

Abstract
Sickle cell disease is the most common inherited hematologic disorder that leads to the irreversible damage of multiple organs. Although sickling of red blood cells and vaso-occlusion are central to the pathophysiology of sickle cell disease the importance of hemolytic anemia and vasculopathy has been recently recognized. Hypercoagulation state is another prominent feature of sickle cell disease and is mediated by activation of both intrinsic and extrinsic coagulation pathways. Growing evidence demonstrates that coagulation may not only contribute to the thrombotic complications, but also to vascular inflammation associated with this disease. This article summarizes the role of vascular inflammation and coagulation activation, discusses potential mechanisms responsible for activation of coagulation and reviews recent data demonstrating the crosstalk between coagulation and vascular inflammation in sickle cell disease.
Keywords: sickle cell, hemolysis, coagulation, tissue factor, inflammation
Introduction
Sickle cell anemia is the most common and severe variant of sickle cell disease. It is a hematologic disorder caused by a single nucleotide mutation that substitutes glutamic acid with valine at the sixth position of the β-globin gene (Frenette and Atweh 2007, Hebbel, et al 2009, Rees, et al 2010). Homozygosity for this mutation leads to abnormal polymerization of hemoglobin tetramers within erythrocytes under hypoxic conditions. Aggregation of abnormally large hemoglobin polymers results in the formation of sickled red blood cells that are less flexible, adhere to the endothelium and are prone to hemolysis (Frenette and Atweh 2007, Hebbel, et al 2009, Rees, et al 2010). Clinical manifestations of sickle cell disease include recurrent painful crises, chronic hemolytic anemia, acute chest syndrome, pulmonary hypertension, stroke, kidney failure, priapism, leg ulcers, osteonecrosis and cardiac disease (Frenette and Atweh 2007, Hebbel, et al 2009, Rees, et al 2010). Sickling of red blood cells results in the two primary pathologic events in sickle cell disease: vaso-occlusion mediated ischemia-reperfusion injury and hemolytic anemia. Both of these are thought to lead to increased vascular inflammation and activation of coagulation (Frenette and Atweh 2007, Hebbel, et al 2009, Rees, et al 2010). The interactions between vascular inflammation and coagulation and the contribution of coagulation to the pathology of sickle cell disease are the primary focus of this review.
In order to study the complex disease pathology, mouse models of SCD were developed. The first generation of mouse models of SCD were created by homozygous deletion of mouse β-globin and addition of the human sickle β-globin (βS) gene; these HbS mice develop mild to moderate anemia with almost none of the clinical manifestations of sickle cell disease due to the lack of human α-globin and low βS expression (Beuzard 2008). To address this issue, newer mouse models have been developed in which mouse α-globin and β-globin genes are deleted. These and other mouse models are described in detail in a recent review (Beuzard 2008).The Berkley (BERK) model are homozygous knockouts for α-globin and β-globin, and express a transgene containing human α-globin and βS-globin, as well as Gγ- and Aγ-globin to prevent erythrocyte sickling during gestation and fetal death (Paszty, et al 1997). The Townes mouse model of SCD also involves homozygous deletion of mouse α-globin and β-globin gene, and knock-in of a transgene containing human α-globin, Aγ-globin, and either βS-globin or healthy βA-globin (Wu, et al 2006). Embryonic lethality is prevented because mice express fetal Hb (HbF), which persists until approximately one month of age when the switch to HbS is complete. The advantage to these mice is that healthy AA mice can be generated as littermate controls with SS mice, by breeding heterozygous βA/βS pairs. The BERK and Townes models of SCD have severe anemia, leukocytosis and multi-organ damage consistent with the human pathology (Paszty, et al 1997, Wu, et al 2006).
The pathology of sickle cell disease
Vaso-occlusion and ischemia reperfusion injury
The primary pathological event in sickle cell disease is the sickling of red blood cells, which adhere to both the vascular endothelium and white blood cells (Frenette and Atweh 2007, Rees, et al 2010). Healthy mature red blood cells have low to nonexistent expression of adhesion molecules on their surface. However, anemia stimulates the bone marrow to release immature red blood cells (reticulocytes). These reticulocytes express high levels of adhesion molecules such as α4β1 integrin and CD36, which facilitate interactions with adhesion molecules on endothelial cells and leukocytes (Joneckis, et al 1993). It has been reported that sickle red blood cells adhere to vascular cell adhesion molecule (VCAM) on endothelial cells via interaction with α4β1 integrin (Ataga, et al 2008, Gee and Platt 1995).
The vascular endothelium is also activated in sickle cell disease, and its role in the pathology of this disease has been extensively reviewed (Hebbel, et al 2004). There is up-regulation of adhesion molecules, such as VCAM, P-selectin, and E-selectin, which bind adhesion molecules on red blood cells and on polymorphonuclear lymphocytes (Hebbel, et al 2004). Biomarkers of endothelial activation, such as soluble (s)VCAM, sE-selectin, and sP-selectin are elevated in patients with sickle cell anemia compared to controls (Ataga, et al 2008, Setty, et al 2012). Endothelial P-selectin interacts with P-selectin glycoprotein ligand 1 (PSGL-1) expressed on almost all hematopoietic cells (Luo, et al 2012), and also contributes to the interaction of sickle red blood cells with the endothelium (Gutsaeva, et al 2011). Luo and colleagues recently demonstrated that inhibition of PSGL-1 in sickle cell mice reduced leukocyte adhesion and rolling, and markers of endothelial activation. PSGL-1 inhibition also reduced inflammation and liver injury (Luo, et al 2012). Endothelial E-selectin interacts with E-selectin ligand 1 (ESL-1) on activated leukocytes (Mohan, et al 2005) in sickle cell patients, as described below. Activated PMNs can bind to red blood cells via interactions between CD11b/CD18 with various receptors on red blood cells (Hofstra, et al 1996). Indeed, red blood cells isolated from sickle cell patients are reported to adhere to PMNs and activate the respiratory burst; dense red blood cells that had undergone repeated sickling events were more adherent (Hofstra, et al 1996). Adherence was due in part to interactions between integrins on red cells and PMNs (Hofstra, et al 1996). In the humanized Berkley (BERK) mouse model of sickle cell disease, intravital microscopy revealed enhanced leukocyte rolling in sickle cell mice compared to controls after an inflammatory stimulus. Interestingly, circulating sickle red blood cells adhered more frequently to leukocytes than the endothelium (Turhan, et al 2002). Inhibition of leukocyte adhesion to the endothelium, through the use of E-selectin and P-selectin knockout mice transplanted with sickle bone marrow also reduced sickle red blood cell adhesion (Turhan, et al 2002). Furthermore, the frequency of red blood cell-PMN interactions is highly elevated and blood flow in the cremaster microcirculation is decreased in sickle cell mice compared to control (Hidalgo, et al 2009). In E-selectin knockout mice that were transplanted with sickle cell bone marrow, the frequency of red blood cell-PMN interactions decreased and blood flow in the microcirculation was improved, suggesting that signaling from E-selectin to PMNs contributes to red blood cell-PMN interactions (Hidalgo, et al 2009). Indeed, E-selectin was demonstrated to promote increased expression and activity of CD11b/CD18 integrin on PMNs from sickle cell mice (Hidalgo, et al 2009). The complex interactions between sickle RBCs, leukocytes and endothelial cells involve multiple adhesion molecules (Figure 1). In essence, these interactions result in recurrent vaso-occlusive events in postcapillary venules that lead to cyclic ischemia-reperfusion injury in multiple organs (Frenette 2002, Hebbel, et al 2009, Rees, et al 2010).
Figure 1.
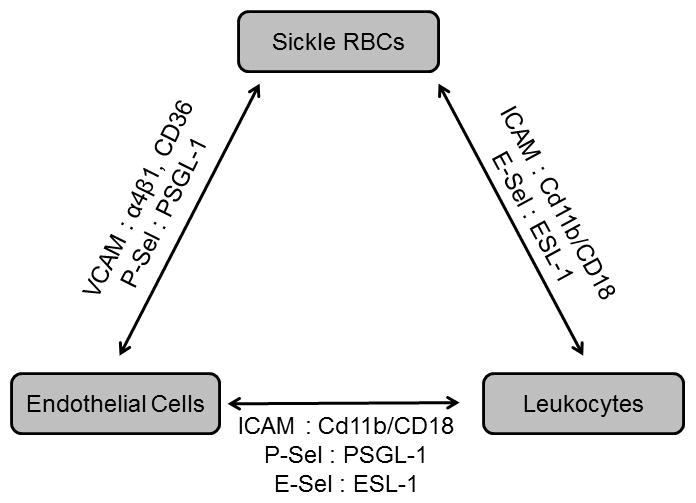
Interactions between sickle RBCs, leukocytes and endothelial cells are mediated by multiple adhesion molecules.
Ischemia-reperfusion injury activates xanthine oxidase leading to the production of free radicals (O2− and H2O2), which incite oxidative stress in endothelial cells (Carden and Granger 2000). Ischemia-reperfusion injury also enhances the expression of adhesion molecules, such as VCAM, intercellular adhesion molecule (ICAM), P-selectin, and E-selectin on endothelial cells. Moreover, oxidative stress stimulates the release of platelet activating factor (PAF) and leukotriene B4 (LTB4) from endothelial cells and these in turn stimulate expression of CD11b/CD18 and CD62L (L-selectin) on PMNs (Carden and Granger 2000). Finally, activated PMNs and monocytes also contribute to oxidative stress in sickle cell disease by generating reactive oxygen species during the respiratory burst (Carden and Granger 2000, Hofstra, et al 1996).
Vascular inflammation
Patients with sickle cell anemia have chronic leukocytosis, particularly an increase in monocytes and polymorphonuclear neutrophils (PMNs) (Qari, et al 2012, Vichinsky, et al 1997). Monocytes from sickle cell patients are highly activated, and express more interleukin (IL)-1β and tumor necrosis factor (TNF)-α compared to monocytes from healthy control subjects (Belcher, et al 2000, Wun, et al 2002). Interestingly, monocytes from sickle cell patients can induce E-selectin, ICAM and VCAM expression on human endothelial cells in culture (Belcher, et al 2000), indicating that endothelial cell-monocyte interactions are highly interdependent. PMNs from sickle cell patients are also activated compared to control subjects, marked by decreased surface CD62L expression and shedding of CD62L into the plasma (Lard, et al 1999). Plasma levels of lactoferrin and elastase are also higher in sickle cell patients, indicating degranulation of specific and azurophilic granules in PMNs, respectively (Lard, et al 1999).
In addition to an activated endothelium and leukocytosis, the inflammatory phenotype of sickle cell disease is also characterized by high levels of acute phase proteins and cytokines. Sickle cell patients have higher levels of tumor necrosis factor (TNF)-α, interleukin (IL)-1β, IL-6, IL-8, interferon (IFN)-γ, C-reactive protein (CRP), macrophage inflammatory protein (MIP)-1α and monocyte chemotactic protein (MCP)-1 than healthy controls (Pathare, et al 2004, Qari, et al 2012). These inflammatory markers are chronically elevated in sickle patients at “steady state”, and are often increased further after acute painful crises (Pathare, et al 2004).
Mouse models of sickle disease share this inflammatory phenotype. Belcher and colleagues (Belcher, et al 2003) reported elevated levels of various inflammatory markers in four transgenic mouse models of sickle cell disease (NY-S, Berkley-SAntilles, NY-S/SAntilles, and Berkley- mice). All strains of sickle mice had leukocytosis, predominantly of neutrophils, monocytes and lymphocytes. Furthermore, plasma levels of serum amyloid protein (SAP, the murine analog of human CRP) and IL-6 were elevated. Additionally, there was an enhanced expression of VCAM-1, ICAM-1 and platelet endothelial cell adhesion molecule (PECAM) in the lungs of sickle mice compared to non-sickle controls. The expression of these adhesion molecules was dependent on the transcription factor NF-κB (Belcher, et al 2003). We have recently confirmed these data in the Berkley mice and showed a similar pro-inflammatory phenotype in the Townes mouse model of sickle cell disease. In addition, an enhanced accumulation of neutrophils in the lung and liver, and increased expression of the chemokines MCP-1 and KC was observed in sickle cell mice in both models (Chantrathammachart, et al 2012a).
Hemolytic anemia and NO depletion
Other important components in sickle cell disease pathology are hemolytic anemia and decreased nitric oxide (NO) bioavailability (Kato, et al 2007). Intravascular hemolysis results in release of cell-free hemoglobin at a rate of up to 30 g per day, which then liberates heme into the plasma. Plasma heme levels average 4.3 ± 1.1 μM in sickle cell patients, whereas it is undetectable in healthy controls (Reiter, et al 2002). This excess amount of hemoglobin and heme saturate their respective scavenging molecules, haptoglobin and hemopexin (Reiter, et al 2002), the levels of which are decreased in sickle cell patients (Muller-Eberhard, et al 1968). Hemoglobin and free heme consume NO to generate methemoglobin and nitrate (Kato, et al 2007). The production of NO is also depleted in sickle cell disease; sickle red blood cells release arginase, which degrades L-arginine, the substrate for NO production by endothelial nitric oxide synthase (eNOS) (Kato, et al 2007, Morris, et al 2005). Reactive oxygen species react with NO to produce peroxynitrite (ONOO−) which nitrosylates various proteins causing their dysfunction (Kato, et al 2007). The loss of NO in sickle cell disease impairs its homeostatic functions, such as vasodilation and inhibition of platelet activation. Furthermore, NO plays an important role in regulating endothelial cell activation and cytokine production (Reiter, et al 2002), both of which are major contributors to the pathogenesis of sickle cell disease.
Free heme liberated from hemoglobin during hemolysis is also pro-inflammatory by directly activating endothelial cells and macrophages. Heme induced TNF-α and keratinocyte chemoattractant (KC) expression in macrophages depends on toll-like receptor 4 (TLR4) signaling (Wagener, et al 2001). TLR-4 signaling has also been implicated in heme-mediated acute lung injury in a sickle cell mouse model (Ghosh, et al 2011). Heme also incites oxidative stress and heme oxygenase (HO)-1 expression in macrophages (Figueiredo, et al 2007). Further, free heme can also contribute to vascular inflammation via endothelial cell dysfunction and activation. Heme generates intracellular ROS and increased expression of VCAM, ICAM and E-selectin on endothelial cells in vitro (Wagener, et al 1997). Injection of heme also increases ICAM-1 expression in livers of mice, and leukocyte migration into various tissues. In addition to upregulation of adhesion molecules and chemokine production, heme can stimulate PMN migration by directly activating chemokine receptors in vitro and in vivo (Porto, et al 2007). Furthermore, free heme activates respiratory burst in PMNs (Porto, et al 2007).
Hypercoagulation and thrombosis in sickle cell disease
The coagulation cascade can be divided into the extrinsic, intrinsic and common pathways (Mackman 2012, Pawlinski, et al 2004). The extrinsic pathway consists of the transmembrane receptor tissue factor (TF) and plasma factor VII/VIIa (FVII/FVIIa). TF binds FVII/FVIIa and forms a TF:FVIIa complex that activates FX to FXa (Mackman 2012, Pawlinski, et al 2004). The intrinsic pathway provides an alternative way to activate coagulation via FXIIa and FXIa. It also amplifies the generation of FXa via the FVIIIa/FIXa intrinsic tenase complex. Coagulation activation results in thrombin generation, fibrin deposition and activation of platelets (Mackman 2012, Pawlinski, et al 2004).
Activation of coagulation in sickle cell disease has been well documented (Ataga 2009, Ataga and Key 2007, De Franceschi, et al 2011). Excessive thrombin generation in sickle cell patients is indicated by increased plasma levels of prothrombin fragment 1.2 (F1.2) and thrombin anti-thrombin (TAT) complexes (Ataga, et al 2008, Peters, et al 1994, Stuart and Setty 2001, van Beers, et al 2008). Higher thrombin peak height, higher rates of thrombin formation, and higher endogenous thrombin potential has been reported in platelet-poor plasma of sickle cell patients compared to age-matched controls using calibrated automated thrombogram method of measuring thrombin generation (Noubouossie, et al 2011). In addition, increased plasma levels of D-dimers, fibrinopeptide A, fibrin-fibrinogen peptide E and plasmin-antiplasmin complexes indicate ongoing thrombin-dependent fibrinogen cleavage, clot formation and subsequent fibrinolysis (Adam, et al 2009, Ataga, et al 2008, Leslie, et al 1975, Tomer, et al 2001a, Tomer, et al 2001b, van Beers, et al 2008, Westerman, et al 1999). Moreover, the levels of protein C and protein S are decreased in patients with sickle cell disease, presumably due to increased consumption of these natural anticoagulants (el-Hazmi, et al 1993, Francis 1988, Green and Scott 1986, Tam 1997, Westerman, et al 1999). In contrast, plasma levels of tissue factor pathway inhibitor, a natural inhibitor of TF, were not changed in sickle cell patients (Key, et al 1998). Finally, patients with sickle cell disease have a propensity to develop thrombotic complications. In situ thrombosis of small pulmonary vessels is a common finding during autopsy studies (Adedeji, et al 2001, Kirkpatrick and Haynes 1994, Oppenheimer and Esterly 1971). The incidence of inpatient pulmonary embolism is higher in the sickle cell disease population than in the non- sickle cell disease population (Novelli, et al 2012). It has been shown that sickle cell patients have a higher prevalence of pulmonary thrombosis but not deep vein thrombosis compared to age- and race-matched controls, suggesting that pulmonary thrombosis occurs as a primary rather than secondary embolic event in this disease (Stein, et al 2006). However, a recent cross-sectional study demonstrated that sickle cell patients had equally high rates of both deep venous thrombosis and pulmonary embolism. Importantly, non catheter-related venous thromboembolism was an independent risk factor for death in this cohort of patients. Mekontso Dessap and coworkers demonstrated pulmonary artery thrombosis in a sickle cell patient during acute chest syndrome (Mekontso Dessap, et al 2011). Sickle cell disease is also a significant risk factor for pregnancy-related venous thromboembolism (James, et al 2006). Furthermore, thrombosis has been reported within the vena cava and hepatic vein of sickle cell patients (Ng and Ashari 2003, Singh, et al 2010). Clinical evidence also points to the role of thrombosis in triggering stroke (Hillery and Panepinto 2004, Prengler, et al 2002). Ischemic stroke commonly occurs in sickle cell patients, with a risk of 0.5 – 1% per year (Adams 2007); strokes are often precipitated by painful crises, when markers of coagulation and inflammation are elevated (Prengler, et al 2002). Consistent with these clinical observations, we and others have shown that plasma levels of thrombin-antithrombin complexes are also increased in mouse models of sickle cell disease (Chantrathammachart, et al 2012a, Guo, et al 2008). Furthermore, microthrombi and increased fibrin deposition were observed in multiple organs in sickle mice including lung, liver, kidneys and brain (Guo, et al 2008, Trudel, et al 1994). Exposing sickle cell mice to hypoxic conditions resulted in further increases in plasma TAT levels and thrombosis within the pulmonary vasculature (de Franceschi, et al 2003, Guo, et al 2008). Interestingly, it was recently reported that biomarkers of coagulation activation (TAT and D-dimer) correlated with a history of stroke, retinopathy and acute chest syndrome in a cohort of 52 sickle cell disease patients (Ataga, et al 2012), indicating that coagulation might contribute to these pathologies.
Activation of coagulation in sickle cell disease
The hypercoagulable state and thrombotic complications of sickle cell disease are well documented (Ataga and Key 2007, De Franceschi, et al 2011, Singer and Ataga 2008), but the stimulus for coagulation activation is yet to be determined. Potential causes of coagulation activation in sickle cell disease are discussed below.
Increased TF expression
TF is the primary activator of the extrinsic coagulation pathway (Bach 1988, Edgington, et al 1991). It is constitutively expressed by perivascular cells such as vascular smooth muscle cells, adventitial fibroblasts, and pericytes. TF expressed by these cells is normally sequestered from blood, and it forms a hemostatic envelope to reduce blood loss upon vascular injury (Drake, et al 1989, Fleck, et al 1990, Mackman, et al 2007). In addition, inducible TF expression is observed in vascular cells during many pathologic conditions, including sickle cell disease (Mackman, et al 2007).
Sickle cell patients have increased TF expression on monocytes (Setty, et al 2012) with elevated whole blood TF procoagulant activity (Key, et al 1998). Furthermore, circulating endothelial cells isolated from sickle cell patients showed increased levels of TF mRNA, antigen, and activity (Solovey, et al 1998). TF expression in whole blood and circulating endothelial cells was similarly increased in patients in pain crisis and those with steady-state disease (Key, et al 1998, Solovey, et al 1998). TF expression was also increased in circulating monocytes and in the endothelium of the lung microvasculature in mouse models of sickle cell disease (Solovey, et al 2004). Hypoxia/reoxygenation challenge further increases TF staining in both cell types (Solovey, et al 2004). We have recently shown that TF expression was also increased in neutrophils isolated from sickle cell mice (Chantrathammachart, et al 2012a). It has been proposed that the interaction of TF-positive neutrophils with endothelial cells is a critical step for initiating thrombosis in the mouse cremaster arterioles in a laser injury model (Darbousset, et al 2012). Perivascular cells can also be a source of TF contributing to the activation of coagulation as shown in a mouse model of endotoxemia (Pawlinski, et al 2010). Endothelial cell injury and increased vascular permeability observed in sickle cell disease can make perivascular TF accessible to circulating clotting factors and activate coagulation (Ghosh, et al 2012, Polanowska-Grabowska, et al 2010, Wallace, et al 2009). Importantly, we have demonstrated that inhibition of TF in sickle mice (Berkley and Townes mice) reduces plasma levels of TAT to the baseline levels observed in control mice, indicating a critical role of TF in activation of coagulation (Chantrathammachart, et al 2012a). The relative contribution of different cellular sources of TF to the activation of coagulation is currently being investigated in our laboratory.
Sickle Red Blood Cells and Activation of Coagulation
In normal red blood cells, procoagulant anionic phospholipids, such as phosphatidylserine (PS), are almost completely restricted to the inner leaflet of the membrane (Connor and Schroit 1991). Repeated cycles of sickling result in increased exposure of PS on the surface of sickle red blood cells (Franck, et al 1985). PS exposure increases procoagulant activity by providing a negatively charged surface which facilitates assembly of the clotting cascade components. This process is mediated via an electrostatic interaction between negatively charged PS and positively charged γ-carboxyglutamic acid domains present in the clotting proteins, including FVII, FIX, FX and prothrombin (Owens and Mackman 2011). Analysis of blood from sickle cell patients demonstrated a positive correlation between PS positive red blood cells and prothrombin fragment 1.2, D-dimers, and plasmin-antiplasmin complex (Setty, et al 2000, Setty, et al 2001). Interestingly, no positive correlation has been found between PS positive platelets and these markers of coagulation activation, which suggests that sickle red blood cells, rather than platelets, contribute to the hypercoagulable state in sickle cell disease (Setty, et al 2001).
Circulating Microparticles
Microparticles (MPs) are small membrane vesicles released from activated or apoptotic cells. The procoagulant properties of MPs are due to the presence of PS and TF on their surface (Owens and Mackman 2011). Two recent papers demonstrated that MPs derived from different cellular sources contribute to thrombin and fibrin generation via different mechanisms (Aleman, et al 2011, Van Der Meijden, et al 2012). Platelet- and erythrocyte-derived MPs, which are TF negative, failed to induce thrombin generation in FXII-deficient plasma, and inhibition of FVII had no effect on thrombin generation (Van Der Meijden, et al 2012). The majority of circulating MPs in sickle cell patients originate from erythrocytes and platelets (Allan, et al 1982, Wun, et al 1998). These MPs are TF negative and demonstrate increased exposure of PS on their surface (Shet, et al 2003). Consistent with the observation made by Van Der Meijden and coworkers, it has been shown that erythrocyte-derived MPs isolated from the blood of sickle cell patients contribute to thrombin generation via a FXI dependent but not FVII dependent mechanism (van Beers, et al 2009). In contrast, thrombin generation induced by TF-positive monocyte-derived MPs requires FVII but not FXII (Van Der Meijden, et al 2012). A recent study demonstrated that thrombospondin-1 mediated release of erythrocyte MPs facilitates endothelial cell injury and vaso-occlusion in sickle cell mice (Camus, et al 2012). It has been shown that the blood of sickle cell patients does contain a small fraction of monocyte- and endothelial cell-derived MPs, which have been identified as TF positive (Shet, et al 2003). In contrast to this observation, a recent study reported no evidence for the presence of TF positive MPs in sickle cell patients (van Beers, et al 2009). Furthermore, Setty and coworkers demonstrated that MP-associated TF procoagulant activity was not elevated in the plasma of young children with sickle cell disease compared to age-matched controls, and MP-associated TF procoagulant activity did not correlate with markers of coagulation activation (Setty, et al 2012). A similar lack of correlation was observed in adult sickle cell patients (Ataga, et al 2012) and in our recent mouse study (Chantrathammachart, et al 2012a). Together, these data suggest that in sickle cell disease, MPs contribute to the activation of coagulation most likely via PS-dependent mechanism that requires activation of the intrinsic coagulation pathway.
Platelets
A moderate increase in platelet numbers is observed in older children and adults with SCD in steady state (Francis 1991). Circulating platelets in these patients are chronically activated and platelet aggregation is increased (Kenny, et al 1980, Westwick, et al 1983). This may be attributed to the increased numbers of young, metabolically active platelets or increased plasma levels of platelets agonists, such as epinephrine, adenosine diphosphate or thrombin, in the blood of sickle cell patients. Platelet levels of P-selectin and CD40 ligand are also increased in sickle cell patients compared to healthy control subjects (Garrido, et al 2012, Lee, et al 2006, Tomer, et al 2001a). An increased number of platelet-monocyte aggregates has been observed in both sickle cell patients and a mouse model of sickle cell disease (Polanowska-Grabowska, et al 2010). The formation of these aggregates was mediated via platelet P-selectin (Polanowska-Grabowska, et al 2010). Interestingly, platelet P-selectin and CD40 ligand have been shown to induce rapid expression of TF in monocytes (Lindmark, et al 2000). Furthermore, inhibition of CD40 ligand receptor reduced monocyte TF expression induced by the plasma from sickle cell patients (Lee, et al 2006). Platelets from sickle cell patients also express higher levels of the pro-inflammatory cytokine LIGHT (TNFSF14), which promotes endothelial activation and inflammation (Garrido, et al 2012) and demonstrated increased αIIbβ3-depenent adhesion to fibrinogen (Proenca-Ferreira, et al 2010). Therefore platelets might participate to vaso-occlusive events by binding at the site of inflammation with consequent release of inflammatory mediators.
Ultralarge von Willebrand factor (vWF) multimers
Sickle cell patients demonstrate increased levels of circulating ultralarge vWF multimers in their blood (Chen, et al 2011, Krishnan, et al 2008). This increase in ultralarge vWF multimers can result from the combination of increased secretion, defective clearance, and impaired cleavage by ADAMTS13. Recent studies demonstrate that impaired processing of ultralarge vWF multimers by ADAMTS13 in sickle cell patients is not the result of ADAMTS13 deficiency, but might reflect resistance of vWF to proteolysis. The resistance could result from either the oxidation of Met1606 at the ADAMTS13 cleavage site, or the binding of free hemoglobin to the A2-domain of vWF (Chen, et al 2010, Zhou, et al 2009). Since a portion of ultralarge vWF multimers remains anchored to the endothelium upon activation of endothelial cells (Dong, et al 2002), it is likely that the increase observed in circulation reflects a similar increase in the endothelium-bound ultralarge vWF multimers in sickle cell patients. It has been shown that ultralarge vWF multimers mediate the adhesion of sickle red blood cells to the endothelium (Wick, et al 1987) and platelets aggregations (Ruggeri 2007). Therefore, the endothelium-bound ultralarge vWF multimers may contribute to both vaso-occlusion and generation of thrombogenic surface on the endothelium in SCD.
Regulation of TF expression and activation of coagulation in sickle cell disease
Several mouse studies have examined the mechanism and regulation of TF expression in sickle cell disease. Using a bone marrow transplantation approach, Kollander and colleagues demonstrated that endothelial TF expression is highly dependent on NF-κB(p50) in peripheral blood mononuclear cells, but not in the vessel wall, of sickle cell mice (Kollander, et al 2010). These data suggest that peripheral blood mononuclear cells indirectly promote endothelial TF expression via a NF-κB(p50)-dependent mechanism.
An in vitro study demonstrated that heme, a product of intravascular hemolysis, induces the expression of functionally active TF on both micro- and macro-vascular endothelial cells (Setty, et al 2008). Consistent with these results, multiple regression analyses identified an association between markers of hemolysis and markers of coagulation activation in sickle cell patients (Setty, et al 2012). Recently, we have shown that heme can induce activation of coagulation in mice in a TF-dependent manner (Sparkenbaugh, et al 2012). Endothelial nitric oxide synthase (eNOS) negatively regulates endothelial cell TF expression via generation of NO (Solovey, et al 2010). Consistent with that observation, inhalation of NO attenuated increased endothelial TF expression and reduced the number of thrombi in small lung vessels in sickle mice subjected to hypoxia/reoxygenation injury (de Franceschi, et al 2003, Solovey, et al 2010). Furthermore, lovastatin treatment also reduced the increased TF staining observed in pulmonary endothelial cells of sickle cell mice (Solovey, et al 2004). Interestingly, lovastatin treatment in sickle cell mice did not attenuate inducible TF expression in monocytes or constitutive TF expression in perivascular cells (Solovey, et al 2004). However, a short term use of simvastatin had only a modest effect on plasma levels of TF antigen in sickle cell patients (Hoppe, et al 2011). In addition, the histone deacetylase inhibitors trichostatin A and suberoylanilide hydroxamic acid also reduced endothelial TF expression in Berkley mice after a hypoxia/reoxygenation challenge (Hebbel, et al 2010). The reduction may result from either direct inhibition of TF expression (Wang, et al 2007) or could be an indirect result of the increased expression of fetal hemoglobin and overall attenuation of disease severity (Bradner, et al 2010, Sangerman, et al 2006).
Finally, hydroxyurea treatment has been shown to reduce many markers of coagulation activation. A recent study by Colella and colleagues showed that sickle cell patients on hydroxyurea treatment had significant reduction in leukocyte TF mRNA expression, plasma levels of TF protein, as well as plasma levels of F1.2 fragments and TAT complexes (Colella, et al 2012). Treatment with hydroxyurea was also associated with a decrease in the formation of red blood cell derived PS-positive MPs and subsequent attenuation of thrombin generation (Gerotziafas, et al 2012). Coincidentally, hydroxyurea therapy is also associated with decreased mortality and morbidity, reduced painful crises and chest syndrome (Steinberg, et al 2003), and fewer vaso-occlusive events (Charache, et al 1995).
Contribution of coagulation to the pathology of sickle cell disease
Several clinical studies have investigated the effect of different antiplatelet and anticoagulant agents in sickle cell disease. The thromboxane inhibitor aspirin or the ADP receptor antagonist ticlopidine had either modest or no effects on the duration or frequency of acute pain crisis in sickle cell patients (Cabannes, et al 1984, Chaplin, et al 1980, Greenberg, et al 1983, Osamo, et al 1981, Semple, et al 1984, Zago, et al 1984). Recently, a small phase 1 study demonstrated that the platelet inhibitor prasugrel significantly reduced ex vivo platelet reactivity and was well tolerated in sickle cell patients (Jakubowski, et al 2012). The impact of anticoagulation on the frequency or severity of vaso-occlusive events has been investigated using warfarin, acenocoumarol or heparins (Chaplin, et al 1989, Salvaggio, et al 1963, Schnog, et al 2001, Wolters, et al 1995). Most of these studies were not randomized, and were performed on a small number of patients. They demonstrated modest effects at best. Notably, the only adequately powered and placebo-controlled study examining the effect of low molecular weight heparin in sickle cell disease showed a significant reduction in the duration of pain crisis and time of hospitalization (Qari, et al 2007). However it is difficult to say if the protective effects were mediated by anticoagulant properties of low molecular weight heparin, or resulted from the inhibition of P-selectin-mediated cellular interactions (Kutlar, et al 2012, Matsui, et al 2002). Since all of the clinical studies investigating the role of anticoagulation in sickle cell disease focused primarily on the frequency or severity of vaso-occlusive events, it is still unknown if activation of coagulation contributes to the pathology of sickle cell disease or is just a secondary event. Some insight into that question comes from the recent studies using mouse models of sickle cell disease. Guo and co-workers demonstrated that the treatment of Berkley mice with enoxaparin for two weeks not only reduced vascular congestion in the lung but also attenuated endothelial cell injury measured by plasma levels of sVCAM-1 (Guo, et al 2010). The same group showed that a genetic deficiency of TF in non-hematopoietic cells reduces vascular congestion in the livers of sickle cell mice (Hillery, et al 2004). Inhibition of TF or thrombin also attenuated the enhanced thrombosis in cerebral microvessels of mice expressing the sickle form of hemoglobin (Gavins, et al 2011). Importantly, we have demonstrated that TF not only promotes activation of coagulation but also contributes to vascular inflammation in two mouse models of sickle cell disease (Berkley and Townes mice) (Chantrathammachart, et al 2012a). Inhibition of TF reduced leukocytosis and attenuated plasma levels of IL-6, SAP and sVCAM-1. In addition, TF inhibition also reduced expression of MPO and chemokines MCP-1 and KC in the lungs of Berkley mice. Furthermore, TF inhibition had no effect on plasma intravascular hemolysis, indicating that TF contributes to the vascular inflammation downstream of intravascular hemolysis (Chantrathammachart, et al 2012a) (Figure 2). It is still unclear if the TF:FVIIa complex promotes inflammation in sickle cell disease directly or via generation of downstream coagulation proteases. We have shown that endothelial cell-specific deletion of the TF gene reduced plasma levels of IL-6 but not TAT (Chantrathammachart, et al 2012a). This intriguing observation strongly suggests that endothelial cell TF contributes to the expression of IL-6 independent of thrombin generation, most likely via FVIIa– and/or FXa–dependent activation of protease-activated receptor-2 (PAR-2) (Camerer, et al 2000, Rao and Pendurthi 2005). TF/FVIIa–dependent activation of PAR-2 has been shown to promote inflammation in various mouse models (Badeanlou, et al 2011, Redecha, et al 2008). Furthermore, we have observed attenuation of vascular inflammation, including reduction of plasma levels of IL-6, in sickle cell mice lacking PAR-2 in all non-hematopoietic cells (Chantrathammachart, et al 2012b).
Figure 2. Cross-talk between coagulation and vascular inflammation in SCD.
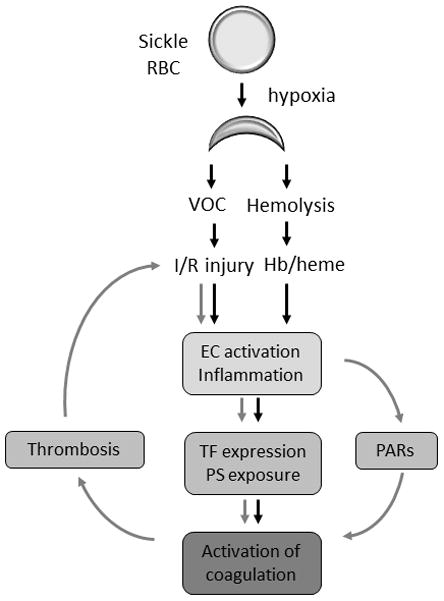
Vaso-occlusion (VOC) and hemolysis-dependent endothelial cell (EC) activation and inflammation lead to the increased expression of tissue factor (TF) by various cell types, phosphatidylserine exposure and subsequent activation of coagulation. We propose that activation of coagulation creates a positive feed-back loop that further enhances VOC, EC activation and inflammation via protease activated receptors (PARs) and thrombosis dependent mechanisms.
Little is known about the contribution of downstream coagulation proteases and fibrinogen to the pathology of sickle cell disease. Inhibition of thrombin abrogated microvascular thrombosis in mice expressing the sickle form of human β-globin. In addition, platelets isolated from these mice exhibited enhanced aggregation after stimulation with thrombin but not ADP (Gavins, et al 2011). PAR-1 is the main thrombin receptor. Injection of a PAR-1 agonist peptide into sickle cell mice increased the expression of endothelial P-selectin and reduced microvascular blood flow by enhancing the adhesion of red blood cells to the endothelium (Embury, et al 2004). Surprisingly, a complete deficiency in fibrinogen was associated with increased mortality in the milder mouse model of SCD (SAD mice) (Roszell, et al 2007).The authors did not investigate the mechanism underlying this observation. However, the pathologic effects of fibrinogen deficiency observed in sickle cell mice may be due to an exacerbation of EC injury caused by increased thrombin signaling. Supporting this hypothesis, sickle cell mice lacking fibrinogen demonstrated increased plasma levels of TAT and sVCAM-1 (Hillery, et al 2004). Increased levels of circulating thrombin are also observed in patients with afibrinogenemia (Dupuy, et al 2001). Further studies are needed to investigate the mechanism by which TF and downstream coagulation proteases, including FXa and thrombin, contribute to vascular inflammation. Possible mechanisms may involve coagulation protease-dependent activation of PARs as well as thrombin-dependent fibrin generation which contributes to local tissue inflammation via activation of leukocytes and promoting infiltration of these cells into inflamed tissues (Figure 3) (Flick, et al 2004a, Flick, et al 2004b, Petzelbauer, et al 2005).
Figure 3.
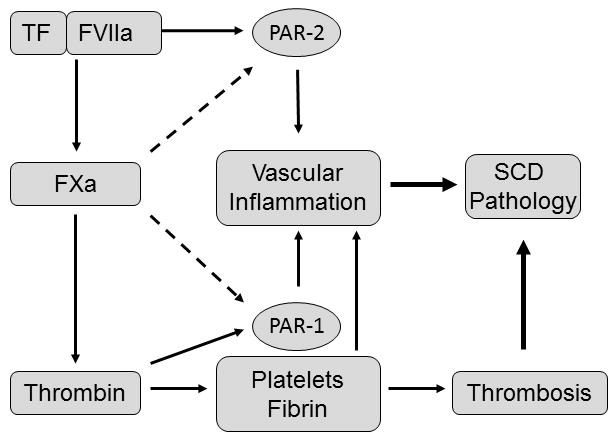
Proposed mechanism by which TF, FXa and thrombin contribute to the pathology of SCD.
Conclusions
Complex interactions between sickle red cells, leukocytes and endothelial cells lead to vaso-occlusive crises, recurring episodes of ischemia-reperfusion injury and chronic hemolysis. It is evident that these processes are highly interdependent, can influence one another and lead to multiple pathologic manifestations of sickle cell disease, including vascular inflammation and activation of coagulation (Belcher, et al 2000, Hidalgo, et al 2009, Wun, et al 2002). Hypercoagulation is a prominent feature of sickle cell disease and leads to the prothrombotic complications in sickle cell patients. Activation of both extrinsic and intrinsic coagulation pathways contributes to the procoagulant state associated with this disease and is mediated by increased TF expression and increased PS exposure. Given the chronic activation of coagulation and our recent data demonstrating cross-talk between coagulation and inflammation in mouse models of sickle cell disease, further and more definitive evaluation of the potential therapeutic benefit of anticoagulants in sickle cell patients is warranted. More studies are necessary to determine the optimal target(s) in the coagulation and PAR-dependent signaling pathways, defined as that which will maximally inhibit coagulation and vascular inflammation while minimally predisposing patients to complications, such as bleeding. These studies should pave the way to the design of future trials examining the effect of anti-coagulant treatment on clinically relevant endpoints in SCD, such as vasoocclusive crisis, venous thromboembolism and pulmonary hypertension. The new oral anticoagulants targeting FXa or thrombin, which have been recently approved for clinical use (Eikelboom and Weitz 2010), provide a potential starting point for these studies.
References
- Adam SS, Key NS, Greenberg CS. D-dimer antigen: current concepts and future prospects. Blood. 2009;113:2878–2887. doi: 10.1182/blood-2008-06-165845. [DOI] [PubMed] [Google Scholar]
- Adams RJ. Big strokes in small persons. Arch Neurol. 2007;64:1567–1574. doi: 10.1001/archneur.64.11.1567. [DOI] [PubMed] [Google Scholar]
- Adedeji MO, Cespedes J, Allen K, Subramony C, Hughson MD. Pulmonary thrombotic arteriopathy in patients with sickle cell disease. Arch Pathol Lab Med. 2001;125:1436–1441. doi: 10.5858/2001-125-1436-PTAIPW. [DOI] [PubMed] [Google Scholar]
- Aleman MM, Gardiner C, Harrison P, Wolberg AS. Differential contributions of monocyte- and platelet-derived microparticles towards thrombin generation and fibrin formation and stability. J Thromb Haemost. 2011;9:2251–2261. doi: 10.1111/j.1538-7836.2011.04488.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan D, Limbrick AR, Thomas P, Westerman MP. Release of spectrin-free spicules on reoxygenation of sickled erythrocytes. Nature. 1982;295:612–613. doi: 10.1038/295612a0. [DOI] [PubMed] [Google Scholar]
- Ataga KI. Hypercoagulability and thrombotic complications in hemolytic anemias. Haematologica. 2009;94:1481–1484. doi: 10.3324/haematol.2009.013672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataga KI, Brittain JE, Desai P, May R, Jones S, Delaney J, Strayhorn D, Hinderliter A, Key NS. Association of coagulation activation with clinical complications in sickle cell disease. PLoS One. 2012;7:e29786. doi: 10.1371/journal.pone.0029786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ataga KI, Key NS. Hypercoagulability in sickle cell disease: new approaches to an old problem. Hematology Am Soc Hematol Educ Program. 2007:91–96. doi: 10.1182/asheducation-2007.1.91. [DOI] [PubMed] [Google Scholar]
- Ataga KI, Moore CG, Hillery CA, Jones S, Whinna HC, Strayhorn D, Sohier C, Hinderliter A, Parise LV, Orringer EP. Coagulation activation and inflammation in sickle cell disease-associated pulmonary hypertension. Haematologica. 2008;93:20–26. doi: 10.3324/haematol.11763. [DOI] [PubMed] [Google Scholar]
- Bach RR. Initiation of coagulation by tissue factor. CRC Crit Rev Biochem. 1988;23:339–368. doi: 10.3109/10409238809082548. [DOI] [PubMed] [Google Scholar]
- Badeanlou L, Furlan-Freguia C, Yang G, Ruf W, Samad F. Tissue factor-protease-activated receptor 2 signaling promotes diet-induced obesity and adipose inflammation. Nat Med. 2011;17:1490–1497. doi: 10.1038/nm.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcher JD, Bryant CJ, Nguyen J, Bowlin PR, Kielbik MC, Bischof JC, Hebbel RP, Vercellotti GM. Transgenic sickle mice have vascular inflammation. Blood. 2003;101:3953–3959. doi: 10.1182/blood-2002-10-3313. [DOI] [PubMed] [Google Scholar]
- Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451–2459. [PubMed] [Google Scholar]
- Beuzard Y. Mouse models of sickle cell disease. Transfus Clin Biol. 2008;15:7–11. doi: 10.1016/j.tracli.2008.04.001. [DOI] [PubMed] [Google Scholar]
- Bradner JE, Mak R, Tanguturi SK, Mazitschek R, Haggarty SJ, Ross K, Chang CY, Bosco J, West N, Morse E, Lin K, Shen JP, Kwiatkowski NP, Gheldof N, Dekker J, DeAngelo DJ, Carr SA, Schreiber SL, Golub TR, Ebert BL. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proc Natl Acad Sci U S A. 2010;107:12617–12622. doi: 10.1073/pnas.1006774107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabannes R, Lonsdorfer J, Castaigne JP, Ondo A, Plassard A, Zohoun I. Clinical and biological double-blind-study of ticlopidine in preventive treatment of sickle-cell disease crises. Agents Actions Suppl. 1984;15:199–212. [PubMed] [Google Scholar]
- Camerer E, Huang W, Coughlin SR. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc Natl Acad Sci U S A. 2000;97:5255–5260. doi: 10.1073/pnas.97.10.5255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus SM, Gausseres B, Bonnin P, Loufrani L, Grimaud L, Charue D, De Moraes JA, Renard JM, Tedgui A, Boulanger CM, Tharaux PL, Blanc-Brude OP. Erythrocyte microparticles can induce kidney vaso-occlusions in a murine model of sickle cell disease. Blood. 2012 doi: 10.1182/blood-2012-02-413138. [DOI] [PubMed] [Google Scholar]
- Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190:255–266. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Chantrathammachart P, Mackman N, Sparkenbaugh E, Wang JG, Parise LV, Kirchhofer D, Key NS, Pawlinski R. Tissue factor promotes activation of coagulation and inflammation in a mouse model of sickle cell disease. Blood. 2012a;120:636–646. doi: 10.1182/blood-2012-04-424143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chantrathammachart P, Sparkenbaugh EM, Mackman N, Key NS, Pawlinski R. Protease Activated Receptor 2 (PAR-2) Promotes Vascular Inflammation in a Mouse Model of Sickle Cell Disease. ASH Annual Meeting Abstracts. 2012b;120:375. [Google Scholar]
- Chaplin H, Jr, Alkjaersig N, Fletcher AP, Michael JM, Joist JH. Aspirin-dipyridamole prophylaxis of sickle cell disease pain crises. Thromb Haemost. 1980;43:218–221. [PubMed] [Google Scholar]
- Chaplin H, Jr, Monroe MC, Malecek AC, Morgan LK, Michael J, Murphy WA. Preliminary trial of minidose heparin prophylaxis for painful sickle cell crises. East Afr Med J. 1989;66:574–584. [PubMed] [Google Scholar]
- Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert SV, McMahon RP, Bonds DR. Effect of hydroxyurea on the frequency of painful crises in sickle cell anemia. Investigators of the Multicenter Study of Hydroxyurea in Sickle Cell Anemia. N Engl J Med. 1995;332:1317–1322. doi: 10.1056/NEJM199505183322001. [DOI] [PubMed] [Google Scholar]
- Chen J, Fu X, Wang Y, Ling M, McMullen B, Kulman J, Chung DW, Lopez JA. Oxidative modification of von Willebrand factor by neutrophil oxidants inhibits its cleavage by ADAMTS13. Blood. 2010;115:706–712. doi: 10.1182/blood-2009-03-213967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Hobbs WE, Le J, Lenting PJ, de Groot PG, Lopez JA. The rate of hemolysis in sickle cell disease correlates with the quantity of active von Willebrand factor in the plasma. Blood. 2011;117:3680–3683. doi: 10.1182/blood-2010-08-302539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colella MP, De Paula EV, Conran N, Machado-Neto JA, Annicchino-Bizzacchi JM, Costa FF, Saad ST, Traina F. Hydroxyurea is associated with reductions in hypercoagulability markers in sickle cell anemia. J Thromb Haemost. 2012;10:1967–1970. doi: 10.1111/j.1538-7836.2012.04861.x. [DOI] [PubMed] [Google Scholar]
- Connor J, Schroit AJ. Transbilayer movement of phosphatidylserine in erythrocytes. Inhibitors of aminophospholipid transport block the association of photolabeled lipid to its transporter. Biochim Biophys Acta. 1991;1066:37–42. doi: 10.1016/0005-2736(91)90247-6. [DOI] [PubMed] [Google Scholar]
- Darbousset R, Thomas GM, Mezouar S, Frere C, Bonier R, Mackman N, Renne T, Dignat-George F, Dubois C, Panicot-Dubois L. Tissue factor-positive neutrophils bind to injured endothelial wall and initiate thrombus formation. Blood. 2012;120:2133–2143. doi: 10.1182/blood-2012-06-437772. [DOI] [PubMed] [Google Scholar]
- de Franceschi L, Baron A, Scarpa A, Adrie C, Janin A, Barbi S, Kister J, Rouyer-Fessard P, Corrocher R, Leboulch P, Beuzard Y. Inhaled nitric oxide protects transgenic SAD mice from sickle cell disease-specific lung injury induced by hypoxia/reoxygenation. Blood. 2003;102:1087–1096. doi: 10.1182/blood-2002-07-2135. [DOI] [PubMed] [Google Scholar]
- De Franceschi L, Cappellini MD, Olivieri O. Thrombosis and sickle cell disease. Semin Thromb Hemost. 2011;37:226–236. doi: 10.1055/s-0031-1273087. [DOI] [PubMed] [Google Scholar]
- Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, Schade AJ, McIntire LV, Fujikawa K, Lopez JA. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. 2002;100:4033–4039. doi: 10.1182/blood-2002-05-1401. [DOI] [PubMed] [Google Scholar]
- Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134:1087–1097. [PMC free article] [PubMed] [Google Scholar]
- Dupuy E, Soria C, Molho P, Zini JM, Rosenstingl S, Laurian C, Bruneval P, Tobelem G. Embolized ischemic lesions of toes in an afibrinogenemic patient: possible relevance to in vivo circulating thrombin. Thromb Res. 2001;102:211–219. doi: 10.1016/s0049-3848(01)00247-x. [DOI] [PubMed] [Google Scholar]
- Edgington TS, Mackman N, Brand K, Ruf W. The structural biology of expression and function of tissue factor. Thromb Haemost. 1991;66:67–79. [PubMed] [Google Scholar]
- Eikelboom JW, Weitz JI. New anticoagulants. Circulation. 2010;121:1523–1532. doi: 10.1161/CIRCULATIONAHA.109.853119. [DOI] [PubMed] [Google Scholar]
- el-Hazmi MA, Warsy AS, Bahakim H. Blood proteins C and S in sickle cell disease. Acta Haematol. 1993;90:114–119. doi: 10.1159/000204390. [DOI] [PubMed] [Google Scholar]
- Embury SH, Matsui NM, Ramanujam S, Mayadas TN, Noguchi CT, Diwan BA, Mohandas N, Cheung AT. The contribution of endothelial cell P-selectin to the microvascular flow of mouse sickle erythrocytes in vivo. Blood. 2004;104:3378–3385. doi: 10.1182/blood-2004-02-0713. [DOI] [PubMed] [Google Scholar]
- Figueiredo RT, Fernandez PL, Mourao-Sa DS, Porto BN, Dutra FF, Alves LS, Oliveira MF, Oliveira PL, Graca-Souza AV, Bozza MT. Characterization of heme as activator of Toll-like receptor 4. The Journal of Biological Chemistry. 2007;282:20221–20229. doi: 10.1074/jbc.M610737200. [DOI] [PubMed] [Google Scholar]
- Fleck RA, Rao LV, Rapaport SI, Varki N. Localization of human tissue factor antigen by immunostaining with monospecific, polyclonal anti-human tissue factor antibody. Thromb Res. 1990;59:421–437. doi: 10.1016/0049-3848(90)90148-6. [DOI] [PubMed] [Google Scholar]
- Flick MJ, Du X, Degen JL. Fibrin(ogen)-alpha M beta 2 interactions regulate leukocyte function and innate immunity in vivo. Exp Biol Med (Maywood) 2004a;229:1105–1110. doi: 10.1177/153537020422901104. [DOI] [PubMed] [Google Scholar]
- Flick MJ, Du X, Witte DP, Jirouskova M, Soloviev DA, Busuttil SJ, Plow EF, Degen JL. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. 2004b;113:1596–1606. doi: 10.1172/JCI20741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis RB., Jr Protein S deficiency in sickle cell anemia. J Lab Clin Med. 1988;111:571–576. [PubMed] [Google Scholar]
- Francis RB., Jr Platelets, coagulation, and fibrinolysis in sickle cell disease: their possible role in vascular occlusion. Blood Coagul Fibrinolysis. 1991;2:341–353. doi: 10.1097/00001721-199104000-00018. [DOI] [PubMed] [Google Scholar]
- Franck PF, Bevers EM, Lubin BH, Comfurius P, Chiu DT, Op den Kamp JA, Zwaal RF, van Deenen LL, Roelofsen B. Uncoupling of the membrane skeleton from the lipid bilayer. The cause of accelerated phospholipid flip-flop leading to an enhanced procoagulant activity of sickled cells. J Clin Invest. 1985;75:183–190. doi: 10.1172/JCI111672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol. 2002;9:101–106. doi: 10.1097/00062752-200203000-00003. [DOI] [PubMed] [Google Scholar]
- Frenette PS, Atweh GF. Sickle cell disease: old discoveries, new concepts, and future promise. J Clin Invest. 2007;117:850–858. doi: 10.1172/JCI30920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido VT, Proenca-Ferreira R, Dominical VM, Traina F, Bezerra MA, de Mello MR, Colella MP, Araujo AS, Saad ST, Costa FF, Conran N. Elevated plasma levels and platelet-associated expression of the pro-thrombotic and pro-inflammatory protein, TNFSF14 (LIGHT), in sickle cell disease. Br J Haematol. 2012;158:788–797. doi: 10.1111/j.1365-2141.2012.09218.x. [DOI] [PubMed] [Google Scholar]
- Gavins FN, Russell J, Senchenkova EL, De Almeida Paula L, Damazo AS, Esmon CT, Kirchhofer D, Hebbel RP, Granger DN. Mechanisms of enhanced thrombus formation in cerebral microvessels of mice expressing hemoglobin-S. Blood. 2011;117:4125–4133. doi: 10.1182/blood-2010-08-301366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee BE, Platt OS. Sickle reticulocytes adhere to VCAM-1. Blood. 1995;85:268–274. [PubMed] [Google Scholar]
- Gerotziafas GT, Van Dreden P, Chaari M, Galea V, Khaterchi A, Lionnet F, Stankovic-Stojanovic K, Blanc-Brude O, Woodhams B, Maier-Redelsperger M, Girot R, Hatmi M, Elalamy I. The acceleration of the propagation phase of thrombin generation in patients with steady-state sickle cell disease is associated with circulating erythrocyte-derived microparticles. Thromb Haemost. 2012;107:1044–1052. doi: 10.1160/TH11-10-0689. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Adisa O, Yang Y, Tan F, Ofori-Acquah SF. Toll-Like Receptor 4 Mediates Heme Induced Acute Lung Injury: Preclinical Study of Resatorvid in Sickle Cell Disease. Blood. 2011;118:926–926. [Google Scholar]
- Ghosh S, Tan F, Ofori-Acquah SF. Spatiotemporal dysfunction of the vascular permeability barrier in transgenic mice with sickle cell disease. Anemia. 2012;2012:582018. doi: 10.1155/2012/582018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green D, Scott JP. Is sickle cell crisis a thrombotic event? Am J Hematol. 1986;23:317–321. doi: 10.1002/ajh.2830230403. [DOI] [PubMed] [Google Scholar]
- Greenberg J, Ohene-Frempong K, Halus J, Way C, Schwartz E. Trial of low doses of aspirin as prophylaxis in sickle cell disease. J Pediatr. 1983;102:781–784. doi: 10.1016/s0022-3476(83)80258-3. [DOI] [PubMed] [Google Scholar]
- Guo Y, Uy T, Wandersee N, Scott JP, Weiler H, Holzhauer S, Retherford D, Foster T, Hillery C. The Protein C Pathway in Human and Murine Sickle Cell Disease: Alterations in Protein C, Thrombomodulin (TM), and Endothelial Protein C Receptor (EPCR) at Baseline and during Acute Vaso-Occlusion. Blood. 2008;112:202–202. [Google Scholar]
- Guo YHH, Field J, Foster TD, Scott JP, Wandersee N, Hillery CA. Low Molecular Weight Heparin Reduces sVCAM-1 and Lung Congestion In a Murine Model of Sickle Cell Disease. Blood. 2010;116:686–687. [Google Scholar]
- Gutsaeva DR, Parkerson JB, Yerigenahally SD, Kurz JC, Schaub RG, Ikuta T, Head CA. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential therapeutic agent for sickle cell disease. Blood. 2011;117:727–735. doi: 10.1182/blood-2010-05-285718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbel RP, Osarogiagbon R, Kaul D. The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation. 2004;11:129–151. [PubMed] [Google Scholar]
- Hebbel RP, Vercellotti G, Nath KA. A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi-modality chemo-prophylaxsis. Cardiovasc Hematol Disord Drug Targets. 2009;9:271–292. doi: 10.2174/1871529x10909040271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebbel RP, Vercellotti GM, Pace BS, Solovey AN, Kollander R, Abanonu CF, Nguyen J, Vineyard JV, Belcher JD, Abdulla F, Osifuye S, Eaton JW, Kelm RJ, Jr, Slungaard A. The HDAC inhibitors trichostatin A and suberoylanilide hydroxamic acid exhibit multiple modalities of benefit for the vascular pathobiology of sickle transgenic mice. Blood. 2010;115:2483–2490. doi: 10.1182/blood-2009-02-204990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo A, Chang J, Jang JE, Peired AJ, Chiang EY, Frenette PS. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nat Med. 2009;15:384–391. doi: 10.1038/nm.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillery CA, Foster TD, Holzhauer SL, Scott JP, Panepinto JA, Mohandas N, Mackman N, Wandersee NJ. Tissue factor deficiency decreases sickle cell-induced vascular stasis in a hematopoietic stem cell transplant model of murine sickle cell disease. Blood. 2004;104:71a–71a. [Google Scholar]
- Hillery CA, Panepinto JA. Pathophysiology of stroke in sickle cell disease. Microcirculation. 2004;11:195–208. doi: 10.1080/10739680490278600. [DOI] [PubMed] [Google Scholar]
- Hofstra TC, Kalra VK, Meiselman HJ, Coates TD. Sickle erythrocytes adhere to polymorphonuclear neutrophils and activate the neutrophil respiratory burst. Blood. 1996;87:4440–4447. [PubMed] [Google Scholar]
- Hoppe C, Kuypers F, Larkin S, Hagar W, Vichinsky E, Styles L. A pilot study of the short-term use of simvastatin in sickle cell disease: effects on markers of vascular dysfunction. Br J Haematol. 2011;153:655–663. doi: 10.1111/j.1365-2141.2010.08480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski JA, Zhou C, Small DS, Winters KJ, Lachno DR, Frelinger AL, 3rd, Howard J, Mant TG, Jurcevic S, Payne CD. A Phase 1 Study of Prasugrel in Patients with Sickle Cell Disease: Pharmacokinetics and Effects on ex Vivo Platelet Reactivity. Br J Clin Pharmacol. 2012 doi: 10.1111/bcp.12042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James AH, Jamison MG, Brancazio LR, Myers ER. Venous thromboembolism during pregnancy and the postpartum period: incidence, risk factors, and mortality. Am J Obstet Gynecol. 2006;194:1311–1315. doi: 10.1016/j.ajog.2005.11.008. [DOI] [PubMed] [Google Scholar]
- Joneckis CC, Ackley RL, Orringer EP, Wayner EA, Parise LV. Integrin alpha 4 beta 1 and glycoprotein IV (CD36) are expressed on circulating reticulocytes in sickle cell anemia. Blood. 1993;82:3548–3555. [PubMed] [Google Scholar]
- Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood reviews. 2007;21:37–47. doi: 10.1016/j.blre.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny MW, George AJ, Stuart J. Platelet hyperactivity in sickle-cell disease: a consequence of hyposplenism. J Clin Pathol. 1980;33:622–625. doi: 10.1136/jcp.33.7.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Key NS, Slungaard A, Dandelet L, Nelson SC, Moertel C, Styles LA, Kuypers FA, Bach RR. Whole blood tissue factor procoagulant activity is elevated in patients with sickle cell disease. Blood. 1998;91:4216–4223. [PubMed] [Google Scholar]
- Kirkpatrick MB, Haynes J. Sickle-Cell Disease and the Pulmonary Circulation. Seminars in Respiratory and Critical Care Medicine. 1994;15:473–481. [Google Scholar]
- Kollander R, Solovey A, Milbauer LC, Abdulla F, Kelm RJ, Jr, Hebbel RP. Nuclear factor-kappa B (NFkappaB) component p50 in blood mononuclear cells regulates endothelial tissue factor expression in sickle transgenic mice: implications for the coagulopathy of sickle cell disease. Transl Res. 2010;155:170–177. doi: 10.1016/j.trsl.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan S, Siegel J, Pullen G, Jr, Hevelow M, Dampier C, Stuart M. Increased von Willebrand factor antigen and high molecular weight multimers in sickle cell disease associated with nocturnal hypoxemia. Thromb Res. 2008;122:455–458. doi: 10.1016/j.thromres.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutlar A, Ataga KI, McMahon L, Howard J, Galacteros F, Hagar W, Vichinsky E, Cheung AT, Matsui N, Embury SH. A potent oral P-selectin blocking agent improves microcirculatory blood flow and a marker of endothelial cell injury in patients with sickle cell disease. Am J Hematol. 2012;87:536–539. doi: 10.1002/ajh.23147. [DOI] [PubMed] [Google Scholar]
- Lard LR, Mul FP, de Haas M, Roos D, Duits AJ. Neutrophil activation in sickle cell disease. J Leukoc Biol. 1999;66:411–415. doi: 10.1002/jlb.66.3.411. [DOI] [PubMed] [Google Scholar]
- Lee SP, Ataga KI, Orringer EP, Phillips DR, Parise LV. Biologically active CD40 ligand is elevated in sickle cell anemia: potential role for platelet-mediated inflammation. Arterioscler Thromb Vasc Biol. 2006;26:1626–1631. doi: 10.1161/01.ATV.0000220374.00602.a2. [DOI] [PubMed] [Google Scholar]
- Leslie J, Langler D, Serjeant GR, Serjeant BE, Desai P, Gordon YB. Coagulation changes during the steady state in homozygous sickle-cell disease in Jamaica. Br J Haematol. 1975;30:159–166. doi: 10.1111/j.1365-2141.1975.tb00530.x. [DOI] [PubMed] [Google Scholar]
- Lindmark E, Tenno T, Siegbahn A. Role of platelet P-selectin and CD40 ligand in the induction of monocytic tissue factor expression. Arterioscler Thromb Vasc Biol. 2000;20:2322–2328. doi: 10.1161/01.atv.20.10.2322. [DOI] [PubMed] [Google Scholar]
- Luo W, Campbell A, Wang H, Guo C, Bradley K, Wang J, Eitzman DT. P-selectin glycoprotein ligand-1 inhibition blocks increased leukocyte-endothelial interactions associated with sickle cell disease in mice. Blood. 2012;120:3862–3864. doi: 10.1182/blood-2012-07-444455. [DOI] [PubMed] [Google Scholar]
- Mackman N. New insights into the mechanisms of venous thrombosis. J Clin Invest. 2012;122:2331–2336. doi: 10.1172/JCI60229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol. 2007;27:1687–1693. doi: 10.1161/ATVBAHA.107.141911. [DOI] [PubMed] [Google Scholar]
- Matsui NM, Varki A, Embury SH. Heparin inhibits the flow adhesion of sickle red blood cells to P-selectin. Blood. 2002;100:3790–3796. doi: 10.1182/blood-2002-02-0626. [DOI] [PubMed] [Google Scholar]
- Mekontso Dessap A, Deux JF, Abidi N, Lavenu-Bombled C, Melica G, Renaud B, Godeau B, Adnot S, Brochard L, Brun-Buisson C, Galacteros F, Rahmouni A, Habibi A, Maitre B. Pulmonary artery thrombosis during acute chest syndrome in sickle cell disease. Am J Respir Crit Care Med. 2011;184:1022–1029. doi: 10.1164/rccm.201105-0783OC. [DOI] [PubMed] [Google Scholar]
- Mohan JS, Lip GY, Wright J, Bareford D, Blann AD. Plasma levels of tissue factor and soluble E-selectin in sickle cell disease: relationship to genotype and to inflammation. Blood coagulation & fibrinolysis: an international journal in haemostasis and thrombosis. 2005;16:209–214. doi: 10.1097/01.mbc.0000164431.98169.8f. [DOI] [PubMed] [Google Scholar]
- Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris SM, Jr, Gladwin MT. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA: the journal of the American Medical Association. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-Eberhard U, Javid J, Liem HH, Hanstein A, Hanna M. Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood. 1968;32:811–815. [PubMed] [Google Scholar]
- Ng PC, Ashari L. Portal vein thrombosis following laparoscopic surgery in a patient with sickle cell disease. Surg Endosc. 2003;17:831. doi: 10.1007/s00464-002-4529-2. [DOI] [PubMed] [Google Scholar]
- Noubouossie DF, Le PQ, Corazza F, Debaugnies F, Rozen L, Ferster A, Demulder A. Thrombin generation reveals high procoagulant potential in the plasma of sickle cell disease children. Am J Hematol. 2011 doi: 10.1002/ajh.22206. [DOI] [PubMed] [Google Scholar]
- Novelli EM, Huynh C, Gladwin MT, Moore CG, Ragni MV. Pulmonary embolism in sickle cell disease: a case-control study. J Thromb Haemost. 2012;10:760–766. doi: 10.1111/j.1538-7836.2012.04697.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppenheimer EH, Esterly JR. Pulmonary changes in sickle cell disease. Am Rev Respir Dis. 1971;103:858–859. doi: 10.1164/arrd.1971.103.6.858. [DOI] [PubMed] [Google Scholar]
- Osamo NO, Photiades DP, Famodu AA. Therapeutic effect of aspirin in sickle cell anaemia. Acta Haematol. 1981;66:102–107. doi: 10.1159/000207105. [DOI] [PubMed] [Google Scholar]
- Owens AP, 3rd, Mackman N. Microparticles in hemostasis and thrombosis. Circ Res. 2011;108:1284–1297. doi: 10.1161/CIRCRESAHA.110.233056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszty C, Brion CM, Manci E, Witkowska HE, Stevens ME, Mohandas N, Rubin EM. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science. 1997;278:876–878. doi: 10.1126/science.278.5339.876. [DOI] [PubMed] [Google Scholar]
- Pathare A, Al Kindi S, Alnaqdy AA, Daar S, Knox-Macaulay H, Dennison D. Cytokine profile of sickle cell disease in Oman. Am J Hematol. 2004;77:323–328. doi: 10.1002/ajh.20196. [DOI] [PubMed] [Google Scholar]
- Pawlinski R, Pedersen B, Erlich J, Mackman N. Role of tissue factor in haemostasis, thrombosis, angiogenesis and inflammation: lessons from low tissue factor mice. Thromb Haemost. 2004;92:444–450. doi: 10.1160/TH04-05-0309. [DOI] [PubMed] [Google Scholar]
- Pawlinski R, Wang JG, Owens AP, 3rd, Williams J, Antoniak S, Tencati M, Luther T, Rowley JW, Low EN, Weyrich AS, Mackman N. Hematopoietic and nonhematopoietic cell tissue factor activates the coagulation cascade in endotoxemic mice. Blood. 2010;116:806–814. doi: 10.1182/blood-2009-12-259267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M, Plaat BE, ten Cate H, Wolters HJ, Weening RS, Brandjes DP. Enhanced thrombin generation in children with sickle cell disease. Thromb Haemost. 1994;71:169–172. [PubMed] [Google Scholar]
- Petzelbauer P, Zacharowski PA, Miyazaki Y, Friedl P, Wickenhauser G, Castellino FJ, Groger M, Wolff K, Zacharowski K. The fibrin-derived peptide Bbeta15–42 protects the myocardium against ischemia-reperfusion injury. Nature Medicine. 2005;11:298–304. doi: 10.1038/nm1198. [DOI] [PubMed] [Google Scholar]
- Polanowska-Grabowska R, Wallace K, Field JJ, Chen L, Marshall MA, Figler R, Gear AR, Linden J. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arterioscler Thromb Vasc Biol. 2010;30:2392–2399. doi: 10.1161/ATVBAHA.110.211615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porto BN, Alves LS, Fernandez PL, Dutra TP, Figueiredo RT, Graca-Souza AV, Bozza MT. Heme induces neutrophil migration and reactive oxygen species generation through signaling pathways characteristic of chemotactic receptors. J Biol Chem. 2007;282:24430–24436. doi: 10.1074/jbc.M703570200. [DOI] [PubMed] [Google Scholar]
- Prengler M, Pavlakis SG, Prohovnik I, Adams RJ. Sickle cell disease: the neurological complications. Ann Neurol. 2002;51:543–552. doi: 10.1002/ana.10192. [DOI] [PubMed] [Google Scholar]
- Proenca-Ferreira R, Franco-Penteado CF, Traina F, Saad ST, Costa FF, Conran N. Increased adhesive properties of platelets in sickle cell disease: roles for alphaIIb beta3-mediated ligand binding, diminished cAMP signalling and increased phosphodiesterase 3A activity. Br J Haematol. 2010;149:280–288. doi: 10.1111/j.1365-2141.2010.08087.x. [DOI] [PubMed] [Google Scholar]
- Qari MH, Aljaouni SK, Alardawi MS, Fatani H, Alsayes FM, Zografos P, Alsaigh M, Alalfi A, Alamin M, Gadi A, Mousa SA. Reduction of painful vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a double-blind randomized trial. Thromb Haemost. 2007;98:392–396. [PubMed] [Google Scholar]
- Qari MH, Dier U, Mousa SA. Biomarkers of inflammation, growth factor, and coagulation activation in patients with sickle cell disease. Clin Appl Thromb Hemost. 2012;18:195–200. doi: 10.1177/1076029611420992. [DOI] [PubMed] [Google Scholar]
- Rao LV, Pendurthi UR. Tissue factor-factor VIIa signaling. Arterioscler Thromb Vasc Biol. 2005;25:47–56. doi: 10.1161/01.ATV.0000151624.45775.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redecha P, Franzke CW, Ruf W, Mackman N, Girardi G. Neutrophil activation by the tissue factor/Factor VIIa/PAR2 axis mediates fetal death in a mouse model of antiphospholipid syndrome. J Clin Invest. 2008;118:3453–3461. doi: 10.1172/JCI36089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DC, Williams TN, Gladwin MT. Sickle-cell disease. Lancet. 2010;376:2018–2031. doi: 10.1016/S0140-6736(10)61029-X. [DOI] [PubMed] [Google Scholar]
- Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, 3rd, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Medicine. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- Roszell NJ, Danton MJ, Jiang M, Witte D, Daugherty C, Grimes T, Girdler B, Anderson KP, Franco RS, Degen JL, Joiner CH. Fibrinogen deficiency, but not plasminogen deficiency, increases mortality synergistically in combination with sickle hemoglobin SAD in transgenic mice. Am J Hematol. 2007;82:1044–1048. doi: 10.1002/ajh.20982. [DOI] [PubMed] [Google Scholar]
- Ruggeri ZM. The role of von Willebrand factor in thrombus formation. Thromb Res. 2007;120(Suppl 1):S5–9. doi: 10.1016/j.thromres.2007.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvaggio JE, Arnold CA, Banov CH. Long-term anti-coagulation in sickle-cell disease. A clinical study. N Engl J Med. 1963;269:182–186. doi: 10.1056/NEJM196307252690403. [DOI] [PubMed] [Google Scholar]
- Sangerman J, Lee MS, Yao X, Oteng E, Hsiao CH, Li W, Zein S, Ofori-Acquah SF, Pace BS. Mechanism for fetal hemoglobin induction by histone deacetylase inhibitors involves gamma-globin activation by CREB1 and ATF-2. Blood. 2006;108:3590–3599. doi: 10.1182/blood-2006-01-023713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnog JB, Kater AP, Mac Gillavry MR, Duits AJ, Lard LR, van Der Dijs FP, Brandjes DP, ten Cate H, van Eps LW, Rojer RA. Low adjusted-dose acenocoumarol therapy in sickle cell disease: a pilot study. Am J Hematol. 2001;68:179–183. doi: 10.1002/ajh.1175. [DOI] [PubMed] [Google Scholar]
- Semple MJ, Al-Hasani SF, Kioy P, Savidge GF. A double-blind trial of ticlopidine in sickle cell disease. Thromb Haemost. 1984;51:303–306. [PubMed] [Google Scholar]
- Setty BN, Betal SG, Zhang J, Stuart MJ. Heme induces endothelial tissue factor expression: potential role in hemostatic activation in patients with hemolytic anemia. J Thromb Haemost. 2008;6:2202–2209. doi: 10.1111/j.1538-7836.2008.03177.x. [DOI] [PubMed] [Google Scholar]
- Setty BN, Key NS, Rao AK, Gayen-Betal S, Krishnan S, Dampier CD, Stuart MJ. Tissue factor-positive monocytes in children with sickle cell disease: correlation with biomarkers of haemolysis. Br J Haematol. 2012 doi: 10.1111/j.1365-2141.2012.09065.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setty BN, Kulkarni S, Rao AK, Stuart MJ. Fetal hemoglobin in sickle cell disease: relationship to erythrocyte phosphatidylserine exposure and coagulation activation. Blood. 2000;96:1119–1124. [PubMed] [Google Scholar]
- Setty BN, Rao AK, Stuart MJ. Thrombophilia in sickle cell disease: the red cell connection. Blood. 2001;98:3228–3233. doi: 10.1182/blood.v98.12.3228. [DOI] [PubMed] [Google Scholar]
- Shet AS, Aras O, Gupta K, Hass MJ, Rausch DJ, Saba N, Koopmeiners L, Key NS, Hebbel RP. Sickle blood contains tissue factor-positive microparticles derived from endothelial cells and monocytes. Blood. 2003;102:2678–2683. doi: 10.1182/blood-2003-03-0693. [DOI] [PubMed] [Google Scholar]
- Singer ST, Ataga KI. Hypercoagulability in sickle cell disease and beta-thalassemia. Curr Mol Med. 2008;8:639–645. doi: 10.2174/156652408786241366. [DOI] [PubMed] [Google Scholar]
- Singh B, Kuruba M, Singh P, Hernandez CM, Waseemuddin M, Nanda NC. Live/Real time three-dimensional transthoracic echocardiographic assessment of inferior vena cava and hepatic vein thrombosis in sickle cell disease. Echocardiography. 2010;27:594–596. doi: 10.1111/j.1540-8175.2010.01201.x. [DOI] [PubMed] [Google Scholar]
- Solovey A, Gui L, Key NS, Hebbel RP. Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest. 1998;101:1899–1904. doi: 10.1172/JCI1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solovey A, Kollander R, Milbauer LC, Abdulla F, Chen Y, Kelm RJ, Jr, Hebbel RP. Endothelial nitric oxide synthase and nitric oxide regulate endothelial tissue factor expression in vivo in the sickle transgenic mouse. Am J Hematol. 2010;85:41–45. doi: 10.1002/ajh.21582. [DOI] [PubMed] [Google Scholar]
- Solovey A, Kollander R, Shet A, Milbauer LC, Choong S, Panoskaltsis-Mortari A, Blazar BR, Kelm RJ, Jr, Hebbel RP. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood. 2004;104:840–846. doi: 10.1182/blood-2003-10-3719. [DOI] [PubMed] [Google Scholar]
- Sparkenbaugh EM, Chantrathammachart P, Mackman N, Key NS, Pawlinski R. Heme Induces Systemic Activation of Coagulation in Vivo in a Tissue Factor-Dependent Manner. ASH Annual Meeting Abstracts. 2012;120:820. [Google Scholar]
- Stein PD, Beemath A, Meyers FA, Skaf E, Olson RE. Deep venous thrombosis and pulmonary embolism in hospitalized patients with sickle cell disease. Am J Med. 2006;119:897, e897–811. doi: 10.1016/j.amjmed.2006.08.015. [DOI] [PubMed] [Google Scholar]
- Steinberg MH, Barton F, Castro O, Pegelow CH, Ballas SK, Kutlar A, Orringer E, Bellevue R, Olivieri N, Eckman J, Varma M, Ramirez G, Adler B, Smith W, Carlos T, Ataga K, DeCastro L, Bigelow C, Saunthararajah Y, Telfer M, Vichinsky E, Claster S, Shurin S, Bridges K, Waclawiw M, Bonds D, Terrin M. Effect of hydroxyurea on mortality and morbidity in adult sickle cell anemia: risks and benefits up to 9 years of treatment. JAMA. 2003;289:1645–1651. doi: 10.1001/jama.289.13.1645. [DOI] [PubMed] [Google Scholar]
- Stuart MJ, Setty BN. Hemostatic alterations in sickle cell disease: relationships to disease pathophysiology. Pediatr Pathol Mol Med. 2001;20:27–46. [PubMed] [Google Scholar]
- Tam DA. Protein C and protein S activity in sickle cell disease and stroke. J Child Neurol. 1997;12:19–21. doi: 10.1177/088307389701200103. [DOI] [PubMed] [Google Scholar]
- Tomer A, Harker LA, Kasey S, Eckman JR. Thrombogenesis in sickle cell disease. J Lab Clin Med. 2001a;137:398–407. doi: 10.1067/mlc.2001.115450. [DOI] [PubMed] [Google Scholar]
- Tomer A, Kasey S, Connor WE, Clark S, Harker LA, Eckman JR. Reduction of pain episodes and prothrombotic activity in sickle cell disease by dietary n-3 fatty acids. Thromb Haemost. 2001b;85:966–974. [PubMed] [Google Scholar]
- Trudel M, De Paepe ME, Chretien N, Saadane N, Jacmain J, Sorette M, Hoang T, Beuzard Y. Sickle cell disease of transgenic SAD mice. Blood. 1994;84:3189–3197. [PubMed] [Google Scholar]
- Turhan A, Weiss LA, Mohandas N, Coller BS, Frenette PS. Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proc Natl Acad Sci U S A. 2002;99:3047–3051. doi: 10.1073/pnas.052522799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beers EJ, Schaap MC, Berckmans RJ, Nieuwland R, Sturk A, van Doormaal FF, Meijers JC, Biemond BJ Cs group. Circulating erythrocyte-derived microparticles are associated with coagulation activation in sickle cell disease. Haematologica. 2009;94:1513–1519. doi: 10.3324/haematol.2009.008938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beers EJ, Spronk HM, Ten Cate H, Duits AJ, Brandjes DP, van Esser JW, Biemond BJ, Schnog JB. No association of the hypercoagulable state with sickle cell disease related pulmonary hypertension. Haematologica. 2008;93:e42–44. doi: 10.3324/haematol.12632. [DOI] [PubMed] [Google Scholar]
- Van Der Meijden PE, Van Schilfgaarde M, Van Oerle R, Renne T, ten Cate H, Spronk HM. Platelet- and erythrocyte-derived microparticles trigger thrombin generation via factor XIIa. J Thromb Haemost. 2012;10:1355–1362. doi: 10.1111/j.1538-7836.2012.04758.x. [DOI] [PubMed] [Google Scholar]
- Vichinsky EP, Styles LA, Colangelo LH, Wright EC, Castro O, Nickerson B. Acute chest syndrome in sickle cell disease: clinical presentation and course. Cooperative Study of Sickle Cell Disease. Blood. 1997;89:1787–1792. [PubMed] [Google Scholar]
- Wagener FA, Eggert A, Boerman OC, Oyen WJ, Verhofstad A, Abraham NG, Adema G, van Kooyk Y, de Witte T, Figdor CG. Heme is a potent inducer of inflammation in mice and is counteracted by heme oxygenase. Blood. 2001;98:1802–1811. doi: 10.1182/blood.v98.6.1802. [DOI] [PubMed] [Google Scholar]
- Wagener FA, Feldman E, de Witte T, Abraham NG. Heme induces the expression of adhesion molecules ICAM-1, VCAM-1, and E selectin in vascular endothelial cells. Proceedings of the Society for Experimental Biology and Medicine. Society for Experimental Biology and Medicine. 1997;216:456–463. doi: 10.3181/00379727-216-44197. [DOI] [PubMed] [Google Scholar]
- Wallace KL, Marshall MA, Ramos SI, Lannigan JA, Field JJ, Strieter RM, Linden J. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. 2009;114:667–676. doi: 10.1182/blood-2009-02-205492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mahmud SA, Bitterman PB, Huo Y, Slungaard A. Histone deacetylase inhibitors suppress TF-kappaB-dependent agonist-driven tissue factor expression in endothelial cells and monocytes. J Biol Chem. 2007;282:28408–28418. doi: 10.1074/jbc.M703586200. [DOI] [PubMed] [Google Scholar]
- Westerman MP, Green D, Gilman-Sachs A, Beaman K, Freels S, Boggio L, Allen S, Zuckerman L, Schlegel R, Williamson P. Antiphospholipid antibodies, proteins C and S, and coagulation changes in sickle cell disease. J Lab Clin Med. 1999;134:352–362. doi: 10.1016/s0022-2143(99)90149-x. [DOI] [PubMed] [Google Scholar]
- Westwick J, Watson-Williams EJ, Krishnamurthi S, Marks G, Ellis V, Scully MF, White JM, Kakkar VV. Platelet activation during steady state sickle cell disease. J Med. 1983;14:17–36. [PubMed] [Google Scholar]
- Wick TM, Moake JL, Udden MM, Eskin SG, Sears DA, McIntire LV. Unusually large von Willebrand factor multimers increase adhesion of sickle erythrocytes to human endothelial cells under controlled flow. J Clin Invest. 1987;80:905–910. doi: 10.1172/JCI113151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolters HJ, ten Cate H, Thomas LL, Brandjes DP, van der Ende A, van der Heiden Y, Statius van Eps LW. Low-intensity oral anticoagulation in sickle-cell disease reverses the prethrombotic state: promises for treatment? Br J Haematol. 1995;90:715–717. doi: 10.1111/j.1365-2141.1995.tb05607.x. [DOI] [PubMed] [Google Scholar]
- Wu LC, Sun CW, Ryan TM, Pawlik KM, Ren J, Townes TM. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood. 2006;108:1183–1188. doi: 10.1182/blood-2006-02-004812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wun T, Cordoba M, Rangaswami A, Cheung AW, Paglieroni T. Activated monocytes and platelet-monocyte aggregates in patients with sickle cell disease. Clin Lab Haematol. 2002;24:81–88. doi: 10.1046/j.1365-2257.2002.00433.x. [DOI] [PubMed] [Google Scholar]
- Wun T, Paglieroni T, Rangaswami A, Franklin PH, Welborn J, Cheung A, Tablin F. Platelet activation in patients with sickle cell disease. Br J Haematol. 1998;100:741–749. doi: 10.1046/j.1365-2141.1998.00627.x. [DOI] [PubMed] [Google Scholar]
- Zago MA, Costa FF, Ismael SJ, Tone LG, Bottura C. Treatment of sickle cell diseases with aspirin. Acta Haematol. 1984;72:61–64. doi: 10.1159/000206360. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Han H, Cruz MA, Lopez JA, Dong JF, Guchhait P. Haemoglobin blocks von Willebrand factor proteolysis by ADAMTS-13: a mechanism associated with sickle cell disease. Thromb Haemost. 2009;101:1070–1077. [PubMed] [Google Scholar]