

Abstract
DNA transposons are primitive genetic elements which have colonized living organisms from plants to bacteria and mammals. Through evolution such parasitic elements have shaped their host genomes by replicating and relocating between chromosomal loci in processes catalyzed by the transposase proteins encoded by the elements themselves. DNA transposable elements are constantly adapting to life in the genome, and self-suppressive regulation as well as defensive host mechanisms may assist in buffering ‘cut-and-paste’ DNA mobilization until accumulating mutations will eventually restrict events of transposition. With the reconstructed Sleeping Beauty DNA transposon as a powerful engine, a growing list of transposable elements with activity in human cells have moved into biomedical experimentation and preclinical therapy as versatile vehicles for delivery and genomic insertion of transgenes. In this review, we aim to link the mechanisms that drive transposon evolution with the realities and potential challenges we are facing when adapting DNA transposons for gene transfer. We argue that DNA transposon-derived vectors may carry inherent, and potentially limiting, traits of their mother elements. By understanding in detail the evolutionary journey of transposons, from host colonization to element multiplication and inactivation, we may better exploit the potential of distinct transposable elements. Hence, parallel efforts to investigate and develop distinct, but potent, transposon-based vector systems will benefit the broad applications of gene transfer. Insight and clever optimization have shaped new DNA transposon vectors, which recently debuted in the first DNA transposon-based clinical trial. Learning from an evolutionary drive may help us create gene vehicles that are safer, more efficient, and less prone for suppression and inactivation.
Review
Introduction
Since its birth the field of therapeutic gene transfer has travelled on a rough road from initial optimism through hype and disappointment towards scientifically well-founded hope and signs of clinical applicability and success. Indeed, incipient confidence is fuelled by promising preclinical and clinical findings and by an increasing wealth of improved gene transfer technologies. Continuous focus on gene carriers and their biological properties, immune responses, and interactions with host cells already has provided clinical benefit [1-3] and will continue to do so. As one of many recent examples, our improved understanding of determinants that control viral gene insertion – being the result of the combined efforts of virologists and gene therapists - not only explains us why integrating viral vectors tend to insert near and deregulate cellular genes but is likely also to pave the way for vector systems with altered integration profiles.
With the revived Sleeping Beauty DNA transposon in the driver’s seat, transposable DNA elements have emerged as promising nonviral vehicles for persistent gene delivery [4,5]. The simple gene integration machinery of cut-and-paste transposons provides non-viral gene delivery systems – naked plasmid-based DNA vectors as the most primitive system – with the ability to insert transgenes into target cell genomes. Nonviral vector types that have traditionally suffered from short-term expression of transgenes now facilitate long-term therapeutic levels of gene expression due to active genomic insertion of the transgene. Potency of transposon-based vectors in preclinical settings has been demonstrated in liver [5-10], lung [11-16], skin [17], and brain [18,19] of adult mice injected with plasmid DNA. Packaged in synthetic wrappings consisting of cationic polylysines or hydrophobic lipids, transposon vectors may eventually mimic integrating viral vectors, but in a format that is potentially less toxic and less immunogenic than viral vectors and which is well-suited for large-scale vector production.
Mobilizing nucleic acids is the key to every application of gene transfer. This is why gene therapists look for inspiration in families of viruses which have through evolution adapted to transport genetic cargo and in some cases to pick up and mobilize cellular genes. In case of transposable elements, with or without viral origin, mammalian genomes provide the inspiration. The mobility of nucleic acid sequences in discrete genetic segments has incredible impact on the dynamics and evolution of almost any genome. Driving their survival, transposable elements possess a unique ability to multiply within genomes, leading to an abundance of genetic entities with no obvious beneficial effects on the host. These small mobile units are genetic parasites, which have adapted to the gene expression machinery of the host and each typically encodes one or two proteins that catalyze their continuous spread within a genome potentially setting the stage for horizontal transmission.
Transposable elements are remarkably widespread and appear to have colonized almost every living organism. In humans, about four million transposable elements, most of them inactive fossil remnants of once actively transposed sequences, represent a stunning almost 50% of the entire genome [20-22]. With such abundance the mobile elements, active or inactive, will inevitably affect the overall ecology of the genome. Indeed, mutations caused by transposon insertion or by chromosomal recombination between inserted transposable elements may serve to increase genetic variation as a platform for selection and evolutionary change. Hence, the activity of transposons may impact and/or favor the adaptability and evolution of species. Examples of specific insertions having beneficial effects on their hosts are rather few, and the accumulation of mobile elements is in general expected to affect host fitness in a negative direction. In smaller multicellular eukaryotes, like fruit flies, fitness is inversely correlated with transposable element copy number [23-25]. Such elements, in the past referred to as selfish or self-promoting elements, may thus multiply to an extent that is defined and limited by the natural selection against their carriers.
The co-existence of a parasitic mobile element with its host relies on the element possessing an evolutionary stable level of activity. Too active elements may simply replicate too efficiently, resulting in numerous insertions that are harmful to the host and eventually fuel extinction of both host and element. As an expected consequence, transposition may be regulated by self-suppressive mechanisms by the element itself and by defensive host mechanisms. These adaptations that may differ from element to element and from host to host collectively define the functional and biological properties of a transposable element.
Since Yant and co-workers in a pioneering study demonstrated in vivo potency of Sleeping Beauty DNA transposon-based gene delivery to the liver of adult mice, continued efforts have been made to understand and further develop DNA transposon elements as gene carriers in mammalian cells and animals. As efforts to optimize and employ Sleeping Beauty vectors have become more and more frequent, the battery of transposable gene vehicles with activity in mammalian cells and with relevance for gene transfer in humans is rapidly expanding (see Table 1 for overview and specific features). In addition to the Sleeping Beauty transposon which is derived from the genome of white cloud mountain minnow (Tanichthys albonubes) [4], the piggyBac element, isolated from the cabbage looper moth Trichoplusia ni [26,27], has shown high levels of DNA transposition in human cells [28,29]. In addition, Frog Prince derived from the genome of the leopard frog Rana pipiens [30], Himar1 derived from the hornfly Haematobia irritans [31], Tol2 isolated from the genome of the Japanese medaka fish Oryzias latipes [32], and Passport derived from the flatfish Pleuronectes platessa [33] transpose in mammalian cells. Also, the ancient human Hsmar1 transposon is efficiently mobilized in human cells by a reconstructed ancestral Hsmar transposase [34]. Recently described elements with robust mobilization in human cells include piggyBat isolated from the bat Myotis lucifungus [35] and TcBuster from the red flour beetle Tribolium castaneum [36,37]. Common to this growing collection of mobile elements is that transposition appears not to rely on species-specific host factors. Still, host factors like DNA-bending proteins may support the transposition process, as shown for Sleeping Beauty and Frog Prince [38]. As a result, some cell types are easier stably transfected with DNA transposon vectors than others [39]. Accordingly, vector systems based on the distinct elements may be favoured in different cell types making a strong argument that parallel efforts to investigate and develop distinct, but potent, systems will benefit the broad applications of transposon-based gene transfer. However, we do not seek here to review the entire package of biological properties that make transposons like Sleeping Beauty and piggyBac ideal for nonviral gene integration purposes in mammalian cells. Numerous excellent reviews already tell that story [40-49]. Instead, with examples from the distinct families of DNA transposable elements we try here to make direct connections between the mechanisms that drive transposon evolution and some of the challenges in transposon-based gene transfer. Experience with Sleeping Beauty in particular tells us that transposon vehicles are travelling with evolution as a rear-seat passenger. By understanding in detail the evolutionary journey of transposons, from host colonization to element multiplication and inactivation, we may be better prepared for utilizing and optimizing transposon-based gene transfer. We argue here that early generations of DNA transposon-derived vectors may suffer from inherent traits of their mother elements, but that new carefully engineered vector generations will address - and in some cases have addressed - key issues rendering transposon gene vehicles safer, more efficient, and less prone for suppression and inactivation.
Table 1.
DNA transposon vectors and their main characteristics
| Transposon | Transposon family | Target sequence | Integration preference | Cargo capacity | Footprint | Overproduction inhibition | Hyperactive transposase | References |
|---|---|---|---|---|---|---|---|---|
|
Sleeping Beauty |
Tc1/mariner |
TA |
Fairly random (31-39% into genes) |
~ 10 kb |
C(A/T)GTA |
Yes |
SB100X |
[50-54] |
|
Frog Prince |
Tc1/mariner |
TA |
ND |
ND |
C(A/T)GTA |
ND |
None |
[30] |
|
Hsmar1 |
Tc1/mariner |
TA |
Fairly random (44% into genes) |
ND |
T(T/A)A |
Yes |
None |
[34,55,56] |
|
Himar1 |
Tc1/mariner |
TA |
Fairly random |
4 kb |
ACTA |
Yes |
C9 |
[31,57-59] |
|
Passport |
Tc1/mariner |
TA |
Transcriptional units (63% into genes) |
ND |
ND |
Yes |
None |
[33] |
|
Tol2 |
hAT |
8 bp random sequence |
Transcriptional units (39-48% into genes) |
> 11 kb |
8 bp random sequence |
Limited |
None |
[60-62] |
|
TcBuster |
hAT |
8 bp random sequence |
Transcriptional units |
ND |
8 bp random sequence |
ND |
TcBusterCOV596A |
[36,37] |
|
PiggyBac |
PiggyBac |
TTAA |
Transcriptional units (47-67% into genes) |
100 kb |
None |
Conflicting reports |
7pB, hyPBase |
[27-29,63,64] |
| PiggyBat | PiggyBac | TTAA | Transcriptional units | ND | ND | ND | None | [35] |
ND: Not determined.
DNA transposable elements – a zoo of endogenous parasites
The far majority of transposable elements in mammalian genomes replicate essentially like retroviruses (except for the lack of an extracellular phase). These so-called retrotransposons replicate through a copy-and-paste mechanism that involves production of an RNA intermediate transcribed from the donor element. A reverse-transcribed DNA version of the element is re-inserted elsewhere in the genome. Such retrotransposons include (i) long interspersed nucleotide elements (LINEs) with two open reading frames and an internal promoter, (ii) short interspersed elements (SINEs) that are parasitic on the LINE replication machinery as they have none of their own, and (iii) LTR retrotransposons which resemble retroviruses in structure with gag and pol genes flanked by long terminal repeats [65,66].
DNA transposable elements that move around the genome by a cut-and-paste mechanism are usually shorter than retrotransposable elements, typically in the range from 1 to 5 kb, and have terminal inverted repeats (IRs) that contain binding sites for the transposase, which is often the only protein encoded by the transposon. The IRs of different elements have variable lengths but may in some cases be more than 700 bp long [67]). Upon transposase complex formation facilitated by binding of transposase subunits to the IRs, the element is cut out of the genome and inserted elsewhere in the genome, in some cases with preference for sites in the immediate neighborhood of the original donor locus. As opposed to retrotransposons, DNA transposons leave behind none or only a short footprint consisting of a few nucleotides that are copied during integration and a few nucleotides that are inserted during DNA repair after element departure.
Cut-and-paste transposons are defined by their similar structure and mechanism of transposition. All elements are composed of a central transposase-coding region flanked by terminal IRs. Cut-and-paste transposition, first demonstrated for P elements and Tc1/mariner elements [68,69], is catalyzed by the transposase protein, facilitating transesterification and transposon excision after binding to the IRs (reviewed in [70] and [55]). The transposase-bound excised transposon is then inserted elsewhere in the genome (Figure 1). While the overall cut-and-paste mechanism is shared by all the superfamilies, some of the molecular details of the mechanism may vary between elements (see Figure 2 for an overview of transposition by Tc1/mariner piggybac, and hAT transposons). In Tc1/mariners, for example, the element is excised by transposase-mediated double-strand breaks, leaving 2 or 3 bp 3′-overhangs at the transposon ends [69,71]. After excision, the element is inserted elsewhere into at TA-dinucleotide target-site [50,69], and repair of the 3′-overhang generated during excision creates, together with the target site duplication (TSD), a characteristic transposition footprint. In the piggyBac superfamily, in contrast, excision of the element results in hairpin formations at the excised transposon ends, and the 5′-TTAA overhangs created in the flanking DNA after excision anneal in the absence of DNA synthesis, leaving an intact excision site without any transposition footprint [72]. Also the hAT transposons (like Tol2) form hairpin structures during transposition, but in this superfamily the hairpins are formed at the ends of the flanking donor DNA instead of at the ends of the excised element [73].
Figure 1.

Model of cut-and-paste transposition. Transposase proteins (green spheres) recognize the terminal inverted repeats (IRs, orange boxes) and form a circular pre-excision synaptic complex from which the transposon is excised. Formation of the synaptic complex, allowing close association of the two transposon ends, involves the formation of transposase tetramers [74]. Subsequently, the transposition complex recognises a target site and the transposase proteins mediate integration of the transposon. The figure shows the proposed mechanisms for Tc1/mariner-type (e.g. Sleeping Beauty) DNA transposition. Yellow boxes marked with IR represent the terminal inverted repeats.
Figure 2.
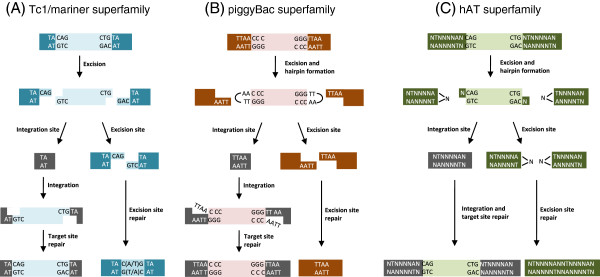
Schematic representation of cut-and-paste transposition. (A) Transposition of Tc1/mariner elements (like Sleeping Beauty and Frog Prince) leads to double-stranded breaks and formation of a 2 or 3 bp 3′-overhang at the excision site (a 3 bp overhang is shown). DNA repair by host-encoded enzymes creates a characteristic footprint at the excision site. Integration occurs at TA dinucleotides which are duplicated upon transposition. The single-stranded gaps are repaired by host-encoded enzymes. (B)PiggyBac-mediated excision is followed by hairpin-formation at the transposon ends. After integration into TTAA target sites that are consequently duplicated, the single-stranded breaks are repaired by ligation. The 5′ TTAA overhangs created at the excision site anneal, thus repairing the double-stranded break without leaving any footprint. (C)hAT transposition creates hairpins at the ends of the flanking donor DNA. Integration is targeted to NTNNNNAN target sites, and as with Tc1/mariner and piggyBac families, hAT-mediated integration creates target-site duplication. Excision site repair leaves a random footprint.
The family of mobile DNA elements also include non-autonomous Miniature inverted-repeat transposable elements (MITEs), whose transposition relies on an in trans supply of transposase protein [75]. Sharing the terminal repeat sequences with those of DNA transposons, MITEs have likely evolved from autonomous elements. The exception to the rule among DNA transposons is the Helitron family of rolling-circle (RC) DNA transposons [76,77]. These atypical elements transpose by a replicative mechanism that they share with circular ssDNA bacteriophages, bacterial plasmids, and geminiviruses.
The evolutionary journey of cut-and-paste DNA transposons
With common mechanisms of transposition and supposedly similar modes of regulation cut-and-paste DNA transposons embark on an evolutionary journey. Following invasion of a new host the newly arrived DNA transposon must proliferate and spread in the host population. After establishment and initial spreading, however, host- and self-regulation starts limiting transposition and inactivating transposons, eventually hampering mobilization of the element in the host. Without being able to transpose and proliferate, the inactive transposons are left as relics in the host genome and may eventually be lost by genetic drift.
To persist and continue their evolutionary path mobile transposons must colonize new hosts. The transmission of transposons between species, known as horizontal transfer (for excellent review see [78]), has been documented for most types of transposable elements. Most of the reported putative horizontal transfer events, however, involve cut-and-paste DNA transposons, supporting the notion of horizontal transfer being essential for DNA transposon survival [79-82]. Indeed it has been suggested that the cessation of DNA transposon activity in anthropoids around 37 million years ago was caused by sudden inability to perform horizontal transfer to these hosts [83]. Though P elements and Tc1/mariners have spawned the strongest and most numerous horizontal transfer documentations (e.g. [84] and [85]), horizontal transfer of elements from other DNA transposon superfamilies has been reported, including Tol2 [86] of the hAT superfamily and piggyBac [87], the latter which was discovered on the basis of horizontal transfer events [88]. Despite the numerous studies trying to identify the vectors that transport mobile DNA elements between species, these remain elusive. Suffering from the lack of clear evidence, viruses [89,90] and various parasites [91,92] have been suggested as carriers of DNA transposons. For successful colonization of a new host the transposon not only needs to transfer to a new host, but needs also to proliferate and spread in the new genome to establish a population [93]. Obviously, the colonizing element is during this phase faced with the challenge of successful replication.
Models of replicative transposition
Replication by transposition from replicated to unreplicated DNA
In consideration of the abundance of DNA transposons and the cut-and-paste mechanism of transposition we are faced with an obvious question: how can elements, which move in a non-replicative way, ever increase in numbers within a genome? Based on genetic studies of the Activator/Dissociation (Ac/Ds) DNA transposable elements in maize, Chen et al. suggested a replicative mode of transposition [94]. According to this model the transposon is replicated during DNA replication, when it transposes from an already replicated site in the genome to a yet unreplicated site (Figure 3A). The model implies a preference for transposition immediately after passage of the replication fork, which has been suggested to rely on DNA methylation [95,96]. Specifically, in a series of methylation studies on Ac/Ds transposition [95], Ros and Kunze showed that full CpG methylation of the Ac/Ds transposon severely inhibited transposition in a way that could be overcome by DNA replication. They revealed by in vitro studies of transposase binding to differently methylated Ac/Ds fragments that the effect of DNA replication relied on selective binding of the transposase to hemimethylated transposase binding sites. In fact, this selectivity was even shown to be specific for one of the two hemimethylated daughter elements produced by replication, making this element over six times as active. Though further mechanistic substantiation is needed, these data, supported by earlier studies on Ac/Ds transposition [94,96,97] and prokaryotic cut-and-paste transposons [97-100], provide an attractive model for replicative transposition of eukaryotic cut-and-paste transposons.
Figure 3.

Models of replicative transposition. (A) After replication, the transposon is excised and integrated into a yet unreplicated genomic site thus duplicating the newly inserted transposon. (B) The double-stranded break created by transposition at newly replicated DNA is repaired using the sister chromatid as a template for homology-directed DNA repair, leading to reconstitution of the excised transposon. IR, terminal inverted repeat.
Additional support for replication of transposons through jumping to unreplicated DNA is provided by early studies on prokaryotic cut-and-paste transposons. Results from the prokaryotic DNA transposons Tn10/IS10 [98] and Tn5/IS50 [97,99] of the IS4 family [100] reveal transpositional regulation very similar to that found for the eukaryotic Ac/Ds elements. Hence, both elements transpose preferentially from hemimethylated DNA with a specific preference for one of the strands. Interestingly, these two elements are, unlike many other prokaryotic DNA transposons, restricted to non-replicative cut-and-paste transposition [101,102]. This also gives reason to the absence of dam (DNA adenine methylase) sites in other prokaryotic DNA transposons like IS1 and IS26 [98,103,104]. However, these elements do not need to tie their transposition to the passage of the replication fork as they can replicate by alternative mechanisms.
Replication by homology-dependent DNA repair
Another model for replication of transposable elements arose from studies by Engels et al. of homolog-dependent high-frequency loss of the P element DNA transposon in Drosophila [105]. The model is based on the formation of a double-stranded DNA break (DSB) at the site of transposon excision and subsequent repair by a homologous DNA template (Figure 3B). If the template is the homologous chromosome (assumed to carry the wild-type sequence at the insertion point) the transposon will be lost, as observed by Engels et al. However, if the template is the sister chromatid, which was shown to be preferred in a study of P elements [106], the transposon will be restored at the excision site, and the transposition is then replicative. Similar results have been obtained in studies of the nematode element Tc1 of the Tc1/mariner superfamily, indicating that this model could be universal [107,108].
Synthesis-dependent strand annealing (SDSA, first described for the T4 phage [109]) has been suggested to be the molecular mechanism underlying this homolog-dependent gap repair [110]. According to this mechanism, 3′-DNA termini left at the DSB independently invade a double-stranded homologous sequence, extend by DNA synthesis using the homologous sequence as template,and after displacement anneal to each other in a region of overlap (Figure 4). Hereafter non-overlapping sequence is removed, and remaining nicks are sealed by ligation. Unlike events of homologous recombination (HR), repair by SDSA does not require cross-overs, and the homologous template is simply copied into the DSB, thus explaining the high deletion and duplication rates observed [105,107]. The SDSA pathway has also been suggested to account for the creation of the non-autonomous Ac/Ds elements by incomplete repair after transposon excision [111], indicating that this model might also apply to the hAT superfamily of cut-and-paste DNA transposons.
Figure 4.

Homology-dependent DNA repair by synthesis-dependent strand annealing (SDSA). Following excision, the 3′-DNA termini from the double-stranded breaks invade the homologous sequence on the sister chromatid. The homologous sequence is then used as template for DNA synthesis and the final, elongated 3′-DNA termini anneal to each other and are joined with the 5′ ends by ligation.
The bigger picture of repair after transposon excision also involves other mechanisms. In non-homologous end-joining (NHEJ; see [112] for recent review), the broken ends in the DSB are joined, resulting in a characteristic footprint (consisting of the overhang sequence flanked by target site duplications) revealing that a transposon was once there. Studies of the repair products of both the P element [113] and the reconstructed Tc1/mariner Sleeping Beauty element [9,114] indicate that NHEJ is a major pathway of DSB repair after transposon excision. Studies on P elements indicated interaction between the P element inverted repeats and the Drosophila homolog of the mammalian protein Ku70 [115] (part of DNA-PK, a main component in NHEJ). Later results revealed direct interactions between the Sleeping Beauty transposase and Ku70 [114], suggesting a role of the transposase in recruiting the DNA-binding Ku70/Ku80 heterodimer subunit of DNA-PK to the DSB to promote NHEJ and ensure genomic stability. Knocking out Ku, however, did not abolish DSB repair. Instead, analysis of the DSB repair products indicated that the homology-dependent SDSA repair pathway was now in place for repair [114].
Studies in Drosophila as the host of P elements reveal a complex scenario in which different mechanisms - NHEJ, SDSA and yet another mechanism referred to as single-strand annealing (SSA) - are major competing or complementary pathways [116-119]) for DSB repair. Factors like genomic context [117], cell cycle phase [119] and developmental stage [118] may determine which pathway is used. Nevertheless, transposons have found ways to influence the process either by directly interacting with factors of the repair pathways, as observed for Sleeping Beauty transposase-Ku70-interaction [114], or by modulating the relevant host factors, as has recently been found to be the case for Sleeping Beauty [120]. Here it was found that the Sleeping Beauty transposase halts cell cycle progression from G1 through interaction with the transcription factor Miz-1 (which regulates several genes involved in cell cycle regulation [121-123], including D1 [124]). Hereby Sleeping Beauty prolongs the G1-phase, possibly favoring transposition in this phase during which NHEJ seems to be favored over homolog-dependent repair [125,126]. The complex balance of competing repair mechanisms probably reflects basic evolutionary selection parameters such as transposon replication (by SDSA), genomic stability (e.g. by avoiding uncontrolled transposon replication), and long-run evolvability enhanced by a certain transposon content in the host genome, illustrating how the different levels of selection are tightly connected in a complex interplay.
Integration site preference of DNA transposon elements
Along with genomic excision transposons are faced with the task of locating new sites in the genome to integrate into. The target site preference of transposons varies between the different transposon families, but common to most elements is that the target site sequence is duplicated upon integration, leaving the inserted transposon flanked by small stretches of identical sequences. Some transposon elements are very strict in their sequence choice, such as the Tc1/mariner elements which always integrate into a TA dinucleotide [127], and piggyBac which always integrate into TTAA tetranucleotides [27]. Other transposon elements, such as the hAT superfamily and P elements, are more flexible and insert into 8-bp integration sequences that may vary in nucleotide composition [73,128].
In addition to the primary target site sequence, several genomic features influence transposon insertion site preference. One feature is the genomic distance from the transposon donor site. In a study of Sleeping Beauty transposition in the mouse germ line it was observed that 27% of the transposition events had occurred within 200 kb of the donor site, and 75% of the transposition events were found to be on the same chromosome [129]. This phenomenon, called local hopping, has been found for numerous other transposable elements such as the Tc1 element [130], P elements [131], the Tol2 element [132], and Ac/Ds elements [133]. As Sleeping Beauty insertion sites are widely distributed in the genome, when transposition occurs from a donor plasmid [50,51], local hopping most likely reflects a tendency of the transposon to select target sites that are physically close to the donor site rather than a preference for some chromosome sequences over others. Interestingly, the piggyBac transposon seems to exhibit no or little local hopping [134,135], suggesting that this element has a different way of reaching its target site relative to the majority of DNA transposable elements.
Another genomic feature that has importance for insertion site preference of several Tc1/mariner elements is DNA bendability. Thus, the flexibility and overall topology may influence the access to the DNA at a given genomic position. Analyses of insertion sites of Tc1, Tc3, Himar1, Sleepng Beauty, and Minos transposons have shown that TA sites in DNA regions with increased bendability are preferred compared to TA sites in more stiff DNA regions [50,51,136,137]. The exact molecular mechanism governing this Tc1/mariner preference still remains to be elucidated, but a possible explanation could be that flexible DNA is more easily attacked by the transposase catalytic site during transposition, and/or that the transposase interacts with cellular proteins associated with bendable DNA. Based on studies demonstrating the sensitivity of Hsmar1 transposition to DNA topology, it was speculated that a certain topology of the targeted sequence enables the transposase to create mechanical strain at the active site by bending the DNA. This may allow structural changes during release of the mechanical strain, making re-excision of the inserted transposon less likely [56].
Retroviruses have been shown to have a non-random integration profile with some genomic locations being favored over others. Lentiviruses, for instance, prefer to integrate into actively transcribed units, while gammaretroviruses have a preference for integrating near the 5′ end of actively transcribed genes [138]. Among the DNA cut-and-paste transposons, some elements appear to have a random integration profile while others have integration profiles that resemble those seen for retroviruses. Analyses of Sleeping Beauty insertion sites from cultured HeLa and K562 cells [50,139], mouse liver [51], NIH 3 T3 mouse fibroblasts [51], and human primary T cells [140], have revealed that the Sleeping Beauty transposon has a fairly random integration profile with no preference for or against genes. The P element, in contrast, has a strong preference for integrating near promoter regions, and analyses of more than 9,000 insertions in the Drosophila genome showed that 73% of P element insertions lie within 500 bp of an annotated 5′ transcription start site [141,142]. A recent study showed that promoters are not randomly targeted. Although 71% of 18,213 insertions analyzed were associated with promoters, only 24% of the total amount of annotated promoters in the Drosophila genome were targeted by an insertion [143]. Furthermore, a strong correlation was seen between P element target sites and Origin Recognition Complex (ORC) binding sites at replication origins. ORC associates with open chromatin and promoters thus explaining the correlation between P element targets and promoters [144]. Also the Tol2 and piggyBac transposons are shown to exhibit nonrandom integration patterns. Of 113 Tol2 insertion sites in HeLa cells, 48% were found to be in transcriptional units and a significant number was observed to be close to transcriptional start sites [145], a preference that was also seen for Tol2 in HEK293 cells [146] and in primary human T cells [140]. Similarly, a bias towards transcriptional start sites and intragenic regions have been observed for the piggyBac transposon in HeLa and HEK293 cells [28,63,146], mouse embryonic stem cells [134], and primary human T cells [140,147]. In addition, piggyBac has been observed to have a preference for integrating into active genes in primary human T cells [147]. The preference of lentiviruses to insert into actively transcribed units has been associated with their binding to the ubiquitously expressed nuclear protein LEDGF/p75 (reviewed in [148]). LEDGF/p75 functions as a transcriptional coactivator, and the lentiviral integrase protein has been observed to bind to the C-terminus of LEDGF/p75, while the N-terminus of LEDGF/p75 can bind to chromatin, thereby tethering the viral integration complex to actively transcribed regions. Interactions between DNA binding proteins associated with transcriptional regions and DNA cut-and-paste transposons have not been observed so far, but perhaps such interactions accounts for the nonrandom integration profile seen for some transposable elements.
Invasion, spread, and regulation in host genomes
In a likely scenario in nature, transposon colonization of the germline of a species is initialized by horizontal transmission between species followed by dramatic multiplication and spread of the mobile DNA element. Over time, the copy number will level out at a steady-state level as the result of the co-evolution of host and transposon. The delicate balance between continuous spread and proliferation of active elements and a decline in host fitness caused by transposon-induced damage to the genome results in selection at the host level against high transposon activity. It also seems reasonable that transposons on this leg of their evolutionary journey travel with regulatory mechanisms that may directly influence the transposition rate and reduce the genetic harm to the host. Although such control mechanisms are vaguely characterized, there is evidence that transposons not only are regulated by host defense pathways, including transcriptional silencing and posttranscriptional silencing by RNA interference (see later), but also that the elements themselves possess autoregulatory control mechanisms with the transposase protein as the key player.
Transposon autoregulation by transposase overproduction inhibition
In attempts to mimic DNA transposition in nature most studies of DNA transposons with activity in eukaryotes rely on genetically engineered transposons and cloned versions of transposases that have been genetically modified to obtain activity. In addition, in functional studies transposase expression is often driven by promoters that facilitate high levels of transposase production. Despite potential discrepancies between actual mechanisms that are in play during genome evolution and in short-term laboratory assays, there is solid evidence that DNA transposition of members of the Tc1/mariner and other transposon superfamilies is inhibited by elevated levels of transposase, indicating, hence, that transposon activity during evolution has been regulated by self-inhibitory regulatory mechanisms. Events of transposase-based autoregulation, collectively referred to as overproduction inhibition (OPI), were first described for eukaryotic transposons by Lohe and Hartl [149], but had seemingly been observed in previous in vitro transposition studies including purified transposase protein derived from the mobile prokaryotic element Tn10[150]. In transposase activity studies based on germline excision of a target mariner transposon present in the genetically engineered white-peach gene of Drosophila mauritiana, Lohe and Hartl found reduced levels of mariner excision as a result of increasing transposase dosage. Hence, homozygous flies carrying two Mos1 transposase-expressing alleles or flies carrying copies of the transposase expression cassette on two different chromosomes showed significantly lower excision rates than flies with a single cassette [149,151]. In addition, Mos1 excision was further reduced by heat-shock treatment of flies containing transposase genes driven by a chimeric heat-shock hsp70-derived promoter, resulting in excision rates that were reduced more than 50% in comparison to untreated flies carrying a single transposase gene [149,152,153]. In potential contradiction to these findings, even though excess amounts of Mos1 impairs paired-end complex assembly in a mobility shift assay, increasing dosages of purified Mos1 transposase expressed in Escherichia coli were found not to cause OPI in an in vitro transposition assay [154,155]. In similar assays, purified transposase derived from the reconstructed Himar1 mariner element was found to induce strong OPI even at low concentrations [57,154]. Transposition studies in HeLa cells and rabbit synovial fibroblasts confirm that increasing amounts of transfected Himar1 transposase-expressing plasmid result in reduced transposition rates [58].
Despite recent evidence for activity of the Himar1 element in human cells [58], the functional properties of the Sleeping Beauty transposase has attracted special attention due to its high activity in human cells [4]. By transiently expressing the Sleeping Beauty transposase from a heat-inducible promoter and from two strong constitutive promoters, Izsvak and coworkers were not able to detect any negative regulatory effects of high transposase production levels in cultured HeLa cells. Hence, within the studied levels of transposase (as detected by western blotting) the transposition rates did not plateau or decrease with increasing transposase expression. [39]. However, by co-transfecting plasmids containing the transposon substrate and the transposase expression cassette, respectively, in a broad range of ratios ranging from 1:17 to 33:1 in HeLa cells (keeping the total amount of DNA constant) dramatic inhibitory effects on transposition were observed with higher doses of transposase [156]. In these experiments, the highest level of transposition was obtained with a transfection mixture containing 0.5 μg transposon plasmid and 0.1 μg of a plasmid containing a CMV-driven transposase expression cassette, but was reduced 12-fold by including 10 times more transposase-encoding plasmid in the transfection mixture. In an identical transfection experiment, performed in HT1080 cells, the Sleeping Beauty transposition rate declined only moderately (about 1.5-fold) [157], indicating that in vitro-documented OPI effects of Tc1/mariner elements may be influenced by factors like cell type and transfection rate.
Based on many reports from the last years, it appears now to be generally accepted that piggyBac, and optimized derivatives of this system, may transpose in human cells with efficiencies that are comparable to improved versions of the Sleeping Beauty transposon. Although a single study shows OPI of piggyBac in HEK-293 cells [60], it appears that these non-Tc1/mariner transposases are generally less sensitive to OPI [60,63,157]. Indeed, in a direct comparison between the Sleeping Beauty, piggyBac and Tol2, the hyperactive SB100X transposase was shown to be more prone to regulation by OPI, reaching optimum transposition conditions at a 1:10 transposase:transposon ratio [145]. Notably, one report stated that molar piggyBac transposase-to-transposon ratios as high as 43:1 did not induce OPI in HEK-293 cells, whereas a similar ratio for Sleeping Beauty caused OPI [63].
OPI-based regulation of transposition represents an additional layer of complexity in transposon-based gene transfer and may have crucial influence on the in vivo use of transposable elements. However, the question remains if negative dosage effects observed in transient transfection studies reflect true biological mechanisms with impact on natural populations and the regulation of DNA transposition through evolution or, perhaps less excitingly, is the result of artificial overproduction of an enzyme with toxic effects on the treated cells. One can easily envisage putative evolutionary implications of OPI as one of many possible ways of regulating genomic transposition. In a likely scenario, low levels of transposase expressed from a relatively low number of integrated elements may provide optimal conditions for mobility. However, as the number of active elements increases over time the overall transposase dosage may reach a certain threshold beyond which transposition is reduced. As of today the mechanisms involved are unknown but may possibly include posttranslational interactions between transposase molecules. Recent studies of a resurrected Hsmar1 mariner transposon support a model by which transposon activity is autoregulated through competition between transposase subunits for binding sites within existing transposon elements [158]. In accordance, it has been suggested that overproduction leads to transposase oligomerization and reduced transpositional activity [149]. In another plausible scenario, a surplus of free transposase molecules form complexes with molecules already bound to the transposon, thereby inhibiting interactions between the inverted repeats during transposition. This ‘quenching’ mechanism would imply most likely that an increase in the amount of transposon substrate in transfections should reduce the extent of OPI at a given level of transposase. However, in at least one Sleeping Beauty study this was found not to be the case [156].
Regulation and silencing by epigenetic modifications
Eukaryotic genomes consist of regions of transcriptionally active euchromatin and transcriptionally inactive heterochromatin. The organization of genomes into such regions is controlled by an interplay between DNA methylation and histone modifications, and these epigenetic modifications have been shown to be closely associated with the transcriptional state of many eukaryotic genes [159,160].
Analyses of DNA methylation patterns in both plant and animal species have revealed that endogenous transposon elements are heavily methylated [161]. Recent genome-wide DNA methylation analyses confirm that this is the case also in human embryonic cells and somatic tissues [162]. Such DNA methylation may correlate with silencing of mobile DNA elements, as increased mobilization of transposons in both plant and animal mutants is linked to abolished DNA methylation. The findings that silent transposable elements can be reactivated upon lack of DNA methylation has led to the belief that epigenetic modifications and heterochromatin formation represent basic defense mechanisms to prevent the harmful activity of mobile DNA elements within genomes [163]. Notably, recent evidence suggests that a minor portion of human transposable elements is hypomethylated in a tissue-specific manner, supporting a model by which transposon elements may possess enhancer-like functions and assist in regulating genes [162].
Studies of transposition in Arabidopsis thaliana mutated in the DDM1 (decrease in DNA methylation) gene revealed increased transposition rates [164,165]. Similar results were observed by Lippman et al. who, by microarray and chromatin immunoprecipitation (ChIP) studies in A. thaliana, showed that heterochromatic CACTA transposons and gypsy-like retrotransposons are activated in DDM1 gene mutants [166]. These results, however, could also be explained by the implication of DDM1 in chromatin remodelling [167]. Nevertheless, in a later study Kato and colleagues showed that transposition of CACTA elements in A. thaliana was enhanced significantly in plants defective in either CG or non-CG DNA methylation[168]. Although transposition assays on hemi-methylated DNA are needed to give firm support from other superfamilies, these studies on CACTA and Mutator indicate profound effects of DNA methylation on transposition – perhaps caused by an underlying replication mechanism based on methylation-state-regulated transposition. A correlation between decreased cytosine methylation and increased transposition of IAP retrotransposons was also observed in mouse embryos deficient of a fully functional Dnmt1 (DNA methyltransferase-1) gene [163].
As an intriguing exception to these findings, it appears that Sleeping Beauty transposition is enhanced by transposon DNA methylation, suggesting that transposon mobilization is supported by CpG methylation [169]. Recent findings have confirmed such supportive role of methylation for mobilization of Sleeping Beauty and Frog Prince elements, leading to the assumption that methylation and a resulting tight chromatin structure is beneficial for transposition of these particular elements [170]. In mouse embryonic stem cell lines, containing a single Sleeping Beauty transposon insertion, excision efficiencies were observed to be 100-fold higher when the Sleeping Beauty transposon was in a heterochromatic conformation compared to control clones without the heterochromatic conformation [171]. Improved transposition efficiency of a methylated Sleeping Beauty transposon in mouse embryonic stem cells was also observed in a study in which transposition efficiency of the piggyBac transposon at the same time was shown to be reduced upon methylation [134]. In accordance, immunostaining and biochemical analyses have shown that the Sleeping Beauty element appears to have an affinity for heterochromatic regions [171]. It is tempting to propose that such mechanism in an evolutionary context would facilitate escape of the transposon from heterochromatic and methylated regions of the genome, but explanations for this observation remain elusive.
The Tol1 and Tol2 transposable elements, both members of the hAT superfamily, are unique among the DNA transposons, as they are the only natural active elements to have been discovered so far in vertebrate genomes. The Tol2 element has had a rapid expansion in the genome of its host, the medaka fish, in the past, but a high spontaneous transposition rate is not observed in the genome of current laboratory fish strains, suggesting that Tol2 has already reached a steady-state level where the transposition frequency is controlled by host mechanisms [172]. Evidence links DNA methylation to host control of Tol2 transposition. Hence, in a study by Iida et al. [173] medaka fish embryos were soaked in 5-azacytidine, a reagent that acts as a false substrate and potent inhibitor of methyltransferases leading to reduction in DNA methylation. Reduced CpG methylation levels together with increased transposition excision frequencies were observed in the treated fish embryos. Interestingly, the 5-azacytidine treatment did not seem to induce increased expression of the transposase gene, suggesting that DNA methylation of the Tol2 transposon did not inhibit transposition by transcriptional silencing of the transposase gene. In studies involving other hAT elements, methylation at the ends of the plant transposon Tam3 (from Antirrhinum) was shown to cause repression of transposition [174], and DNA methylation at the transposase binding sites of the Ac/Ds element was shown to inhibit Ac transposase binding [95]. These findings support the notion that transpositional regulation by DNA methylation in the hAT transposon superfamily is due to inhibited transposase binding to transposon binding sites.
Rather than affecting the process of transposition itself, epigenetic modifications may impact transposase expression and thereby indirectly regulate transposition activity. CpG methylation of the Sleeping Beauty element carrying a transgene, and not the natural transposase gene, was analyzed in transgenic mice containing single-copy transposon insertions [175]. The DNA methylation status of the inserted transgene cassette, which consisted of the ROSA26 promoter and the eGFP gene, was examined by bisulfate-mediated sequencing for six independent insertions. The results showed that Sleeping Beauty transposons inserted into the mouse genome were heavily methylated in the ROSA26 promoter and the eGFP coding sequence. In contrast, the endogenous mouse ROSA26 promoter was devoid of CpG methylation in the transgenic mice. It appears that the host cells were able to distinguish between the endogenous genomic sequence and the exogenous counterpart inserted by Sleeping Beauty, suggesting that the transposon was specifically recognized and targeted by the host cellular epigenetic modification system. In another study, however, Sleeping Beauty transgenic mice containing a transgene cassette comprised of a human K14 promoter and the Agouti reporter gene did not show significant levels of CpG methylation at the inserted transgene [176], indicating that Sleeping Beauty cargo DNA sequences rather than transposon inverted repeat sequences influence CpG methylation status. We have at several occasions observed transcriptional down-regulation and silencing of Sleeping Beauty-inserted transgene cassettes [177-179], but the promoter rather than the carrier seems to a key determinant for such silencing. Nevertheless, in comparative studies performed in human retinal pigment epithelium cells, a transgene cassette delivered by piggyBac was less vulnerable to silencing than a cassette delivered by the Sleeping Beauty counterpart, lending support to the notion that the level of silencing was influenced by the distinct integration profiles [179].
The role of histone modifications in transposon control is under continuous investigation. A recent study in murine embryos showed reactivation of the LINE-1 and IAP retrotransposons in 2-cell embryos. A subsequent loss of expression in 8-cell embryos was shown to correlate with a loss of the activating trimethylation of H3K4 rather than gain of the repressive trimethylation of H3K9 [180]. It is not currently known to which degree DNA transposition is directly affected by histone modifications.
Regulation by RNA interference (RNAi)
To produce transposase proteins that facilitate jumping of transposons to a new location in the genome the transposon open reading frame must first be copied into mRNA. The evolution of host silencing mechanisms, based on the ability of small inhibitory RNA molecules to guide mRNA degradation in a sequence-specific fashion, has allowed higher organisms to defend the genome against mobile elements and harmful effects of insertional mutagenesis and genomic instability. Formation of transposon-derived double-stranded RNA (dsRNA) is a hallmark of RNAi-based suppression of transposition. Figure 5 provides a schematic representation of three potential scenarious for production of transposon-derived dsRNA that may be processed into effectors of a regulatory RNAi response. These include intramolecular basepairing between terminal inverted repeats of read-through RNA transcripts (Figure 5A), basepairing between bidirectional RNA transcripts generated from sense and antisense promoters within the element (Figure 5B), or intermolecular annealing of sense and antisense read-through transcripts (Figure 5C).
Figure 5.

Transposon regulation by RNA interference. (A) An upstream promoter (UP) generates read-through transcripts that fold back on themselves due to intramolecular base pairing of the inverted repeats (IRs), thereby generating dsRNA transcripts that may be processed into RNAi effectors. (B) Upstream and downstream promoters (DP) create bi-directional RNA transcripts that anneal and form dsRNA. (C) Promoter-like activity in the inverted repeats generates sense and antisense transcripts which, as in (B) anneal to form dsRNA.
The lack of easy identifiable regulatory elements in DNA transposons has fueled the idea that expression of the transposase gene in original elements depends on endogenous regulatory sequences flanking the inserted transposon. This may give rise to read-through RNA transcripts that may serve as templates for transposase synthesis. In C. elegans, transcription of Tc1 sequences is initiated in the flanking sequences and not within the inverted repeats [181]. Interestingly, such read-through transcripts may form dsRNA by folding back on themselves, allowing complementary intramolecular inverted repeat sequences to anneal (Figure 5A). These dsRNA regions are processed by the cellular RNAi machinery, triggering silencing of the transposable element by transposon-specific RNA degradation [181]. As Tc1 transposition in wildtype worms occurs only in somatic cells and not in the germ line, identification of mutants with active transposition in germ cells has led to identification of genes that play an active role in the RNAi response in C. elegans[182,183]. With our current knowledge, it is not surprising that these genes, rde-2 and mut-7 among others, encode conserved proteins involved in degrading RNA by RNAi. In agreement with data showing that mutations in these genes relieve a block on transposition [182,183], small RNA effectors of the RNAi machinery (referred to as small interfering RNAs, siRNAs) derived from Tc1 are much less abundant in mutant worms defective in both RNAi and transposon silencing [181].
Fastly accumulating evidence suggests that retrotransposons in wide variety of host organisms are suppressed through the RNAi pathway. In mouse oocytes, repeat-associated siRNAs (ra-siRNAs) derived from retroelements have been identified and are likely effectors in an anti-transposon response through RNAi [184], as supported by findings showing high abundance of retrotransposon-derived transcripts in Dicer-deficient mouse embryonic stem cells [185]. Moreover, RNAi-based Dicer knockdown in preimplantation mouse embryos causes an increase in abundance of transcripts from LTR retrotransposons [186]. In the Drosophila germline, ra-siRNA-directed silencing serves to suppress transposon expression, but through a yet incompletely understood RNA processing pathway that does not involve siRNA production by Dicer. Such ra-siRNA effectors are referred to also as piRNAs due to their association with the Argonaute subfamily of PIWI proteins [187,188] and have been found to suppress activities of both retrotransposons and DNA transposons in the zebrafish and mouse germline [189,190]. Intriguingly, PIWI/piRNA complexes not only exercise their function on a post-transcriptional level, but have recently been shown to enforce transcriptional repression of transposable elements [191,192] and to recruit HP1 to confer repressive chromatin marks when bound to euchromatin [193]. Furthermore, a loss of repressive marks at transposon loci is shown in Drosophila PIWI mutants.
LINE-1 retroelements comprise about 17% of the human genome and an estimated 100 of these elements are fully capable of transposition. Sense and antisense promoters in the 5′ untranslated region of LINE-1 direct production of bi-directional RNA transcripts which can potentially anneal to form double-stranded RNA (Figure 5B). Such dsRNA molecules are processed to siRNAs which can suppress LINE-1 retrotransposition in human cells [194,195]. Notably, loss of the antisense promoter is accompanied by increased LINE-1 transposition, and knockdown of Dicer by synthetic siRNAs causes an increased abundance of endogenous LINE-1 RNA transcripts in cultured human cells [194], both findings suggesting that LINE-1 retrotransposition is suppressed by RNAi.
We previously showed that the terminal IR sequences of the Sleeping Beauty DNA transposon possess moderate gene-regulatory activities [196]. IRs oriented against the center of the transposon stimulate gene expression in transient reporter assays and have a considerable impact on the expression of genes within the integrated transposon. We therefore envisage a scenario reminiscent of RNAi suppression of LINE-1 elements and speculate that opposing transcriptional activities driven by the element itself, or neighbouring regions, may influence transposition by mechanisms that may possibly involve transcriptional interference pathways. These findings suggest that DNA transposable elements during evolution in higher vertebrates have been held in check partially by RNAi-related pathways. This notion is supported by the fact that Sleeping Beauty DNA transposition is enhanced in cells in which the RNAi machinery is suppressed by expression of the P19 protein [197]. It is unclear yet, however, whether the Sleeping Beauty IRs serve as primitive promoters or support gene expression by other mechanisms. Interestingly, similar regulatory activities of the C. elegans Tc3 element [196] could indicate that bidirectional transcription of DNA transposons is a conserved feature of DNA transposable elements. However, elements with shorter inverted repeats, e.g. Tc1, may rely on read-through transcription (Figure 5C), as suggested by the finding that Tc1 inverted repeats do not possess promoter-like activities [196].
Ending the journey – dominant-negative complementation and transposase titration
Transposable elements cannot keep racing evolution forever and eventually run out of gas, as evidenced by the numerous DNA transposons that reside as inactive genetic relics in genomes throughout the animal kingdom. The mechanisms underlying this fate have been addressed mainly for the P element and Tc1/mariner superfamilies, but likely apply to all cut-and-paste DNA transposons. The probability of further spreading of transposable elements and the inherent risk of insertional mutagenesis is reduced over time due to the accumulation of loss-of-function mutations in the transposase gene. While such mutations will be favored by selection at the host level, they will be nearly neutral at the transposon level, as the transposase proteins produced by intact (autonomous) elements cannot distinguish autonomous from non-autonomous elements in choosing its substrate (Figure 6). Mutations will thus over time accumulate in the transposase genes, rendering more and more transposons non-autonomous, a process referred to as ‘vertical inactivation’ [79]. Besides the direct effect on transposition activity by lowering the amount of functional transposase, vertical inactivation promotes at least two indirect mechanisms of transposition regulation. Firstly, inactive transposase protein is thought to interact with active transposase protein to inhibit transposition in a process termed dominant-negative complementation (DNC) [149]. Secondly, the non-autonomous transposons produced by vertical inactivation might still serve as substrate for active transposase, hitching free rides at the cost of autonomous transposons. By this mechanism, called transposase titration [198], the proliferation rate of autonomous elements and, hence, further transposase production is hampered. Vertical inactivation thus continuously lowers transposition activity until all elements present in the genome are inactive. Without transposition activity or selection the population of inactive transposons will slowly be lost from the genome by genetic drift (stochastic loss [79]). At this point all hope is out for the paralyzed and dying transposons. To survive as a group of mobile genetic elements horizontal transfer must occur before all elements are inactivated, as proliferation and maintained transposition activity is required to colonize naive host genomes.
Figure 6.
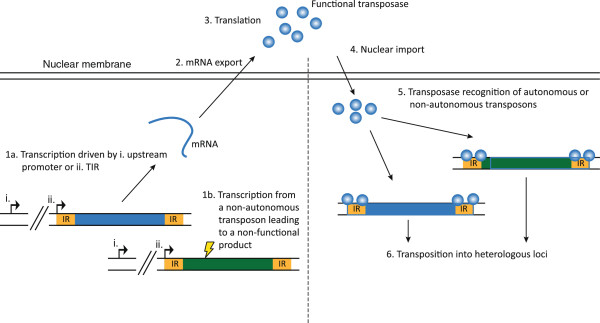
Mobilization of autonomous and non-autonomous transposons. Transcription from autonomous transposons is driven by either an upstream promoter (i) or by the IR (ii) (1a). Non-autonomous transposons comprising loss-of-function mutations (indicated by lightning bolt) are unable to make a functional transposase (1b). mRNA from the autonomous transposon is exported to the cytoplasm (2), translated into functional transposase (3), which is transported into the nucleus (4). The transposase recognizes both autonomous and non-autonomous transposons (5) leading to transposition of both (6).
Mobile DNA elements as therapeutic gene vehicles
Since its discovery in 1997 the Sleeping Beauty transposon has been extensively studied for genetic applications in vertebrates. As we have learned more about the biology of Sleeping Beauty and its advantages and potential drawbacks as a vector for therapeutic gene delivery, several alternative DNA transposons with activities in human cells have appeared on the scene. Although active in human cells, elements like Tc1 and Tc3 from C. elegans, Mos1 and Minos from Drosophila, and Himar1 from hornfly do not appear to transpose with the activity of Sleeping Beauty in human cells [60,170,199] and therefore have not attracted much attention as therapeutically relevant gene vehicles. In contrast, elements like piggyBac, Tol2, Passport, and Frog Prince have shown robust transposition in human cells [29,30,33,60,63,200] and may serve, therefore, as attractive alternatives to Sleeping Beauty-derived gene vectors. Much of the focus, however, has been put on the piggyBac element as an alternative to Sleeping Beauty in mediating therapeutic gene insertion. The therapeutic properties of both Sleeping Beauty and piggyBac systems are covered in several excellent reviews [44-46,201].
Common to these mobile elements is that they - during invasion, genomic spread and regulation - have been shaped by an evolutionary drive for survival and maintenance of high copy numbers in their natural hosts. As a consequence, differences between the systems and their performance in human cells may likely be influenced by how far they have reached on their evolutionary journey. Hence, it can be hypothesized that invading autonomous elements have high activity but also may not yet have adapted to regulatory defense mechanisms of the host. In addition, such elements may not yet have evolved self-regulatory mechanisms which are likely to support a steady-state level of transposition and a balanced number of elements after initial invasion. In any circumstance, each of the isolated elements is the result of a genetic interplay between element and host, and the biological properties are therefore more than likely to vary between the elements. With current interests in employing DNA transposons for gene delivery it is important to make the point that the vehicles, which we create for gene transfer purposes in humans, are derived from elements formed by an ongoing evolutionary journey and therefore may carry different functional properties as result of variable selective pressures. This stresses the necessity of creating vectors based on different mobile elements and, moreover, fully characterizing the biological properties of such vectors. Except for Tol2 and piggyBac, which both encode a functional transposase and are still active in their original host genome, current elements with high activity in human cells originate from fossils which have accumulated mutations, rendering them inactive in their natural hosts. As reawakening of vertically inactivated elements relies on a human touch of site-directed mutagenesis, it is likely also that differences between the systems may originate, at least partially, from different strategies and results of reconstruction. Moreover, it can be argued that elements with a history in vertebrates, like Tol2 and Sleeping Beauty, may have evolved under cellular conditions that may favor their optimized use in human cells as opposed to elements derived from insects or nematodes.
Despite the proven potency of Sleeping Beauty-mediated gene insertion for a range of applications in various tissues, Sleeping Beauty-based vectors do indeed carry traits of their heritage and evolution that may represent potential shortcomings in a therapeutic scenario. Phylogenetic analyses have suggested that Sleeping Beauty ancestors have evolved through reassortment of functional domains between mobile elements and horizontal transmission [202]. Hence, members of Tc1-like transposon family are prevalent in many fish genomes but are inactive due to either (i) accumulation of mutations in the transposase gene or (ii) more severe genetic alterations, like deletions, caused by vertical inactivation. Among Tc1-like elements in fish the salmonid subfamily appears to be the youngest and most recently active [202]. Based on phylogenetics of this subfamily the pioneering reconstruction work performed by Zoltan Ivics and Zsuzsanna Izsvák in the laboratory of Perry Hackett brought a functional ‘consensus’ element, perhaps identical but more likely equivalent to an riginal ancient element, back to life. Since the initial discovery, hyperactive variants of the Sleeping Beauty transposase have been developed by mutagenesis, leading to several versions of the transposase with enhanced transposition properties [52,136,203]. Ultimatively, a high-throughput PCR-based DNA-shuffling strategy was utilized to produce the hyperactive transposase SB100X, which was found to be 100-fold more active than the original Sleeping Beauty transposase (originally designated SB10) under certain experimental conditions [204]. Moreover, higher levels of piggyBac transposition have been achieved by codon optimization of the transposase gene, leading to induced levels of expression [205,206]. Also, a hyperactive piggyBac transposase (HyPBase) was recently identified by screening of a transposase mutant library and combining beneficial mutations in a single transposase variant [64]. By employing mouse liver as an in vivo model, SB100X and HyPBase have been found to increase in vivo efficacy above what has been observed with previous transposase variants [29,53] and currently represent the first choices for therapeutic transposition of genes. Although such variants may prove to be prominent tools for gene insertion in hard-to-transfect cell types or tissues, it should be noted that such transposases with an enhanced gene-inserting potential may also pose an increased risk of causing insertional mutagenesis.
Delivery of transposons and transposases
Conventional DNA transposon systems consist of two plasmids, one helper plasmid carrying the transposase expression cassette and one donor plasmid carrying the DNA transposon (Figure 7, panel a). After transfection, both plasmids find way to the nucleus, allowing production of transposase-encoding RNA from the helper plasmid and subsequent excision of the transposon from the donor plasmid facilitated by transposase subunits imported into the nucleus. This approach can be further refined by placing both the transposase gene and the transposon on a single plasmid, originally referred to as helper-independent transposon-transposase vectors [7]. Such one-plasmid systems do however require a stringent design to avoid OPI. Alternatively, transfected in vitro-transcribed mRNA may serve as a rich source of transposase [207], eliminating the risk of creating cells with prolonged expression of the transposase.
Figure 7.
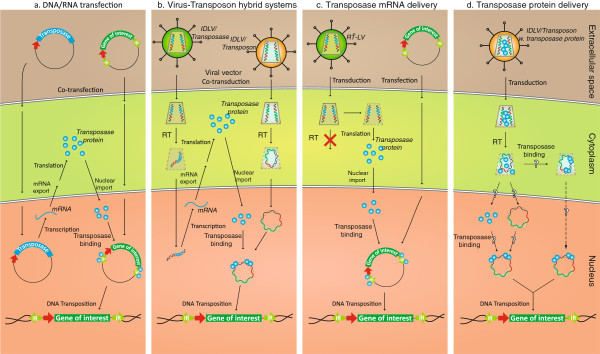
Schematic representation of four different approaches for delivery of components of the DNA transposon-based gene delivery systems. Panel a illustrates the conventional delivery of transposase and transposon by plasmid DNA transfection. This approach relies on nuclear uptake of both helper and donor plasmid DNA allowing transcription of transposase-encoding mRNA, mRNA export, production of transposase in the cytoplasm, and subsequent nuclear import of transposases. Transposases bind to the transposon donor plasmid and facilitates transposition. A variant of this approach is based on transfection of in vitro-transcribed mRNA encoding the transposase (not shown). Panel b represents an emerging approach based on virus-mediated delivery of DNA transposon systems. The example shown demonstrates the use of integrase-defective lentiviral vector (IDLVs) as carriers of the transposase gene (left) and the transposon (right), allowing transposition from reverse-transcribed (RT) lentiviral DNA intermediates (here represented by circular forms). Related approaches have been developed for vectors based on adenoviruses, adeno-associated viruses, and herpes simplex viruses. Panel c illustrates the use of reverse transcription-defective retroviral vectors as carriers of transposase-encoding mRNA. Modifications of the primer binding site, where reverse transcriptions is normally initiated by an annealed tRNA, inhibit reverse transcription and thus facilitating vector RNA delivery, and direct translation into protein. The transposon donor is in this example delivered by plasmid DNA transfection. Panel d demonstrates the possibility of delivering DNA transposon systems in engineered ‘all-in-one’ lentiviral particles that co-deliver both transposase protein and the donor for DNA transposition. Transposase subunits delivered by lentiviral protein transduction are delivered in the virus context and facilitate efficient transposition through mechanisms that may benefit from the close interaction between transposases and the reverse-trancribed donor within the viral pre-integration complex. Question marks indicate that it is currently unknown at which stage transposases bind to the donor to form the synaptic transposition complex.
Alternative means of delivering the components needed in transposon-based gene delivery systems have emerged over the years. In prominent hybrid delivery systems the components of transposon-based vectors are carried and delivered by viral capsids, providing otherwise episomal vectors – like adenoviral [208-210] or herpes simplex virus-based vectors [211] – the ability to integrate genes and establish long-term transgene expression. Combining the best of two worlds the viral coat provides vector stability, tissue-specific transposon delivery and transport across the cellular membrane, while transposons facilitate viral vector integration according to the characteristic integration profile of transposable elements [51,208]. Viral vector-based transfer of the Sleeping Beauty transposon system was first demonstrated in mouse liver with adenoviral vectors [208], and recent studies have demonstrated the applicability of this approach in larger animals [210]. Also, adeno-associated viral vectors have been adapted as carriers of the Sleeping Beauty system [212]. We and others have established Sleeping Beauty DNA transposition from integrase-defective lentiviral vectors (IDLVs) (Figure 7, panel b), providing a new viral platform for delivering components of the Sleeping Beauty transposition system and altering the integration profile of lentivirally delivered transgenes [139,204,213]. Along these lines, retroviral vectors without the ability to undergo reverse transcription have been explored as a source of transposase-encoding mRNA (Figure 7, panel c) [214]. Most recently, we developed lentiviral protein transduction for direct delivery of transposase protein, allowing efficient DNA transposition in lentivirally transduced cells (Figure 7, panel d) [215]. Efforts to combine viral gene delivery with non-viral integration systems in ‘hybrid’ vectors have recently been reviewed [209,216,217].
Preclinical evidence of transposon-based gene delivery
Among the elements with activity in mammalian cells Sleeping Beauty is by far the most studied for gene transfer purposes. Several studies have demonstrated high levels of gene insertion in mouse liver after hydrodynamic injection of naked DNA into the tail vein. Proof-of-principle was initially provided by Yant et al. who obtained persistent levels of human coagulation factor IX (hFIX) after hydrodynamic injection of plasmids carrying the transgene-tagged transposon and the transposase expression cassette, respectively [5]. Delivery of the SB100X transposase variant and hyperactive piggyBac was found later to provide even higher levels of transposition in mouse liver [53]. As this type of treatment was found to be therapeutic in a mouse model of hemophilia B [5], recent findings have demonstrated long-term phenotypic correction in immunotolerized hemophilia A mice treated with a transposon encoding human factor VIII (hFVIII) [8]. In addition, the Sleeping Beauty system has been successful in liver-directed treatment of mucopolysaccharidosis types I and VII [218] and tyrosinemia type I [219], and administration of in vitro synthesized RNA encoding the Sleeping Beauty transposase, as opposed to plasmid DNA, is sufficient to catalyze vector transposition in mouse liver [207,220]. Initial studies of Sleeping Beauty-directed transfer of hFIX showed efficacy but also demonstrated severe OPI in mice treated with too high levels of plasmid DNA with transposase expression driven by a standard cytomegalovirus (CMV) promoter. Hence, whereas a single microgram of transposase-encoding plasmid DNA was found to facilitate high levels of transposition, resulting in correction of the disease in mouse model of hemophilia, injection of 25 μg transposase-encoding plasmid DNA on the other hand did not result in DNA transposition [5]. In subsequent studies using helper-independent transposon-transposase vectors, the transposase expression was balanced by the use of a promoter that were markedly weaker than CMV [7]. It is not clear whether such OPI in mouse liver reflects true transposase-regulating mechanisms that mirror natural regulatory mechanisms of Sleeping Beauty, or rather is caused by toxicity in hepatocytes with high expression of the transposase. In either case, these findings marked the importance of recognizing specific characteristics of the vector technology and balancing the expression of the transposase for each specific application.
By administration of DNA-polyethylenimine complexes, Sleeping Beauty-containing plasmid vectors have been delivered to lung epithelial cells [12], allowing phenotypic correction in hemophilia A mice neonatally injected with a transposon vector encoding the hFVIII gene [16]. Gene expression in the alveolar region of Sleeping Beauty-treated mouse lungs may last as long as 3 months after injection [11]. Promising preclinical effects of Sleeping Beauty have been elegantly demonstrated in human glioblastoma xenografts in mice in which tumor-induced angiogenesis was inhibited by Sleeping Beauty-based co-expression of soluble vascular endothelial growth factor and a angiostatin-endostatin fusion variant [18,19]. Sustained tumor regression of intracranial gliomas was achieved only when functional Sleeping Beauty transposase was present, allowing persistent expression of the two genes.
Conclusion
Mobile elements heading for the clinic
Fifteen years have gone since the Sleeping Beauty element was re-awakened and soon after adapted for in vivo gene delivery facilitating persistent gene expression. Despite the various examples of pre-clinical efficacy for DNA transposon-based in vivo gene therapy, the road to the clinic will wind through additional experimentation and evidence of therapeutic effects in large animal models. As we keep learning more about the properties and details of the different transposable elements and continue developing both nonviral and viral delivery technologies, in vivo applicability could be waiting around the next corner. Until then, the history of mobile elements is helping us refining these engineering tools even further for use in both biomedical experimentation and clinical settings. With the current primary focus on the Sleeping Beauty and piggyBac elements, mechanisms that guide the distinct integration patterns, OPI, gene cargo capacities, putative cell type variations, and sensitivity to epigenetic silencing of these two elements seem to be key focus points for optimizing the performance of these elements further.
The combination of improved transposons, possibly mobilized from DNA minicircles free of bacterial sequences [221], and engineered hyperactive transposase variants has immediate potential in developing ex vivo therapies. Transposon-based gene insertion in plasmid-transfected primary T cells [222-225], hematopoietic stem cells [204,226-228], and human embryonic stem cells [229,230] is creating optimism for the use of DNA transposons in ex vivo genetic treatment. In fact, the first clinical trial based on transposon-directed gene integration aims at inserting a gene encoding a chimeric antigen receptor specific for CD19 in primary T cells [222] and has recently been initiated for treatment of patients with B-lymphoid malignancies with adoptive immunotherapy [231-233]. Through scenes of an evolutionary drive, DNA transposon-based vectors arrived at the clinic, but this is certainly not the final stop.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The manuscript was prepared by KAS, PRA, NS and JGM. All authors read and approved the final manuscript.
Contributor Information
Kristian Alsbjerg Skipper, Email: kas@hum-gen.au.dk.
Peter Refsing Andersen, Email: pra@mb.au.dk.
Nynne Sharma, Email: nynnesharma@hotmail.com.
Jacob Giehm Mikkelsen, Email: giehm@hum-gen.au.dk.
Acknowledgments
Work in the laboratory of Jacob Giehm Mikkelsen has been made possible over the years through support by The Danish Council for Independent Research | Medical Sciences, the Lundbeck Foundation, the Novo Nordisk Foundation, EU FP6 (INTHER), the Danish Cancer Society, the Hørslev Foundation, The Riisfort Foundation, Aase og Ejnar Danielsens Fond, Agnes og Poul Friis Fond, Aage Bangs Fond, Grosserer A. V. Lykfeldt og Hustrus Legat, Else og Mogens Wedell-Wedellsborgs Fond, Fonden af 17-12-1981, Kong Christian den Tiendes Fond, Frits, Georg og Marie Cecilie Gluds Legat, Civilingeniør Frode V. Nyegaard og hustrus Fond, Helga og Peter Kornings Fond, Oda og Hans Svenningsens Fond, Snedkermester Sophus Jacobsen & Hustru Astrid Jacobsens Fond, Kirsten Anthonius Mindelegat, Familien Hede Nielsens Fond, and Grosserer L. F. Foghts Fond. JGM is head of Gene Therapy Initiative Aarhus (GTI-Aarhus) funded by the Lundbeck Foundation and a member of the Aarhus Research Center for Innate Immunology (ARCII) established through funding by the AU-Ideas program at Aarhus University.
References
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, Dionisio F, Calabria A, Giannelli S, Castiello MC. et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;20:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, Baldoli C, Martino S, Calabria A, Canale S. et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;20:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Nathwani AC, Tuddenham EG, Rangarajan S, Rosales C, McIntosh J, Linch DC, Chowdary P, Riddell A, Pie AJ, Harrington C. et al. Adenovirus-associated virus vector-mediated gene transfer in hemophilia B. N Engl J Med. 2011;20:2357–2365. doi: 10.1056/NEJMoa1108046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;20:501–510. doi: 10.1016/S0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- Yant SR, Meuse L, Chiu W, Ivics Z, Izsvak Z, Kay MA. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet. 2000;20:35–41. doi: 10.1038/75568. [DOI] [PubMed] [Google Scholar]
- Ehrhardt A, Xu H, Huang Z, Engler JA, Kay MA. A direct comparison of two nonviral gene therapy vectors for somatic integration: in vivo evaluation of the bacteriophage integrase phiC31 and the Sleeping Beauty transposase. Mol Ther. 2005;20:695–706. doi: 10.1016/j.ymthe.2005.01.010. [DOI] [PubMed] [Google Scholar]
- Mikkelsen JG, Yant SR, Meuse L, Huang Z, Xu H, Kay MA. Helper-Independent Sleeping Beauty transposon-transposase vectors for efficient nonviral gene delivery and persistent gene expression in vivo. Mol Ther. 2003;20:654–665. doi: 10.1016/S1525-0016(03)00216-8. [DOI] [PubMed] [Google Scholar]
- Ohlfest JR, Frandsen JL, Fritz S, Lobitz PD, Perkinson SG, Clark KJ, Nelsestuen G, Key NS, McIvor RS, Hackett PB, Largaespada DA. Phenotypic correction and long-term expression of factor VIII in hemophilic mice by immunotolerization and nonviral gene transfer using the Sleeping Beauty transposon system. Blood. 2005;20:2691–2698. doi: 10.1182/blood-2004-09-3496. [DOI] [PubMed] [Google Scholar]
- Yant SR, Kay MA. Nonhomologous-end-joining factors regulate DNA repair fidelity during Sleeping Beauty element transposition in mammalian cells. Mol Cell Biol. 2003;20:8505–8518. doi: 10.1128/MCB.23.23.8505-8518.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronovich EL, Bell JB, Khan SA, Belur LR, Gunther R, Koniar B, Schachern PA, Parker JB, Carlson CS, Whitley CB. et al. Systemic correction of storage disease in MPS I NOD/SCID mice using the sleeping beauty transposon system. Mol Ther. 2009;20:1136–1144. doi: 10.1038/mt.2009.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belur LR, Frandsen JL, Dupuy AJ, Ingbar DH, Largaespada DA, Hackett PB, Scott McIvor R. Gene insertion and long-term expression in lung mediated by the Sleeping Beauty transposon system. Mol Ther. 2003;20:501–507. doi: 10.1016/S1525-0016(03)00211-9. [DOI] [PubMed] [Google Scholar]
- Liu L, Sanz S, Heggestad AD, Antharam V, Notterpek L, Fletcher BS. Endothelial targeting of the Sleeping Beauty transposon within lung. Mol Ther. 2004;20:97–105. doi: 10.1016/j.ymthe.2004.04.006. [DOI] [PubMed] [Google Scholar]
- Liu H, Liu L, Fletcher BS, Visner GA. Sleeping Beauty-based gene therapy with indoleamine 2,3-dioxygenase inhibits lung allograft fibrosis. Faseb J. 2006;20:2384–2386. doi: 10.1096/fj.06-6228fje. [DOI] [PubMed] [Google Scholar]
- Belur LR, Podetz-Pedersen K, Frandsen J, McIvor RS. Lung-directed gene therapy in mice using the nonviral Sleeping Beauty transposon system. Nat Protoc. 2007;20:3146–3152. doi: 10.1038/nprot.2007.460. [DOI] [PubMed] [Google Scholar]
- Lin EH, Keramidas M, Rome C, Chiu WT, Wu CW, Coll JL, Deng WP. Lifelong reporter gene imaging in the lungs of mice following polyethyleneimine-mediated sleeping-beauty transposon delivery. Biomaterials. 2011;20:1978–1985. doi: 10.1016/j.biomaterials.2010.11.026. [DOI] [PubMed] [Google Scholar]
- Liu L, Mah C, Fletcher BS. Sustained FVIII expression and phenotypic correction of hemophilia A in neonatal mice using an endothelial-targeted sleeping beauty transposon. Mol Ther. 2006;20:1006–1015. doi: 10.1016/j.ymthe.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Ortiz-Urda S, Lin Q, Yant SR, Keene D, Kay MA, Khavari PA. Sustainable correction of junctional epidermolysis bullosa via transposon-mediated nonviral gene transfer. Gene Ther. 2003;20:1099–1104. doi: 10.1038/sj.gt.3301978. [DOI] [PubMed] [Google Scholar]
- Ohlfest JR, Demorest ZL, Motooka Y, Vengco I, Oh S, Chen E, Scappaticci FA, Saplis RJ, Ekker SC, Low WC. et al. Combinatorial antiangiogenic gene therapy by nonviral gene transfer using the sleeping beauty transposon causes tumor regression and improves survival in mice bearing intracranial human glioblastoma. Mol Ther. 2005;20:778–788. doi: 10.1016/j.ymthe.2005.07.689. [DOI] [PubMed] [Google Scholar]
- Ohlfest JR, Lobitz PD, Perkinson SG, Largaespada DA. Integration and long-term expression in xenografted human glioblastoma cells using a plasmid-based transposon system. Mol Ther. 2004;20:260–268. doi: 10.1016/j.ymthe.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Prak ET, Kazazian HH Jr. Mobile elements and the human genome. Nat Rev Genet. 2000;20:134–144. doi: 10.1038/35038572. [DOI] [PubMed] [Google Scholar]
- Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opin Genet Dev. 1999;20:657–663. doi: 10.1016/S0959-437X(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W. et al. Initial sequencing and analysis of the human genome. Nature. 2001;20:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Cooley L, Kelley R, Spradling A. Insertional mutagenesis of the Drosophila genome with single P elements. Science. 1988;20:1121–1128. doi: 10.1126/science.2830671. [DOI] [PubMed] [Google Scholar]
- Mackay TF, Lyman RF, Jackson MS. Effects of P element insertions on quantitative traits in Drosophila melanogaster. Genetics. 1992;20:315–332. doi: 10.1093/genetics/130.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikitin AG, Woodruff RC. Somatic movement of the mariner transposable element and lifespan of Drosophila species. Mutat Res. 1995;20:43–49. doi: 10.1016/0921-8734(95)00010-4. [DOI] [PubMed] [Google Scholar]
- Cary LC, Goebel M, Corsaro BG, Wang HG, Rosen E, Fraser MJ. Transposon mutagenesis of baculoviruses: analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology. 1989;20:156–169. doi: 10.1016/0042-6822(89)90117-7. [DOI] [PubMed] [Google Scholar]
- Fraser MJ, Ciszczon T, Elick T, Bauser C. Precise excision of TTAA-specific lepidopteran transposons piggyBac (IFP2) and tagalong (TFP3) from the baculovirus genome in cell lines from two species of Lepidoptera. Insect Mol Biol. 1996;20:141–151. doi: 10.1111/j.1365-2583.1996.tb00048.x. [DOI] [PubMed] [Google Scholar]
- Burnight ER, Staber JM, Korsakov P, Li X, Brett BT, Scheetz TE, Craig NL, McCray PB Jr. A Hyperactive Transposase Promotes Persistent Gene Transfer of a piggyBac DNA Transposon. Mol Ther Nucleic Acids. 2012;20:e50. doi: 10.1038/mtna.2012.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty JE, Huye LE, Yusa K, Zhou L, Craig NL, Wilson MH. Hyperactive piggyBac gene transfer in human cells and in vivo. Hum Gene Ther. 2012;20:311–320. doi: 10.1089/hum.2011.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskey C, Izsvak Z, Plasterk RH, Ivics Z. The Frog Prince: a reconstructed transposon from Rana pipiens with high transpositional activity in vertebrate cells. Nucleic Acids Res. 2003;20:6873–6881. doi: 10.1093/nar/gkg910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe DJ, Churchill ME, Robertson HM. A purified mariner transposase is sufficient to mediate transposition in vitro. EMBO J. 1996;20:5470–5479. [PMC free article] [PubMed] [Google Scholar]
- Kawakami K, Koga A, Hori H, Shima A. Excision of the tol2 transposable element of the medaka fish, Oryzias latipes, in zebrafish, Danio rerio. Gene. 1998;20:17–22. doi: 10.1016/S0378-1119(98)00537-X. [DOI] [PubMed] [Google Scholar]
- Clark KJ, Carlson DF, Leaver MJ, Foster LK, Fahrenkrug SC. Passport, a native Tc1 transposon from flatfish, is functionally active in vertebrate cells. Nucleic Acids Res. 2009;20:1239–1247. doi: 10.1093/nar/gkn1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miskey C, Papp B, Mates L, Sinzelle L, Keller H, Izsvak Z, Ivics Z. The ancient mariner sails again: transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol Cell Biol. 2007;20:4589–4600. doi: 10.1128/MCB.02027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R, Li X, Kapusta A, Mayhew D, Mitra RD, Feschotte C, Craig NL. Functional characterization of piggyBat from the bat Myotis lucifugus unveils an active mammalian DNA transposon. Proc Natl Acad Sci USA. 2013;20:234–239. doi: 10.1073/pnas.1217548110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Ewis H, Hice RH, Malani N, Parker N, Zhou L, Feschotte C, Bushman FD, Atkinson PW, Craig NL. A resurrected mammalian hAT transposable element and a closely related insect element are highly active in human cell culture. Proc Natl Acad Sci USA. 2013;20:E478–E487. doi: 10.1073/pnas.1121543109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodard LE, Li X, Malani N, Kaja A, Hice RH, Atkinson PW, Bushman FD, Craig NL, Wilson MH. Comparative analysis of the recently discovered hAT transposon TcBuster in human cells. PLoS One. 2012;20:e42666. doi: 10.1371/journal.pone.0042666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayed H, Izsvak Z, Khare D, Heinemann U, Ivics Z. The DNA-bending protein HMGB1 is a cellular cofactor of Sleeping Beauty transposition. Nucleic Acids Res. 2003;20:2313–2322. doi: 10.1093/nar/gkg341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izsvak Z, Ivics Z, Plasterk RH. Sleeping Beauty, a wide host-range transposon vector for genetic transformation in vertebrates. J Mol Biol. 2000;20:93–102. doi: 10.1006/jmbi.2000.4047. [DOI] [PubMed] [Google Scholar]
- Ivics Z, Kaufman CD, Zayed H, Miskey C, Walisko O, Izsvak Z. The Sleeping Beauty transposable element: evolution, regulation and genetic applications. Curr Issues Mol Biol. 2004;20:43–55. [PubMed] [Google Scholar]
- Izsvak Z, Ivics Z. Sleeping beauty transposition: biology and applications for molecular therapy. Mol Ther. 2004;20:147–156. doi: 10.1016/j.ymthe.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Hackett PB, Ekker SC, Largaespada DA, McIvor RS. Sleeping beauty transposon-mediated gene therapy for prolonged expression. Adv Genet. 2005;20:189–232. doi: 10.1016/S0065-2660(05)54009-4. [DOI] [PubMed] [Google Scholar]
- Ammar I, Izsvak Z, Ivics Z. The sleeping beauty transposon toolbox. Methods Mol Biol. 2012;20:229–240. doi: 10.1007/978-1-61779-603-6_13. [DOI] [PubMed] [Google Scholar]
- Aronovich EL, McIvor RS, Hackett PB. The sleeping beauty transposon system: a non-viral vector for gene therapy. Hum Mol Genet. 2011;20:R14–R20. doi: 10.1093/hmg/ddr140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Matteo M, Belay E, Chuah MK, Vandendriessche T. Recent developments in transposon-mediated gene therapy. Expert Opin Biol Ther. 2012;20:841–858. doi: 10.1517/14712598.2012.684875. [DOI] [PubMed] [Google Scholar]
- Ivics Z, Izsvak Z. Nonviral gene delivery with the sleeping beauty transposon system. Hum Gene Ther. 2011;20:1043–1051. doi: 10.1089/hum.2011.143. [DOI] [PubMed] [Google Scholar]
- Swierczek M, Izsvak Z, Ivics Z. The sleeping beauty transposon system for clinical applications. Expert Opin Biol Ther. 2012;20:139–153. doi: 10.1517/14712598.2012.642358. [DOI] [PubMed] [Google Scholar]
- Di Matteo M, Matrai J, Belay E, Firdissa T, Vandendriessche T, Chuah MK. PiggyBac toolbox. Methods Mol Biol. 2012;20:241–254. doi: 10.1007/978-1-61779-603-6_14. [DOI] [PubMed] [Google Scholar]
- Munoz-Lopez M, Garcia-Perez JL. DNA transposons: nature and applications in genomics. Curr Genomics. 2010;20:115–128. doi: 10.2174/138920210790886871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigdal TJ, Kaufman CD, Izsvak Z, Voytas DF, Ivics Z. Common physical properties of DNA affecting target site selection of sleeping beauty and other Tc1/mariner transposable elements. J Mol Biol. 2002;20:441–452. doi: 10.1016/S0022-2836(02)00991-9. [DOI] [PubMed] [Google Scholar]
- Yant SR, Wu X, Huang Y, Garrison B, Burgess SM, Kay MA. High-resolution genome-wide mapping of transposon integration in mammals. Mol Cell Biol. 2005;20:2085–2094. doi: 10.1128/MCB.25.6.2085-2094.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zayed H, Izsvak Z, Walisko O, Ivics Z. Development of hyperactive sleeping beauty transposon vectors by mutational analysis. Mol Ther. 2004;20:292–304. doi: 10.1016/j.ymthe.2003.11.024. [DOI] [PubMed] [Google Scholar]
- Mates L, Chuah MK, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, Grzela DP, Schmitt A, Becker K, Matrai J. et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet. 2009;20:753–761. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- Liu G, Geurts AM, Yae K, Srinivasan AR, Fahrenkrug SC, Largaespada DA, Takeda J, Horie K, Olson WK, Hackett PB. Target-site preferences of Sleeping Beauty transposons. J Mol Biol. 2005;20:161–173. doi: 10.1016/j.jmb.2004.09.086. [DOI] [PubMed] [Google Scholar]
- Claeys Bouuaert C, Chalmers R. Transposition of the human Hsmar1 transposon: rate-limiting steps and the importance of the flanking TA dinucleotide in second strand cleavage. Nucleic Acids Res. 2010;20:190–202. doi: 10.1093/nar/gkp891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeys Bouuaert C, Chalmers R. Hsmar1 transposition is sensitive to the topology of the transposon donor and the target. PLoS One. 2013;20:e53690. doi: 10.1371/journal.pone.0053690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe DJ, Grant TE, Robertson HM. Factors affecting transposition of the Himar1 mariner transposon in vitro. Genetics. 1998;20:179–187. doi: 10.1093/genetics/149.1.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keravala A, Liu D, Lechman ER, Wolfe D, Nash JA, Lampe DJ, Robbins PD. Hyperactive Himar1 transposase mediates transposition in cell culture and enhances gene expression in vivo. Human gene therapy. 2006;20:1006–1018. doi: 10.1089/hum.2006.17.1006. [DOI] [PubMed] [Google Scholar]
- Pelicic V, Morelle S, Lampe D, Nassif X. Mutagenesis of Neisseria meningitidis by in vitro transposition of Himar1 mariner. J of bacteriol. 2000;20:5391–5398. doi: 10.1128/JB.182.19.5391-5398.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Meir YJ, Coates CJ, Handler AM, Pelczar P, Moisyadi S, Kaminski JM. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc Natl Acad Sci USA. 2006;20:15008–15013. doi: 10.1073/pnas.0606979103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga A, Iida A, Kamiya M, Hayashi R, Hori H, Ishikawa Y, Tachibana A. The medaka fish Tol2 transposable element can undergo excision in human and mouse cells. J of hum genet. 2003;20:231–235. doi: 10.1007/s10038-003-0016-4. [DOI] [PubMed] [Google Scholar]
- Urasaki A, Morvan G, Kawakami K. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics. 2006;20:639–649. doi: 10.1534/genetics.106.060244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MH, Coates CJ, George AL Jr. PiggyBac Transposon-mediated Gene Transfer in Human Cells. Mol Ther. 2007;20:139–145. doi: 10.1038/sj.mt.6300028. [DOI] [PubMed] [Google Scholar]
- Yusa K, Zhou L, Li MA, Bradley A, Craig NL. A hyperactive piggyBac transposase for mammalian applications. Proc Natl Acad Sci U S A. 2011;20:1531–1536. doi: 10.1073/pnas.1008322108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy EM, McDonald JF. Long terminal repeat retrotransposons of Mus musculus. Genome Biol. 2004;20:R14. doi: 10.1186/gb-2004-5-3-r14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohshima K, Okada N. SINEs and LINEs: symbionts of eukaryotic genomes with a common tail. Cytogenet Genome Res. 2005;20:475–490. doi: 10.1159/000084981. [DOI] [PubMed] [Google Scholar]
- Jehle JA, Nickel A, Vlak JM, Backhaus H. Horizontal escape of the novel Tc1-like lepidopteran transposon TCp3.2 into Cydia pomonella granulovirus. J Mol Evol. 1998;20:215–224. doi: 10.1007/PL00006296. [DOI] [PubMed] [Google Scholar]
- Kaufman PD, Rio DC. P element transposition in vitro proceeds by a cut-and-paste mechanism and uses GTP as a cofactor. Cell. 1992;20:27–39. doi: 10.1016/0092-8674(92)90116-T. [DOI] [PubMed] [Google Scholar]
- Van Luenen HG, Colloms SD, Plasterk RH. The mechanism of transposition of Tc3 in C. elegans. Cell. 1994;20:293–301. doi: 10.1016/0092-8674(94)90198-8. [DOI] [PubMed] [Google Scholar]
- Craig N, Craigie R, Gellert M, Lambowitz A. Mobile DNA II. Washington, DC: ASM Press; 2002. [Google Scholar]
- Bryan G, Garza D, Hartl D. Insertion and excision of the transposable element mariner in Drosophila. Genetics. 1990;20:103–114. doi: 10.1093/genetics/125.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra R, Fain-Thornton J, Craig NL. piggyBac can bypass DNA synthesis during cut and paste transposition. EMBO J. 2008;20:1097–1109. doi: 10.1038/emboj.2008.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L, Mitra R, Atkinson PW, Hickman AB, Dyda F, Craig NL. Transposition of hAT elements links transposable elements and V(D)J recombination. Nature. 2004;20:995–1001. doi: 10.1038/nature03157. [DOI] [PubMed] [Google Scholar]
- Izsvak Z, Khare D, Behlke J, Heinemann U, Plasterk RH, Ivics Z. Involvement of a bifunctional, paired-like DNA-binding domain and a transpositional enhancer in Sleeping Beauty transposition. J Biol Chem. 2002;20:34581–34588. doi: 10.1074/jbc.M204001200. [DOI] [PubMed] [Google Scholar]
- Fattash I, Rooke R, Wong A, Hui C, Luu T, Bhardwaj P, Yang G. Miniature inverted-repeat transposable elements: discovery, distribution, and activity. Genome. 2013;20:475–486. doi: 10.1139/gen-2012-0174. [DOI] [PubMed] [Google Scholar]
- Kapitonov VV, Jurka J. Helitrons on a roll: eukaryotic rolling-circle transposons. Trends Genet. 2007;20:521–529. doi: 10.1016/j.tig.2007.08.004. [DOI] [PubMed] [Google Scholar]
- Pritham EJ, Feschotte C. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc Natl Acad Sci USA. 2007;20:1895–1900. doi: 10.1073/pnas.0609601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva JC, Loreto EL, Clark JB. Factors that affect the horizontal transfer of transposable elements. Curr Issues Mol Biol. 2004;20:57–71. [PubMed] [Google Scholar]
- Lohe AR, Moriyama EN, Lidholm DA, Hartl DL. Horizontal transmission, vertical inactivation, and stochastic loss of mariner-like transposable elements. Mol Biol Evol. 1995;20:62–72. doi: 10.1093/oxfordjournals.molbev.a040191. [DOI] [PubMed] [Google Scholar]
- Pinsker W, Haring E, Hagemann S, Miller WJ. The evolutionary life history of P transposons: from horizontal invaders to domesticated neogenes. Chromosoma. 2001;20:148–158. doi: 10.1007/s004120100144. [DOI] [PubMed] [Google Scholar]
- Kuraku S, Qiu H, Meyer A. Horizontal transfers of Tc1 elements between teleost fishes and their vertebrate parasites, lampreys. Genome Biol Evol. 2012;20:929–936. doi: 10.1093/gbe/evs069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira SG, Bao W, Martins C, Jurka J. Horizontal transfers of Mariner transposons between mammals and insects. Mob DNA. 2012;20:14. doi: 10.1186/1759-8753-3-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace JK II, Feschotte C. The evolutionary history of human DNA transposons: evidence for intense activity in the primate lineage. Genome Res. 2007;20:422–432. doi: 10.1101/gr.5826307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels SB, Peterson KR, Strausbaugh LD, Kidwell MG, Chovnick A. Evidence for horizontal transmission of the P transposable element between Drosophila species. Genetics. 1990;20:339–355. doi: 10.1093/genetics/124.2.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leaver MJ. A family of Tc1-like transposons from the genomes of fishes and frogs: evidence for horizontal transmission. Gene. 2001;20:203–214. doi: 10.1016/S0378-1119(01)00530-3. [DOI] [PubMed] [Google Scholar]
- Koga A, Shimada A, Shima A, Sakaizumi M, Tachida H, Hori H. Evidence for recent invasion of the medaka fish genome by the Tol2 transposable element. Genetics. 2000;20:273–281. doi: 10.1093/genetics/155.1.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handler AM, McCombs SD. The piggyBac transposon mediates germ-line transformation in the Oriental fruit fly and closely related elements exist in its genome. Insect Mol Biol. 2000;20:605–612. doi: 10.1046/j.1365-2583.2000.00227.x. [DOI] [PubMed] [Google Scholar]
- Fraser MJ, Smith GE, Summers MD. Acquisition of Host Cell DNA Sequences by Baculoviruses: Relationship Between Host DNA Insertions and FP Mutants of Autographa californica and Galleria mellonella Nuclear Polyhedrosis Viruses. J Virol. 1983;20:287–300. doi: 10.1128/jvi.47.2.287-300.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser MJ, Brusca JS, Smith GE, Summers MD. Transposon-mediated mutagenesis of a baculovirus. Virology. 1985;20:356–361. doi: 10.1016/0042-6822(85)90172-2. [DOI] [PubMed] [Google Scholar]
- Miller DW, Miller LK. A virus mutant with an insertion of a copia-like transposable element. Nature. 1982;20:562–564. doi: 10.1038/299562a0. [DOI] [PubMed] [Google Scholar]
- Yoshiyama M, Tu Z, Kainoh Y, Honda H, Shono T, Kimura K. Possible horizontal transfer of a transposable element from host to parasitoid. Mol Biol Evol. 2001;20:1952–1958. doi: 10.1093/oxfordjournals.molbev.a003735. [DOI] [PubMed] [Google Scholar]
- Houck MA, Clark JB, Peterson KR, Kidwell MG. Possible horizontal transfer of Drosophila genes by the mite Proctolaelaps regalis. Science. 1991;20:1125–1128. doi: 10.1126/science.1653453. [DOI] [PubMed] [Google Scholar]
- Kidwell MG. Horizontal transfer of P elements and other short inverted repeat transposons. Genetica. 1992;20:275–286. doi: 10.1007/BF00133726. [DOI] [PubMed] [Google Scholar]
- Chen J, Greenblatt IM, Dellaporta SL. Molecular analysis of Ac transposition and DNA replication. Genetics. 1992;20:665–676. doi: 10.1093/genetics/130.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ros F, Kunze R. Regulation of activator/dissociation transposition by replication and DNA methylation. Genetics. 2001;20:1723–1733. doi: 10.1093/genetics/157.4.1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Heinlein M, Kunze R. Methylation pattern of Activator transposase binding sites in maize endosperm. Plant Cell. 1996;20:747–758. doi: 10.1105/tpc.8.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZH, Liu DP, Wang J, Guo ZC, Yin WX, Liang CC. Inversion and transposition of Tc1 transposon of C. elegans in mammalian cells. Somat Cell Mol Genet. 1998;20:363–369. doi: 10.1023/A:1024494508878. [DOI] [PubMed] [Google Scholar]
- Robertson HM, Zumpano KL, Lohe AR, Hartl DL. Reconstructing the ancient mariners of humans. Nat Genet. 1996;20:360–361. doi: 10.1038/ng0496-360. [DOI] [PubMed] [Google Scholar]
- Dodson KW, Berg DE. Factors affecting transposition activity of IS50 and Tn5 ends. Gene. 1989;20:207–213. doi: 10.1016/0378-1119(89)90161-3. [DOI] [PubMed] [Google Scholar]
- Rezsohazy R, Hallet B, Delcour J, Mahillon J. The IS4 family of insertion sequences: evidence for a conserved transposase motif. Mol Microbiol. 1993;20:1283–1295. doi: 10.1111/j.1365-2958.1993.tb01258.x. [DOI] [PubMed] [Google Scholar]
- Bender J, Kleckner N. Genetic evidence that Tn10 transposes by a nonreplicative mechanism. Cell. 1986;20:801–815. doi: 10.1016/0092-8674(86)90555-6. [DOI] [PubMed] [Google Scholar]
- Haniford DB, Benjamin HW, Kleckner N. Kinetic and structural analysis of a cleaved donor intermediate and a strand transfer intermediate in Tn10 transposition. Cell. 1991;20:171–179. doi: 10.1016/0092-8674(91)90218-N. [DOI] [PubMed] [Google Scholar]
- Biel SW, Berg DE. Mechanism of IS1 transposition in E. coli: choice between simple insertion and cointegration. Genetics. 1984;20:319–330. doi: 10.1093/genetics/108.2.319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iida S, Mollet B, Meyer J, Arber W. Functional characterization of the prokaryotic mobile genetic element IS26. Mol Gen Genet. 1984;20:84–89. doi: 10.1007/BF00328705. [DOI] [PubMed] [Google Scholar]
- Engels WR, Johnson-Schlitz DM, Eggleston WB, Sved J. High-frequency P element loss in Drosophila is homolog dependent. Cell. 1990;20:515–525. doi: 10.1016/0092-8674(90)90016-8. [DOI] [PubMed] [Google Scholar]
- Engels WR, Preston CR, Johnson-Schlitz DM. Long-range cis preference in DNA homology search over the length of a Drosophila chromosome. Science. 1994;20:1623–1625. doi: 10.1126/science.8128250. [DOI] [PubMed] [Google Scholar]
- Plasterk RH. The origin of footprints of the Tc1 transposon of Caenorhabditis elegans. Embo J. 1991;20:1919–1925. doi: 10.1002/j.1460-2075.1991.tb07718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasterk RH, Groenen JT. Targeted alterations of the Caenorhabditis elegans genome by transgene instructed DNA double strand break repair following Tc1 excision. Embo J. 1992;20:287–290. doi: 10.1002/j.1460-2075.1992.tb05051.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Formosa T, Alberts BM. DNA synthesis dependent on genetic recombination: characterization of a reaction catalyzed by purified bacteriophage T4 proteins. Cell. 1986;20:793–806. doi: 10.1016/0092-8674(86)90522-2. [DOI] [PubMed] [Google Scholar]
- Nassif N, Penney J, Pal S, Engels WR, Gloor GB. Efficient copying of nonhomologous sequences from ectopic sites via P-element-induced gap repair. Mol Cell Biol. 1994;20:1613–1625. doi: 10.1128/mcb.14.3.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin E, Levy AA. Abortive gap repair: underlying mechanism for Ds element formation. Mol Cell Biol. 1997;20:6294–6302. doi: 10.1128/mcb.17.11.6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefferin ML, Tomkinson AE. Mechanism of DNA double-strand break repair by non-homologous end joining. DNA Repair (Amst) 2005;20:639–648. doi: 10.1016/j.dnarep.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Staveley BE, Heslip TR, Hodgetts RB, Bell JB. Protected P-element termini suggest a role for inverted-repeat-binding protein in transposase-induced gap repair in Drosophila melanogaster. Genetics. 1995;20:1321–1329. doi: 10.1093/genetics/139.3.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izsvak Z, Stuwe EE, Fiedler D, Katzer A, Jeggo PA, Ivics Z. Healing the wounds inflicted by sleeping beauty transposition by double-strand break repair in mammalian somatic cells. Mol Cell. 2004;20:279–290. doi: 10.1016/S1097-2765(03)00524-0. [DOI] [PubMed] [Google Scholar]
- Beall EL, Admon A, Rio DC. A Drosophila protein homologous to the human p70 Ku autoimmune antigen interacts with the P transposable element inverted repeats. Proc Natl Acad Sci USA. 1994;20:12681–12685. doi: 10.1073/pnas.91.26.12681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McVey M, Radut D, Sekelsky JJ. End-joining repair of double-strand breaks in Drosophila melanogaster is largely DNA ligase IV independent. Genetics. 2004;20:2067–2076. doi: 10.1534/genetics.104.033902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston CR, Engels W, Flores C. Efficient repair of DNA breaks in Drosophila: evidence for single-strand annealing and competition with other repair pathways. Genetics. 2002;20:711–720. doi: 10.1093/genetics/161.2.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preston CR, Flores CC, Engels WR. Differential usage of alternative pathways of double-strand break repair in Drosophila. Genetics. 2006;20:1055–1068. doi: 10.1534/genetics.105.050138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Min B, Rio DC. P element excision and repair by non-homologous end joining occurs in both G1 and G2 of the cell cycle. DNA Repair. 2005;20:171–181. doi: 10.1016/j.dnarep.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Walisko O, Izsvak Z, Szabo K, Kaufman CD, Herold S, Ivics Z. Sleeping Beauty transposase modulates cell-cycle progression through interaction with Miz-1. Proc Natl Acad Sci USA. 2006;20:4062–4067. doi: 10.1073/pnas.0507683103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kime L, Wright SC. Mad4 is regulated by a transcriptional repressor complex that contains Miz-1 and c-Myc. Biochem J. 2003;20:291–298. doi: 10.1042/BJ20021679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, Moroy T, Bartek J, Massague J, Hanel F, Eilers M. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001;20:392–399. doi: 10.1038/35070076. [DOI] [PubMed] [Google Scholar]
- Wu S, Cetinkaya C, Munoz-Alonso MJ, von der Lehr N, Bahram F, Beuger V, Eilers M, Leon J, Larsson LG. Myc represses differentiation-induced p21CIP1 expression via Miz-1-dependent interaction with the p21 core promoter. Oncogene. 2003;20:351–360. doi: 10.1038/sj.onc.1206145. [DOI] [PubMed] [Google Scholar]
- Peukert K, Staller P, Schneider A, Carmichael G, Hanel F, Eilers M. An alternative pathway for gene regulation by Myc. Embo J. 1997;20:5672–5686. doi: 10.1093/emboj/16.18.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Mitchell RA, Cheng A, Hendrickson EA. Evidence for DNA-PK-dependent and -independent DNA double-strand break repair pathways in mammalian cells as a function of the cell cycle. Mol Cell Biol. 1997;20:1425–1433. doi: 10.1128/mcb.17.3.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. Embo J. 1998;20:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Luenen HG, Plasterk RH. Target site choice of the related transposable elements Tc1 and Tc3 of Caenorhabditis elegans. Nucleic acids Res. 1994;20:262–269. doi: 10.1093/nar/22.3.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare K, Rubin GM. Structures of P transposable elements and their sites of insertion and excision in the Drosophila melanogaster genome. Cell. 1983;20:25–35. doi: 10.1016/0092-8674(83)90133-2. [DOI] [PubMed] [Google Scholar]
- Horie K, Yusa K, Yae K, Odajima J, Fischer SE, Keng VW, Hayakawa T, Mizuno S, Kondoh G, Ijiri T. et al. Characterization of Sleeping Beauty transposition and its application to genetic screening in mice. Mol Cell Biol. 2003;20:9189–9207. doi: 10.1128/MCB.23.24.9189-9207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer SE, Wienholds E, Plasterk RH. Continuous exchange of sequence information between dispersed Tc1 transposons in the Caenorhabditis elegans genome. Genetics. 2003;20:127–134. doi: 10.1093/genetics/164.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tower J, Karpen GH, Craig N, Spradling AC. Preferential transposition of Drosophila P elements to nearby chromosomal sites. Genetics. 1993;20:347–359. doi: 10.1093/genetics/133.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keng VW, Ryan BJ, Wangensteen KJ, Balciunas D, Schmedt C, Ekker SC, Largaespada DA. Efficient transposition of Tol2 in the mouse germline. Genetics. 2009;20:1565–1573. doi: 10.1534/genetics.109.100768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bancroft I, Dean C. Transposition pattern of the maize element Ds in Arabidopsis thaliana. Genetics. 1993;20:1221–1229. doi: 10.1093/genetics/134.4.1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Lin C, Lu D, Ning Z, Cox T, Melvin D, Wang X, Bradley A, Liu P. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc Natl Acad Sci USA. 2008;20:9290–9295. doi: 10.1073/pnas.0801017105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q, Kong J, Stalker J, Bradley A. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis. 2009;20:404–408. doi: 10.1002/dvg.20508. [DOI] [PubMed] [Google Scholar]
- Baus J, Liu L, Heggestad AD, Sanz S, Fletcher BS. Hyperactive Transposase Mutants of the Sleeping Beauty Transposon. Mol Ther. 2005;20:1148–1156. doi: 10.1016/j.ymthe.2005.06.484. [DOI] [PubMed] [Google Scholar]
- Metaxakis A, Oehler S, Klinakis A, Savakis C. Minos as a genetic and genomic tool in Drosophila melanogaster. Genetics. 2005;20:571–581. doi: 10.1534/genetics.105.041848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry C, Hannenhalli S, Leipzig J, Bushman FD. Selection of target sites for mobile DNA integration in the human genome. PLoS Comput Biol. 2006;20:e157. doi: 10.1371/journal.pcbi.0020157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldt B, Miskey C, Staunstrup NH, Gogol-Doring A, Bak RO, Sharma N, Mates L, Izsvak Z, Chen W, Ivics Z, Mikkelsen JG. Comparative genomic integration profiling of sleeping beauty transposons mobilized with high efficacy from integrase-defective lentiviral vectors in primary human cells. Mol Ther. 2011;20:1499–1510. doi: 10.1038/mt.2011.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Guo H, Tammana S, Jung YC, Mellgren E, Bassi P, Cao Q, Tu ZJ, Kim YC, Ekker SC. et al. Gene transfer efficiency and genome-wide integration profiling of Sleeping Beauty, Tol2, and piggyBac transposons in human primary T cells. Mol Ther. 2010;20:1803–1813. doi: 10.1038/mt.2010.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Stern DM, Kiss I, Roote J, Laverty T, Rubin GM. Gene disruptions using P transposable elements: an integral component of the Drosophila genome project. Proc Natl Acad Sci USA. 1995;20:10824–10830. doi: 10.1073/pnas.92.24.10824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellen HJ, Levis RW, He Y, Carlson JW, Evans-Holm M, Bae E, Kim J, Metaxakis A, Savakis C, Schulze KL. et al. The drosophila gene disruption project: progress using transposons with distinctive site-specificities. Genetics. 2011;20:731–743. doi: 10.1534/genetics.111.126995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC, Bellen HJ, Hoskins RA. Drosophila P elements preferentially transpose to replication origins. Proc Natl Acad Sci USA. 2011;20:15948–15953. doi: 10.1073/pnas.1112960108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacAlpine HK, Gordan R, Powell SK, Hartemink AJ, MacAlpine DM. Drosophila ORC localizes to open chromatin and marks sites of cohesin complex loading. Genome Res. 2010;20:201–211. doi: 10.1101/gr.097873.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabundzija I, Irgang M, Mates L, Belay E, Matrai J, Gogol-Doring A, Kawakami K, Chen W, Ruiz P, Chuah MK. et al. Comparative analysis of transposable element vector systems in human cells. Mol Ther. 2010;20:1200–1209. doi: 10.1038/mt.2010.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meir YJ, Weirauch MT, Yang HS, Chung PC, Yu RK, Wu SC. Genome-wide target profiling of piggyBac and Tol2 in HEK 293: pros and cons for gene discovery and gene therapy. BMC Biotechnol. 2011;20:28. doi: 10.1186/1472-6750-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvan DL, Nakazawa Y, Kaja A, Kettlun C, Cooper LJ, Rooney CM, Wilson MH. Genome-wide mapping of PiggyBac transposon integrations in primary human T cells. J Immunother. 2009;20:837–844. doi: 10.1097/CJI.0b013e3181b2914c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meehan AM, Poeschla EM. Chromatin tethering and retroviral integration: recent discoveries and parallels with DNA viruses. Biochim Biophys Acta. 2009;20:182–191. doi: 10.1016/j.bbagrm.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohe AR, Hartl DL. Autoregulation of mariner transposase activity by overproduction and dominant-negative complementation. Mol Biol Evol. 1996;20:549–555. doi: 10.1093/oxfordjournals.molbev.a025615. [DOI] [PubMed] [Google Scholar]
- Chalmers RM, Kleckner N. Tn10/IS10 transposase purification, activation, and in vitro reaction. J Biol Chem. 1994;20:8029–8035. [PubMed] [Google Scholar]
- Lohe AR, Lidholm DA, Hartl DL. Genotypic effects, maternal effects and grand-maternal effects of immobilized derivatives of the transposable element mariner. Genetics. 1995;20:183–192. doi: 10.1093/genetics/140.1.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl DL, Lozovskaya ER, Nurminsky DI, Lohe AR. What restricts the activity of mariner-like transposable elements. Trends Genet. 1997;20:197–201. doi: 10.1016/S0168-9525(97)01087-1. [DOI] [PubMed] [Google Scholar]
- Lohe AR, Sullivan DT, Hartl DL. Subunit interactions in the mariner transposase. Genetics. 1996;20:1087–1095. doi: 10.1093/genetics/144.3.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi LR, Beverley SM. cis and trans factors affecting Mos1 mariner evolution and transposition in vitro, and its potential for functional genomics. Nucleic acids Res. 2000;20:784–790. doi: 10.1093/nar/28.3.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaillet J, Genty M, Cambefort J, Rouault JD, Auge-Gouillou C. Regulation of mariner transposition: the peculiar case of Mos1. PLoS One. 2012;20:e43365. doi: 10.1371/journal.pone.0043365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts AM, Yang Y, Clark KJ, Liu G, Cui Z, Dupuy AJ, Bell JB, Largaespada DA, Hackett PB. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol Ther. 2003;20:108–117. doi: 10.1016/S1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- Balciunas D, Wangensteen KJ, Wilber A, Bell J, Geurts A, Sivasubbu S, Wang X, Hackett PB, Largaespada DA, McIvor RS, Ekker SC. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS genet. 2006;20:e169. doi: 10.1371/journal.pgen.0020169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claeys Bouuaert C, Lipkow K, Andrews SS, Liu D, Chalmers R. The autoregulation of a eukaryotic DNA transposon. Elife. 2013;20:e00668. doi: 10.7554/eLife.00668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng S, Jacobsen SE, Reik W. Epigenetic reprogramming in plant and animal development. Science. 2010;20:622–627. doi: 10.1126/science.1190614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;20(Suppl):245–254. doi: 10.1038/ng1089. [DOI] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;20:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- Xie M, Hong C, Zhang B, Lowdon RF, Xing X, Li D, Zhou X, Lee HJ, Maire CL, Ligon KL. et al. DNA hypomethylation within specific transposable element families associates with tissue-specific enhancer landscape. Nat Genet. 2013;20:836–841. doi: 10.1038/ng.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh CP, Bestor TH. Cytosine methylation and mammalian development. Genes Dev. 1999;20:26–34. doi: 10.1101/gad.13.1.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura A, Yonebayashi S, Watanabe K, Toyama T, Shimada H, Kakutani T. Mobilization of transposons by a mutation abolishing full DNA methylation in Arabidopsis. Nature. 2001;20:212–214. doi: 10.1038/35075612. [DOI] [PubMed] [Google Scholar]
- Gray JV, Petsko GA, Johnston GC, Ringe D, Singer RA, Werner-Washburne M. “Sleeping beauty”: quiescence in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 2004;20:187–206. doi: 10.1128/MMBR.68.2.187-206.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman Z, Gendrel AV, Black M, Vaughn MW, Dedhia N, McCombie WR, Lavine K, Mittal V, May B, Kasschau KD. et al. Role of transposable elements in heterochromatin and epigenetic control. Nature. 2004;20:471–476. doi: 10.1038/nature02651. [DOI] [PubMed] [Google Scholar]
- Brzeski J, Jerzmanowski A. Deficient in DNA methylation 1 (DDM1) defines a novel family of chromatin-remodeling factors. J Biol Chem. 2003;20:823–828. doi: 10.1074/jbc.M209260200. [DOI] [PubMed] [Google Scholar]
- Kato M, Miura A, Bender J, Jacobsen SE, Kakutani T. Role of CG and non-CG methylation in immobilization of transposons in Arabidopsis. Curr Biol. 2003;20:421–426. doi: 10.1016/S0960-9822(03)00106-4. [DOI] [PubMed] [Google Scholar]
- Yusa K, Takeda J, Horie K. Enhancement of Sleeping Beauty transposition by CpG methylation: possible role of heterochromatin formation. Mol Cell Biol. 2004;20:4004–4018. doi: 10.1128/MCB.24.9.4004-4018.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jursch T, Izsvak Z, Ivics Z. Regulation of DNA transposition by CpG methylation and chromatin structure in human cells. Mob DNA. 2013;20:15. doi: 10.1186/1759-8753-4-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda R, Kokubu C, Yusa K, Keng VW, Horie K, Takeda J. Sleeping beauty transposase has an affinity for heterochromatin conformation. Mol Cell Biol. 2007;20:1665–1676. doi: 10.1128/MCB.01500-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga A, Hori H. Homogeneity in the structure of the medaka fish transposable element Tol2. Genet Res. 1999;20:7–14. doi: 10.1017/S0016672398003620. [DOI] [PubMed] [Google Scholar]
- Iida A, Shimada A, Shima A, Takamatsu N, Hori H, Takeuchi K, Koga A. Targeted reduction of the DNA methylation level with 5-azacytidine promotes excision of the medaka fish Tol2 transposable element. Genet Res. 2006;20:187–193. doi: 10.1017/S0016672306008184. [DOI] [PubMed] [Google Scholar]
- Kitamura K, Hashida SN, Mikami T, Kishima Y. Position effect of the excision frequency of the Antirrhinum transposon Tam3: implications for the degree of position-dependent methylation in the ends of the element. Plant Mol Biol. 2001;20:475–490. doi: 10.1023/A:1011892003996. [DOI] [PubMed] [Google Scholar]
- Park CW, Park J, Kren BT, Steer CJ. Sleeping Beauty transposition in the mouse genome is associated with changes in DNA methylation at the site of insertion. Genomics. 2006;20:204–213. doi: 10.1016/j.ygeno.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Park CW, Kren BT, Largaespada DA, Steer CJ. DNA methylation of Sleeping Beauty with transposition into the mouse genome. Genes Cells. 2005;20:763–776. doi: 10.1111/j.1365-2443.2005.00875.x. [DOI] [PubMed] [Google Scholar]
- Dalsgaard T, Moldt B, Sharma N, Wolf G, Schmitz A, Pedersen FS, Mikkelsen JG. Shielding of sleeping beauty DNA transposon-delivered transgene cassettes by heterologous insulators in early embryonal cells. Mol Ther. 2009;20:121–130. doi: 10.1038/mt.2008.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrison BS, Yant SR, Mikkelsen JG, Kay MA. Postintegrative gene silencing within the Sleeping Beauty transposition system. Mol and cell biol. 2007;20:8824–8833. doi: 10.1128/MCB.00498-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Hollensen AK, Bak RO, Staunstrup NH, Schroder LD, Mikkelsen JG. The impact of cHS4 insulators on DNA transposon vector mobilization and silencing in retinal pigment epithelium cells. PLoS One. 2012;20:e48421. doi: 10.1371/journal.pone.0048421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadloun A, Le Gras S, Jost B, Ziegler-Birling C, Takahashi H, Gorab E, Carninci P, Torres-Padilla ME. Chromatin signatures and retrotransposon profiling in mouse embryos reveal regulation of LINE-1 by RNA. Nat Struct Mol Biol. 2013;20:332–338. doi: 10.1038/nsmb.2495. [DOI] [PubMed] [Google Scholar]
- Sijen T, Plasterk RH. Transposon silencing in the Caenorhabditis elegans germ line by natural RNAi. Nature. 2003;20:310–314. doi: 10.1038/nature02107. [DOI] [PubMed] [Google Scholar]
- Ketting RF, Haverkamp TH, Van Luenen HG, Plasterk RH. Mut-7 of C. elegans, required for transposon silencing and RNA interference, is a homolog of Werner syndrome helicase and RNaseD. Cell. 1999;20:133–141. doi: 10.1016/S0092-8674(00)81645-1. [DOI] [PubMed] [Google Scholar]
- Tabara H, Sarkissian M, Kelly WG, Fleenor J, Grishok A, Timmons L, Fire A, Mello CC. The rde-1 gene, RNA interference, and transposon silencing in C. elegans. Cell. 1999;20:123–132. doi: 10.1016/S0092-8674(00)81644-X. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Takeda A, Tsukiyama T, Mise K, Okuno T, Sasaki H, Minami N, Imai H. Identification and characterization of two novel classes of small RNAs in the mouse germline: retrotransposon-derived siRNAs in oocytes and germline small RNAs in testes. Genes Dev. 2006;20:1732–1743. doi: 10.1101/gad.1425706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulou C, Muljo SA, Kung AL, Ganesan S, Drapkin R, Jenuwein T, Livingston DM, Rajewsky K. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;20:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda P, Stein P, Anger M, Bernstein E, Hannon GJ, Schultz RM. RNAi and expression of retrotransposons MuERV-L and IAP in preimplantation mouse embryos. Dev biol. 2004;20:276–285. doi: 10.1016/j.ydbio.2004.01.028. [DOI] [PubMed] [Google Scholar]
- Zamore PD. RNA silencing: genomic defence with a slice of pi. Nature. 2007;20:864–865. doi: 10.1038/446864a. [DOI] [PubMed] [Google Scholar]
- Brennecke J, Aravin AA, Stark A, Dus M, Kellis M, Sachidanandam R, Hannon GJ. Discrete small RNA-generating loci as master regulators of transposon activity in Drosophila. Cell. 2007;20:1089–1103. doi: 10.1016/j.cell.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Houwing S, Kamminga LM, Berezikov E, Cronembold D, Girard A, van den Elst H, Filippov DV, Blaser H, Raz E, Moens CB. et al. A role for Piwi and piRNAs in germ cell maintenance and transposon silencing in Zebrafish. Cell. 2007;20:69–82. doi: 10.1016/j.cell.2007.03.026. [DOI] [PubMed] [Google Scholar]
- Aravin AA, Sachidanandam R, Bourc’his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell. 2008;20:785–799. doi: 10.1016/j.molcel.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozhkov NV, Hammell M, Hannon GJ. Multiple roles for Piwi in silencing Drosophila transposons. Genes Dev. 2013;20:400–412. doi: 10.1101/gad.209767.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sienski G, Donertas D, Brennecke J. Transcriptional silencing of transposons by Piwi and maelstrom and its impact on chromatin state and gene expression. Cell. 2012;20:964–980. doi: 10.1016/j.cell.2012.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XA, Yin H, Sweeney S, Raha D, Snyder M, Lin H. A major epigenetic programming mechanism guided by piRNAs. Dev Cell. 2013;20:502–516. doi: 10.1016/j.devcel.2013.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Kazazian HH Jr. L1 retrotransposition is suppressed by endogenously encoded small interfering RNAs in human cultured cells. Nat struct & mol biol. 2006;20:763–771. doi: 10.1038/nsmb1141. [DOI] [PubMed] [Google Scholar]
- Soifer HS, Zaragoza A, Peyvan M, Behlke MA, Rossi JJ. A potential role for RNA interference in controlling the activity of the human LINE-1 retrotransposon. Nucleic acids Res. 2005;20:846–856. doi: 10.1093/nar/gki223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldt B, Yant SR, Andersen PR, Kay MA, Mikkelsen JG. Cis-acting gene regulatory activities in the terminal regions of sleeping beauty DNA transposon-based vectors. Hum gene therapy. 2007;20:1193–1204. doi: 10.1089/hum.2007.099. [DOI] [PubMed] [Google Scholar]
- Rauschhuber C, Ehrhardt A. RNA interference is responsible for reduction of transgene expression after Sleeping Beauty transposase mediated somatic integration. PLoS One. 2012;20:e35389. doi: 10.1371/journal.pone.0035389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons MJ, Bucholz LM. Transposase titration in Drosophila melanogaster: a model of cytotype in the P-M system of hybrid dysgenesis. Proc Natl Acad Sci USA. 1985;20:8119–8123. doi: 10.1073/pnas.82.23.8119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schouten GJ, Van Luenen HG, Verra NC, Valerio D, Plasterk RH. Transposon Tc1 of the nematode Caenorhabditis elegans jumps in human cells. Nucleic Acids Res. 1998;20:3013–3017. doi: 10.1093/nar/26.12.3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell. 2005;20:473–483. doi: 10.1016/j.cell.2005.07.013. [DOI] [PubMed] [Google Scholar]
- Izsvak Z, Hackett PB, Cooper LJ, Ivics Z. Translating Sleeping Beauty transposition into cellular therapies: victories and challenges. Bioessays. 2010;20:756–767. doi: 10.1002/bies.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Izsvak Z, Minter A, Hackett PB. Identification of functional domains and evolution of Tc1-like transposable elements. Proc Natl Acad Sci USA. 1996;20:5008–5013. doi: 10.1073/pnas.93.10.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yant SR, Park J, Huang Y, Mikkelsen JG, Kay MA. Mutational analysis of the N-terminal DNA-binding domain of sleeping beauty transposase: critical residues for DNA binding and hyperactivity in mammalian cells. Mol Cell Biol. 2004;20:9239–9247. doi: 10.1128/MCB.24.20.9239-9247.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staunstrup NH, Moldt B, Mates L, Villesen P, Jakobsen M, Ivics Z, Izsvak Z, Mikkelsen JG. Hybrid lentivirus-transposon vectors with a random integration profile in human cells. Mol Ther. 2009;20:1205–1214. doi: 10.1038/mt.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacoste A, Berenshteyn F, Brivanlou AH. An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell. 2009;20:332–342. doi: 10.1016/j.stem.2009.07.011. [DOI] [PubMed] [Google Scholar]
- Cadinanos J, Bradley A. Generation of an inducible and optimized piggyBac transposon system. Nucleic acids Res. 2007;20:e87. doi: 10.1093/nar/gkm446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilber A, Wangensteen KJ, Chen Y, Zhuo L, Frandsen JL, Bell JB, Chen ZJ, Ekker SC, McIvor RS, Wang X. Messenger RNA as a source of transposase for sleeping beauty transposon-mediated correction of hereditary tyrosinemia type I. Mol Ther. 2007. [DOI] [PubMed]
- Yant SR, Ehrhardt A, Mikkelsen JG, Meuse L, Pham T, Kay MA. Transposition from a gutless adeno-transposon vector stabilizes transgene expression in vivo. Nat Biotechnol. 2002;20:999–1005. doi: 10.1038/nbt738. [DOI] [PubMed] [Google Scholar]
- Hausl M, Zhang W, Voigtlander R, Muther N, Rauschhuber C, Ehrhardt A. Development of adenovirus hybrid vectors for Sleeping Beauty transposition in large mammals. Curr Gene Ther. 2011;20:363–374. doi: 10.2174/156652311797415890. [DOI] [PubMed] [Google Scholar]
- Hausl MA, Zhang W, Muther N, Rauschhuber C, Franck HG, Merricks EP, Nichols TC, Kay MA, Ehrhardt A. Hyperactive sleeping beauty transposase enables persistent phenotypic correction in mice and a canine model for hemophilia B. Mol Ther. 2010;20:1896–1906. doi: 10.1038/mt.2010.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers WJ, Mastrangelo MA, Howard DF, Southerland HA, Maguire-Zeiss KA, Federoff HJ. Neuronal precursor-restricted transduction via in utero CNS gene delivery of a novel bipartite HSV amplicon/transposase hybrid vector. Mol Ther. 2006;20:580–588. doi: 10.1016/j.ymthe.2005.11.011. [DOI] [PubMed] [Google Scholar]
- Zhang W, Solanki M, Muther N, Ebel M, Wang J, Sun C, Izsvak Z, Ehrhardt A. Hybrid adeno-associated viral vectors utilizing transposase-mediated somatic integration for stable transgene expression in human cells. PLoS One. 2013;20:e76771. doi: 10.1371/journal.pone.0076771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vink CA, Gaspar HB, Gabriel R, Schmidt M, McIvor RS, Thrasher AJ, Qasim W. Sleeping beauty transposition from nonintegrating lentivirus. Mol Ther. 2009;20:1197–1204. doi: 10.1038/mt.2009.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galla M, Schambach A, Falk CS, Maetzig T, Kuehle J, Lange K, Zychlinski D, Heinz N, Brugman MH, Gohring G. et al. Avoiding cytotoxicity of transposases by dose-controlled mRNA delivery. Nucleic Acids Res. 2011;20:7147–7160. doi: 10.1093/nar/gkr384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Bak RO, Krogh LB, Staunstrup NH, Moldt B, Corydon TJ, Schrøder LD, Mikkelsen JG. DNA transposition by protein transduction of the piggyBac transposase from lentiviral Gag precursors. Nucleic acids Res. 2013. doi: 10.1093/nar/gkt1163. [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- Bak RO, Mikkelsen JG. Mobilization of DNA transposable elements from lentiviral vectors. Mob Genet Elem. 2011;20:139–144. doi: 10.4161/mge.1.2.17062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staunstrup NH, Mikkelsen JG. Integrase-defective lentiviral vectors–a stage for nonviral integration machineries. Curr Gene Ther. 2011;20:350–362. doi: 10.2174/156652311797415881. [DOI] [PubMed] [Google Scholar]
- Aronovich EL, Bell JB, Belur LR, Gunther R, Koniar B, Erickson DC, Schachern PA, Matise I, McIvor RS, Whitley CB, Hackett PB. Prolonged expression of a lysosomal enzyme in mouse liver after Sleeping Beauty transposon-mediated gene delivery: implications for non-viral gene therapy of mucopolysaccharidoses. J Gene Med. 2007;20:403–415. doi: 10.1002/jgm.1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montini E, Held PK, Noll M, Morcinek N, Al-Dhalimy M, Finegold M, Yant SR, Kay MA, Grompe M. In vivo correction of murine tyrosinemia type I by DNA-mediated transposition. Mol Ther. 2002;20:759–769. doi: 10.1006/mthe.2002.0812. [DOI] [PubMed] [Google Scholar]
- Wilber A, Frandsen JL, Geurts JL, Largaespada DA, Hackett PB, McIvor RS. RNA as a source of transposase for sleeping beauty-mediated gene insertion and expression in somatic cells and tissues. Mol Ther. 2006;20:625–630. doi: 10.1016/j.ymthe.2005.10.014. [DOI] [PubMed] [Google Scholar]
- Sharma N, Cai Y, Bak RO, Jakobsen MR, Schroder LD, Mikkelsen JG. Efficient sleeping beauty DNA transposition from DNA minicircles. Mol Ther Nucleic Acids. 2013;20:e74. doi: 10.1038/mtna.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Manuri PR, Olivares S, Dara N, Dawson MJ, Huls H, Hackett PB, Kohn DB, Shpall EJ, Champlin RE, Cooper LJ. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;20:2961–2971. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Guo H, Kang J, Choi S, Zhou TC, Tammana S, Lees CJ, Li ZZ, Milone M, Levine BL. et al. Sleeping Beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol Ther. 2008;20:580–589. doi: 10.1038/sj.mt.6300404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wilber A, McIvor RS, Zhou X. DNA transposons for modification of human primary T lymphocytes. Methods in mol biol. 2009;20:115–126. doi: 10.1007/978-1-59745-409-4_9. [DOI] [PubMed] [Google Scholar]
- Jin Z, Maiti S, Huls H, Singh H, Olivares S, Mates L, Izsvak Z, Ivics Z, Lee DA, Champlin RE, Cooper LJ. The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 2011;20:849–856. doi: 10.1038/gt.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollis RP, Nightingale SJ, Wang X, Pepper KA, Yu XJ, Barsky L, Crooks GM, Kohn DB. Stable gene transfer to human CD34(+) hematopoietic cells using the Sleeping Beauty transposon. Exp Hematol. 2006;20:1333–1343. doi: 10.1016/j.exphem.2006.05.023. [DOI] [PubMed] [Google Scholar]
- Sumiyoshi T, Holt NG, Hollis RP, Ge S, Cannon PM, Crooks GM, Kohn DB. Stable transgene expression in primitive human CD34+ hematopoietic stem/progenitor cells, using the Sleeping Beauty transposon system. Hum Gene Ther. 2009;20:1607–1626. doi: 10.1089/hum.2009.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue X, Huang X, Nodland SE, Mates L, Ma L, Izsvak Z, Ivics Z, LeBien TW, McIvor RS, Wagner JE, Zhou X. Stable gene transfer and expression in cord blood-derived CD34+ hematopoietic stem and progenitor cells by a hyperactive Sleeping Beauty transposon system. Blood. 2009;20:1319–1330. doi: 10.1182/blood-2009-03-210005. [DOI] [PubMed] [Google Scholar]
- Wilber A, Linehan JL, Tian X, Woll PS, Morris JK, Belur LR, McIvor RS, Kaufman DS. Efficient and stable transgene expression in human embryonic stem cells using transposon-mediated gene transfer. Stem Cells. 2007;20:2919–2927. doi: 10.1634/stemcells.2007-0026. [DOI] [PubMed] [Google Scholar]
- Orban TI, Apati A, Nemeth A, Varga N, Krizsik V, Schamberger A, Szebenyi K, Erdei Z, Varady G, Karaszi E. et al. Applying a “double-feature” promoter to identify cardiomyocytes differentiated from human embryonic stem cells following transposon-based gene delivery. Stem Cells. 2009;20:1077–1087. doi: 10.1002/stem.45. [DOI] [PubMed] [Google Scholar]
- Williams DA. Sleeping beauty vector system moves toward human trials in the United States. Mol ther: the j of the Am Soc of Gene Ther. 2008;20:1515–1516. doi: 10.1038/mt.2008.169. [DOI] [PubMed] [Google Scholar]
- Kebriaei P, Huls H, Jena B, Munsell M, Jackson R, Lee DA, Hackett PB, Rondon G, Shpall E, Champlin RE, Cooper LJ. Infusing CD19-directed T cells to augment disease control in patients undergoing autologous hematopoietic stem-cell transplantation for advanced B-lymphoid malignancies. Hum Gene Ther. 2012;20:444–450. doi: 10.1089/hum.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, Huls H, Miller JC, Kebriaei P, Rabinovitch B. et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;20:5697–5705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]