crizotinib through Steric Interference with Drug Binding.

In Panel A, the alignments of amino acid sequences show that G2032 (red asterisk) is highly conserved among all 13 ROS1 paralogs and among many other clinically important tyrosine and serine–threonine kinases. Identical residues are highlighted in black, and conserved substitutions are highlighted in gray. Panel B shows the results of the transient transfection of 293T cells with expression plasmids containing either nonmutated CD74–ROS1 or G2032R CD74–ROS1. The transfected cells were treated with increasing concentrations of crizotinib (top) or TAE684 (bottom) for 6 hours. Lysates were prepared and Western blot analyses were performed with the use of the indicated antibodies. The 293T cells were also transfected with a plasmid-expressing green fluorescence protein as a negative control. GAPDH denotes glyceraldehyde-3-phosphate-dehydrogenase. In Panel C, in vitro enzymatic assays show a marked decrease in the ability of crizotinib to inhibit kinase activity for the G2032R ROS1 mutant as compared with the nonmutant. Vi and Vo are the initial reaction rates in the presence and absence of crizotinib, respectively. Panel D shows the crystal structure of crizotinib bound to the nonmutant ROS1 kinase domain (at left) and a model of the G2032R mutant (at right), in which there is a predicted steric clash with crizotinib. In this atom-coloring scheme, ROS1 carbon is green, crizotinib carbon purple, oxygen red, nitrogen blue, chlorine gold, and fluorine brown. For emphasis, residue 2032 is shown in a space-filling representation. The red asterisks indicate the L2026 gatekeeper residue of ROS1.
In vitro enzymatic assays also showed that the crizotinib concentration needed to achieve 50% enzyme inhibition (Kiapp) was increased by a factor of 270 for the G2032R mutant kinase as compared with nonmutant ROS1 (Kiapp 570±29 nM vs. Kiapp 2.1±0.1 nM) (Fig. 3C). Furthermore, the ATP concentration needed to achieve half-maximal enzyme velocity (Kmapp) for the G2032R mutant was reduced by a factor of 3 as compared with that for nonmutant ROS1 (Kmapp 22 μM vs. Kmapp 65 μM) (Fig. S5 in the Supplementary Appendix) — perhaps contributing to the increased activity of the mutant kinase. However, in soft-agar assays, the transforming capacities of non-mutant and mutant CD74–ROS1 were similar (Fig. S6 in the Supplementary Appendix).
To elucidate the molecular basis of G2032R-mediated resistance, we determined the crystal structure of the phosphorylated nonmutant ROS1 kinase domain bound to crizotinib (Fig. 3D, left). Like ALK and c-MET (also known as the hepatocyte growth-factor receptor), ROS1 binds crizotinib at the ATP-binding site in the cleft between the N-terminal and C-terminal domains of the kinase.20 The G2032 residue sits at the solvent front in the distal end of the kinase hinge and creates a turn, putting the G2032 alpha carbon in position to engage in a van der Waals interaction with the pyrazole ring of crizotinib. An arginine at position 2032 has been modeled into an empty ROS1 ATP–inhibitor binding site (Fig. 3D) and is believed to sterically clash with the piperidine ring of crizotinib while still allowing for ATP binding (Fig. S7 in the Supplementary Appendix). These data provide a biochemical and structural basis for the resistance to crizotinib conferred by the G2032R mutation.
Discussion
In this study, we report an acquired mutation for crizotinib resistance in a cancer driven by an oncogenic ROS1 fusion. We also present the crystal structure of crizotinib bound to the ROS1 kinase domain. Unlike the classic gatekeeper mutations for drug resistance that have been identified in ABL,21 EGFR,22 and ALK,14 the G2032R ROS1 mutation is located in the solvent front of the kinase domain and is analogous to the G1202R ALK mutation identified in crizotinib-resistant ALK-rearranged lung cancers15 (Fig. S8 in the Supplementary Appendix). Whereas the L1196M ALK gatekeeper mutant may still be sensitive to newer ALK inhibitors, such as CH5424802,18 we previously found that G1202R ALK confers high-level resistance to crizotinib and to all the next-generation ALK inhibitors that were examined.15 In light of these observations, it may be necessary to identify novel compounds that specifically target the G1202R ALK or G2032R ROS1 mutant, to overcome the development of crizotinib resistance in these cancers.
At the time of disease progression, when the patient was taking crizotinib, a repeat biopsy was performed to identify the mechanism of drug resistance. The biopsy was performed in accordance with a clinical protocol that was approved by the institutional review board at our institution specifically for patients in whom resistance has developed to targeted therapies such as crizotinib and erlotinib. Repeat biopsies have emerged as an invaluable tool for discovering clinically relevant resistance mechanisms, including secondary mutations within the target, activation of alternative signaling pathways, and in the case of EGFR-mutant lung cancer, small-cell transformation.23 Elucidating these mechanisms has helped to guide the development of new treatment strategies designed to overcome resistance.
The autopsy performed in this case revealed the presence of the CD74–ROS1 gene translocation at all sites of disease. Despite the genetic heterogeneity that has been identified at various metastatic sites in an individual patient,24 our findings support the notion that some oncogenic drivers are present in founder clones25 and therefore may be present at all sites of metastasis. It is also noteworthy that the same mechanism of acquired resistance — the G2032R mutation — was identified at all the sites of disease that were examined, and no other ROS1 kinase mutations were identified by deep sequencing. Thus, it appears that this mutation occurred early in the development of resistance and suggests that a potent inhibitor of this mutant kinase may have been clinically effective after the failure of crizotinib.
Supplementary Material
Acknowledgments
Supported by Pfizer, by grants from the National Cancer Institute (5R01CA164273-02, to Drs. Shaw and Engelman; R01CA137008 and Lung SPORE P50CA090578, to Dr. Engelman; and a1R21CA161590-01, to Dr. Iafrate), by a V Foundation Translational Research Grant (to Drs. Shaw and Engelman), by Be a Piece of the Solution, and by the Evan Spirito Memorial Foundation.
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Druker BJ, Talpaz M, Resta DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–7. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- 2.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non–small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 3.Paez JG, Jänne PA, Lee JC, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 4.Herbst RS, Heymach JV, Lippman SM. Lung cancer. N Engl J Med. 2008;359:1367–80. doi: 10.1056/NEJMra0802714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–6. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 6.Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 7.Demetri GD, Reichardt P, Kang YK, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013;381:295–302. doi: 10.1016/S0140-6736(12)61857-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome–positive leukemias. N Engl J Med. 2012;367:2075–88. doi: 10.1056/NEJMoa1205127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non–small-cell lung cancer. N Engl J Med. 2010;363:1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Camidge DR, Bang YJ, Kwak EL, et al. Activity and safety of crizotinib in patients with ALK-positive non-small-cell lung cancer: updated results from a phase 1 study. Lancet Oncol. 2012;13:1011–9. doi: 10.1016/S1470-2045(12)70344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bergethon K, Shaw AT, Ou SHI, et al. ROS1 rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–70. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davies KD, Le AT, Theodoro MF, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clin Cancer Res. 2012;18:4570–9. doi: 10.1158/1078-0432.CCR-12-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shaw AT, Camidge DR, Engelman JA, et al. Clinical activity of crizotinib in advanced non-small cell lung cancer (NSCLC) harboring ROS1 gene rearrangement; Presented at the Annual Meeting of the American Society of Clinical Oncology; Chicago. June 1–5, 2012; abstract. [Google Scholar]
- 14.Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–9. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 15.Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–82. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108:7535–40. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19:679–90. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 19.Becker-Herman S, Arie G, Medve-dovsky H, Kerem A, Shachar I. CD74 is a member of the regulated intramembrane proteolysis-processed protein family. Mol Biol Cell. 2005;16:5061–9. doi: 10.1091/mbc.E05-04-0327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cui JJ, Tran-Dubé M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J Med Chem. 2011;54:6342–63. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 21.Gorre ME, Mohammed M, Ellwood K, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 22.Kobayashi S, Boggon TJ, Dayaram T, et al. EGFR mutation and resistance of non–small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–92. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 23.Sequist LV, Waltman BA, Dias-Santa-gata D, et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. Erratum, N Engl J Med 2012;367:976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Govindan R, Ding L, Griffith M, et al. Genomic landscape of non-small cell lung cancer in smokers and never-smokers. Cell. 2012;150:1121–34. doi: 10.1016/j.cell.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.